
Abstract
Immune checkpoint inhibitors have become the mainstay of treatment for hepatocellular carcinoma (HCC). However, they are ineffective in some cases. Previous studies have reported that genetic alterations in oncogenic pathways such as Wnt/β-catenin are the important triggers in HCC for primary refractoriness. T-cell exhaustion has been reported in various tumors and is likely to play a prominent role in the emergence of HCC due to chronic inflammation and cirrhosis-associated immune dysfunction. Immunosuppressive cells including regulatory T-cells and tumor-associated macrophages infiltrating the tumor are associated with hyperprogressive disease in the early stages of immune checkpoint inhibitor treatment. In addition, stellate cells and tumor-associated fibroblasts create an abundant desmoplastic environment by producing extracellular matrix. This strongly contributes to epithelial to mesenchymal transition via signaling activities including transforming growth factor beta, Wnt/β-catenin, and Hippo pathway. The abundant desmoplastic environment has been demonstrated in pancreatic ductal adenocarcinoma and cholangiocarcinoma to suppress cytotoxic T-cell infiltration, PD-L1 expression, and neoantigen expression, resulting in a highly immunosuppressive niche. It is possible that a similar immunosuppressive environment is created in HCC with advanced fibrosis in the background liver. Although sufficient understanding is required for the establishment of immune therapies of HCC, further investigations are still required in this field:
Keywords
Introduction
Cancer immunotherapies that target immunosuppressive checkpoint receptors, including cytotoxic T-lymphocyte-associated protein-4 (CTLA-4), programmed cell death 1 (PD-1), and programmed death ligand 1 (PD-L1), have changed the landscape of anticancer immunotherapy in patients with various types of cancer, including malignant melanoma, non-small cell lung cancer (NSCLC), colorectal cancer, esophageal gastric cancer, and hepatocellular carcinoma (HCC).1–5 Immune checkpoint inhibitors (ICIs) have a long-lasting response and improve overall survival (OS). 6 Systemic therapies for unresectable HCC have improved significantly, and molecular targeted agents (MTAs) such as sorafenib7,8 and lenvatinib 9 as well as combination therapies, including anti-PD-1/PD-L1 checkpoint blockade and vascular endothelial growth factor (VEGF) antibody, have been approved as first-line drugs.10–12 However, 82–85% of patients are resistant to monotherapy with anti-PD-1/PD-L1,11,13 there is an urgent need to clarify the characteristics of tumors that can be treated using this method.
Although much of our understanding of ICI resistance exists due to research on malignant melanoma and NSCLC, analysis of the tumor microenvironment in cases of HCC is also progressing. Tumor-infiltrating cytotoxic T-lymphocytes (TILs) and expression levels of PD-L1 in tumor cells are assumed to be involved in primary resistance to anti-PD-1/PD-L1 antibody, similar to other types of cancer. 14 Constitutive activation of Wnt/β-catenin signaling also has a negative impact on anti-PD-1/PD-L1 antibody treatment in cases of HCC, 15 suggesting that certain driver gene mutations significantly affect the tumor microenvironment.
It has been reported that hyperprogressive disease (HPD) induced by tumor-infiltrating regulatory T (Treg) cells and macrophage is important in HCC because they induce unexpected refractoriness to ICIs 16 ; microenvironment prone to HPD may also be a consequence of overadaptation to antitumor immunity in HCC. 17 It is also possible that characteristics of tumor microenvironment are regulated by the balance of active and suppressive immune cells, such as infiltration of CD8+ T-cell and Treg, tumor-associated macrophage (TAM), myeloid-derived suppressor cell (MDSC), and other stromal cells, which may be affected by the activation of oncogenic pathway unique to cancer type. 18 Other studies have shown that chronic viral infections precede HCC emergence cause exhaustion and the loss of function of CD8+ T-cells, which is associated with the expression of PD-1 inhibitory receptors.19–22 T-cell exhaustion was also reportedly influenced by nutritional competition between tumor cells and effector T-lymphocytes. 23 In pancreatic ductal adenocarcinoma and intrahepatic cholangiocarcinoma, fibrosis caused by cancer-associated fibroblasts (CAFs), stellate cells, or myofibroblast plays an immunosuppressive role in the tumor microenvironment. These tolerances due to adaptation of the immunosuppressive niche can be termed non-genetic refractoriness.
On the other hand, the strong relationship between liver carcinogenesis and advanced fibrosis of background liver is well described.24,25 Regardless of the etiology of liver injury, the emergence of HCC is strongly affected by the persistent damage of parenchymal cells and subsequent fibrosis due to the interaction between damaged hepatocytes and stromal cells; thus, approximately 80–90% of HCC cases have underlying fibrosis.24,25 However, the role of liver fibrosis in the promotion of HCC has not been clarified, especially in the context of the establishment of an immunological tumor microenvironment and how it affects the efficacy of ICI treatment.
HCC has some unique aspects compared with other cancers: diversity among genes mutated in HCC due to multicentric carcinogenesis, chronic viral infections which are strongly involved in carcinogenesis, severe fibrosis characterizing the liver background, and a variety of metabolites of glucose, amino acids, and lipids that could be involved in carcinogenesis.
However, the establishment of immunosuppressive niche in HCC remains largely unknown, and few studies have examined the direct and indirect effects of background organ fibrosis on the therapeutic efficacy of PD-1/PD-L1 antibodies thus far. This review aims to give an overview for the factors that influence the immunosuppressive microenvironment of HCC and to provide a new perspective on the immunosuppressive niche involving liver fibrosis.
Body
The Role of Cancer-immunity Cycle in Acquired Resistance
It is widely known that the cancer-immunity cycle helps to determine the refractoriness to cancer immunotherapy; the failure of this cycle may induce an immunosuppressive microenvironment. It is defined as a series of functional stepwise events that proceed and expand iteratively. 26 This cycle can be divided into the following seven steps: Step 1. Release of neoantigens formed during oncogenesis. Step 2. Cancer antigen presentation on major histocompatibility complex (MHC) class I and II molecules. Step 3. Priming and activation of effector T-cells; Step 4. Trafficking of T-cells to tumors. Step 5. Infiltration of cytotoxic T-cells into tumors. Step 6. Specific recognition of cancer cells by T-cells. Step 7. Killing of cancer cells, which releases additional tumor-associated antigens; this cycle expands iteratively.
In cancer patients, the cancer-immunity cycle does not proceed optimally because tumors employ multiple strategies to attenuate the attack of cytotoxic T-cells for survival. 27 This immune escape mechanism can partially be explained by tumor-specific somatic mutations that affect the antigenicity of cancer cells and the induction of immune suppressive cytokines, chemokines, growth factors, and metabolites.
Effects of Oncogenic Pathways Implicated in HCC on the Immunological Microenvironment
Several mutations in the genes involved in cellular signaling pathways in which constitutive activation and inactivation play a critical role in carcinogenesis, including in that of HCC, have been reported,28–31 such as the telomerase reverse transcriptase signaling pathway,31–33 tumor protein p53 (TP53) pathway, 29 retinoblastoma pathway, 29 Wnt/β-catenin pathway,34,35 phosphatidylinositol 3 kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) pathway, 29 RAS/mitogen-activated protein kinase (MAPK) pathway, Janus kinase (JAK)/signal transducers and activators of transcription (STAT) signaling pathways, 36 chromatin remodeling pathways involving AT-rich interactive domain-containing protein 1A, 29 and the Hedgehog pathway. It has been reported that TP53 was rarely positive in the Wnt/β-catenin signaling-related marker-positive HCC. 37 In addition, nearly half of the cases of HCC with high Treg cell infiltration are positive for TP53. 18
Even though cancer is caused by the accumulation of multiple genomic abnormalities, oncogene addiction is defined as the heightened dependency of tumor cells on signal transduction and protein production due to the activated single oncogene or loss of single tumor suppressor. The involvement of these driver and passenger mutations in primary resistance to cancer immunotherapy will be explained 17 (Fig. 1).
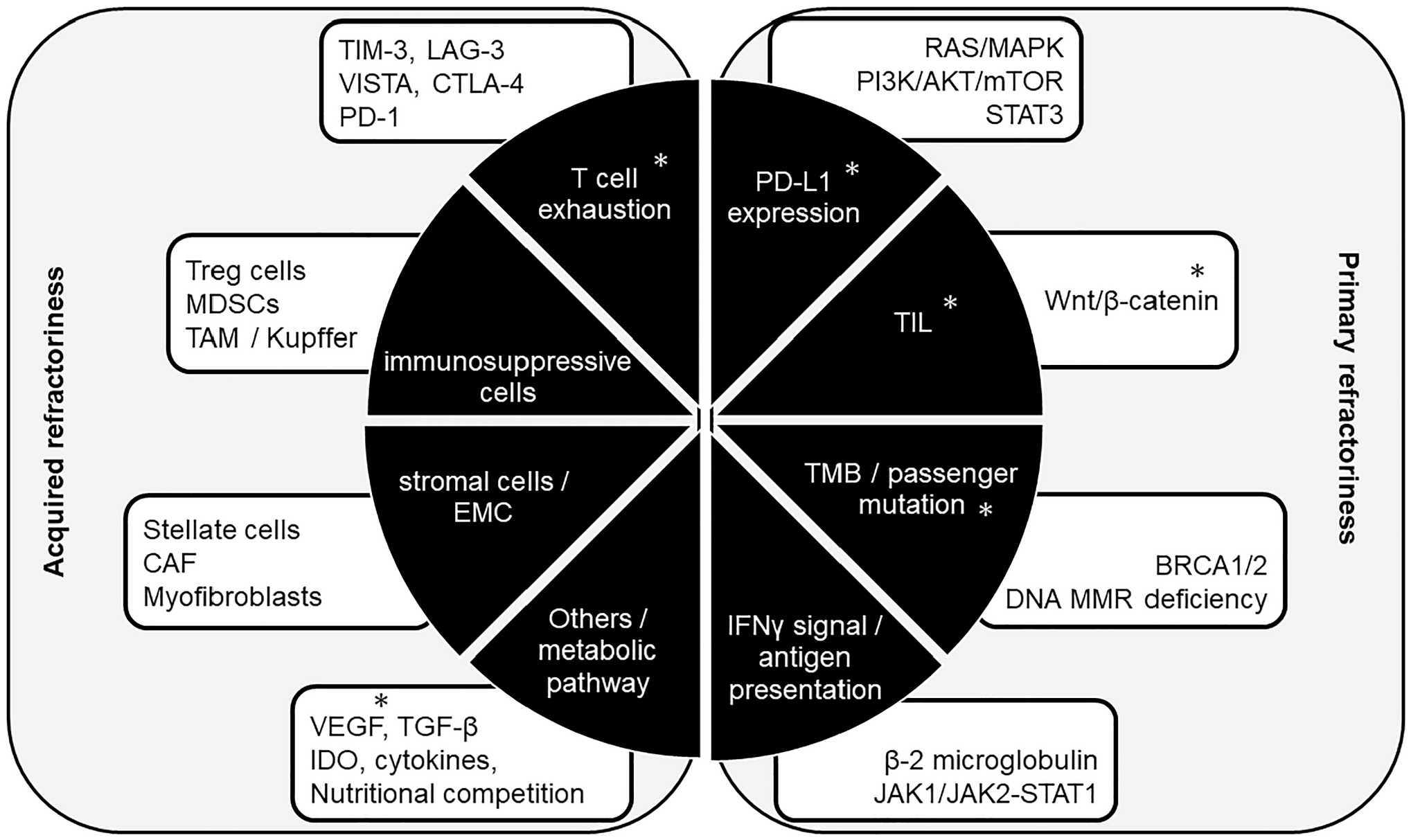
Refractoriness to cancer immunotherapy of anti-PD-1/PD-L1 antibody. Tumor factors associated with primary resistance to anti-PD-1/PD-L1 antibodies are listed. Genetic mutations that may affect the primary resistance are described. Particularly important factors are PD-L1 expression, TILs, and TMB. Other genetic mutations related to antigen presentation and IFN-γ signaling pathway have also been reported to be induced anti-PD-1/PD-L1 resistance. Acquired resistance usually arises from adaptation over time and creates an immunosuppressive niche in the tumor microenvironment. T-cell exhaustion may lead to cross talk between severe fibrosis and immunosuppressive niches. High amount of infiltrating regulatory T (Treg) cells and tumor-associated macrophages (TAMs) play an important role in hyperprogression of tumor during anti-PD-1/PD-L1 treatment. Stromal cells including stellate cells and cancer-associated fibroblasts (CAF) create an abundant desmoplastic environment by producing extracellular matrix. Factors identified in HCC are given an “*.” Abbreviations: PD-1, programmed cell death 1; PD-L1, programmed cell death ligand 1; TILs, tumor-infiltrating cytotoxic T-lymphocytes; TMB, tumor mutational burden; IFN, interferon; MAPK, mitogen-activated protein kinase; PI3K, phosphatidylinositol 3 kinase; mTOR, mammalian target of rapamycin; STAT, signal transducers and activators of transcription; DNA, deoxyribonucleic acid; MMR, mismatch repair; BRCA, breast cancer susceptibility gene; HLA, human leukocyte antigen; TIM-3, T-cell immunoglobulin and mucin domain-containing molecule-3; LAG-3, lymphocyte activation gene-3; VISTA, V-domain Ig suppressor of T-cell activation; CTLA-4, cytotoxic T-lymphocyte-associated protein-4; MDSCs, myeloid-derived suppressor cells; VEGF, vascular endothelial growth factor; TGF-β, transforming growth factor beta; IDO, indoleamine 2,3-dioxygenase; JAK, Janus kinase; HCC, hepatocellular carcinoma.
Driver Mutations Involved in PD-L1 Expression
RAS/MAPK signaling and STAT3 signaling pathways induce PD-L1 expression in NSCLC cells. 38 Although it is difficult to determine whether the increase in PD-L1 expression is attributed entirely to the activation of these cellular signals pathways in human cancer, low expression levels of PD-L1 on tumor cells39–44 or PD-1 on antigen-specific CD8+ T-cells 45 are likely to result in poor therapeutic effects of ICIs on cancers. PD-L1 expression on tumor cells and/or tumor-infiltrating immune cells is reportedly associated with an increase in OS in cases of advanced NSCLC, 46 esophageal squamous cell carcinoma,43,44,47 malignant melanoma, 48 and unresectable HCC. 49 In cases of gastrointestinal cancer, such as HCC, the combined positive scores, including expression levels of PD-L1 in tumor cells and antigen-presenting cells, also correlate to the response to anti-PD-1/PD-L1 antibody.
Driver Mutations Correlating With the Presence of TILs
Second, Wnt/β-catenin signal activating mutations reportedly inhibit TILs in malignant melanoma 50 and urothelial bladder cancer. 51 Activation of the canonical Wnt/β-catenin signaling pathway decreases chemokine (c-c motif) ligand 4 (CCL4) expression in malignant melanoma 50 and CCL5 expression in HCC. A reduction in the recruitment of CD103+ dendritic cells decreases the infiltration rate of CD8+ T-lymphocytes into tumor tissues,52,53 where the establishment of an “immune cold tumor” with an absence of antitumor cytotoxic T-cells and primary resistance to ICIs occurs.15,53,54 The number of TILs correlates with the OS and the efficacy of anti-PD-1/PD-L1 therapy in various cancers.42,55- 57 It has been reported that Wnt/β-catenin mutations are present in approximately 11–37% of HCC cases. 58 Harding et al. 15 reported that activating mutation of the Wnt/β-catenin pathway is associated with progressive disease and exhibit a short median progression-free survival in patients with HCC who were treated with ICIs.
In HCC, an intense T-cell infiltrate is sometimes observed within the tumor tissues, where intercellular adhesion molecule-1 and vascular adhesion protein-1, reportedly, are strongly expressed on the tumor endothelium. 59 HCCs with a large number of TILs are reportedly positively correlated with hepatitis C virus (HCV) but negatively correlated with hepatitis B virus (HBV) infection; generally, HCC related to HCV infection exhibits reduced vascular invasion, a low recurrence rate, and a high 5-year survival rate. 60
Hugo et al. 61 identified factors that may influence innate sensitivity or resistance to anti-PD-1 therapy, which were aptly named innate anti-PD-1 resistance signatures. The MAPK signaling cascade, which often contains mutations in cases of melanoma, is associated with T-cell exclusion and subsequent resistance to PD-1/PD-L1 blockade. Inhibition of this cascade has been shown to improve CD8+ T-cell infiltration within tumors and sensitize these cells to anti-PD-1 antibodies. 62
Passenger Mutations and Tumor Mutational Burden
A low tumor mutational burden (TMB) causes reduced responsiveness to cancer immunotherapy due to the lack of antigenicity of cancer to the immune response. Solid tumors with high TMB exhibit an active T helper 1 (Th1)/cytotoxic T-lymphocyte (CTL) immune response and CD8+ T-cell infiltration into tumor cells, probably due to the recognition of a variety of neoantigens.63,64 Microsatellite instability (MSI)-high solid tumors had a significantly higher TMB than microsatellite stable or MSI-low solid tumors.65,66 Recently, it was reported that loss-of-function mutations in BRCA1/2 genes, which play a crucial role in homologous recombination during deoxyribonucleic acid (DNA) repair, also lead to a high TMB, and are associated with the positive outcome of treatment with ICIs. 67
Following malignant melanoma and NSCLC, a high degree of somatic mutation has been reported in esophageal adenocarcinoma, gastric cancer, and colorectal cancer. 68 It has been reported that ICI efficacy is correlated with a higher proportion of DNA repair pathway mutations, TMB, and neoantigen burden.69–74 Tumors with few somatic mutations, such as pancreatic and prostate cancers, are generally more resistant to anti-PD-1/PD-L1 antibody treatment.75,76 ICI treatment is more effective in cases of malignant melanoma and NSCLC with a high TMB.71,72 Regarding HCC, some studies reported an association between a large number of passenger mutations and the efficacy of ICI. 77 However, the TMB value as an objective biomarker has not been explored in HCC cases. TMB is also the expected biomarker for the treatment of ICIs; however, the cutoff values vary among the types of cancer, and it has not yet been decided whether to evaluate the values using whole genome sequencing or cancer gene panel inspection, which should also be assessed in cases of HCC.
Gene Mutations Impairing Antigen Presentation and Interferon-γ Signaling
β-2 microglobulin is the beta-chain of MHC class I molecules, which contain one immunoglobulin-like domain. Mutations in β-2 microglobulin impair antigen presentation and induce a poor immune response.78–80 Whole-exome sequencing of patients with malignant melanoma 81 and NSCLC revealed β-2 microglobulin mutations which impaired antigen presentation of MHC class I expression. In patients with NSCLC, acquired homozygous loss of β-2 microglobulin caused a lack of MHC class I expression on the cell surface of the tumor and a matched patient-derived xenograft. 82 Another study revealed that the loss of expression of the chromosome 6 haplotype encoding the MHC class I molecule on CD8+ cells targeting the mutated KRAS gene provided a direct mechanism of acquired resistance in metastatic colorectal cancer. 83 These reports indicate that MHC class I antigen processing and presentation machinery disruption can mediate escape from ICIs. Mutations in β-2 microglobulin have been reported in patients with HCC.84–87 In addition, β-2 microglobulin expression differed significantly between patients with and without HBV infection. 87 In contrast, exome-derived mutated human leukocyte antigen class I ligands are rarely present in HCC cases. 88
Alterations in the JAK-STAT pathway that affect interferon (IFN)-γ signaling89,90 are also promising predictive biomarkers of ICI responsiveness. Loss-of-function mutations in the JAK1/JAK2-STAT1 signaling pathway induce the loss of sensitivity of cancer cells to INF-γ, resulting in a poor response to ICIs. Activation of JAK signaling to STAT is a characteristic of HCC progression via mutations associated with response to drug sensitivity in HCC. 91 The acquired resistance to anti-PD-1 antibody therapy in patients with malignant melanoma is associated with defects in the JAK1 or JAK2-(STAT)1 signaling pathway. IFN-γ secreted by CD8+ T-cell causes the upregulation of PD-L1 expression in tumors, increased antigen production, and the increased release of T-cell-attracting chemokines.42,92,93 mRNA expression profile analyses revealed that T-cell-inflamed microenvironment contained IFN-γ-responsive genes is a useful pan-tumor determinant of anti PD-1/PD-L1 therapy response. 94
Additional Signaling and Metabolic Pathways That Affect the Tumor Microenvironment
The tumor microenvironment is defined as the tumor cell population in a complex mixture of surrounding stroma, which include fibroblasts, endothelial cells, pericytes, immune cells, and proteins such as extracellular matrix (ECM) elements, cytokines, chemokines, and enzymes, where the cross talk of cells in the tumor is mediated by several humoral factors, including transforming growth factor beta (TGF-β), indoleamine 2,3-dioxygenase (IDO), VEGF, arginase, and several cytokines, namely CCL5, CCL17, CCL22, CXCL8, and CXCL12. 95
It has been reported that the constitutive activation of the MAPK signaling pathway causes the production of the immunosuppressive cytokines VEGF and interleukin 8 (IL-8), which inhibit T-cell recruitment to and functionality in tumors.96,97 IL-6 reportedly affects tumor cells directly through the JAK2/STAT3, RAS/MAPK, and PI3K/Akt pathways 98 and suppresses CD4+ T-cells, inhibiting their differentiation to Th1 cells 99 ; therefore, it is considered a poor prognostic factor.100,101 Loss of the tumor suppressor phosphatase and tensin homolog, which induces the activation of PI3K signaling, has also been associated with increased VEGF production, reduced CD8+ T-cell infiltration of tumors, and subsequent resistance to anti-PD-1 antibody treatment. 102
Other evidence has suggested that metabolic changes to the tumor microenvironment also affected the composition and function of tumor cells and immune cells, including CD8+ T-cells. The local environment of the tumor is hypoxic, with a low pH due to a high lactate concentration and low nutrition. Nutrient competition for glucose between cancer cells and effector T-lymphocytes may influence tumor progression. 103 Cham and Gajewski 104 stated that optimal induction of IFN-γ transcription is a glucose-dependent process and that glucose deprivation strongly inhibits multiple key gene expression events, including IFN-γ gene expression and effector functions of CD8+ T-cells.23,104,105 Effector immune cells, which are nutritionally deprived due to competition with tumor cells, gradually lose their function due to the reduction of mTOR activity, glycolytic capacity, and IFN-γ production.
Hung et al. 106 reported that the tumor methionine recycling pathway plays a central role as the driver of T-cell exhaustion in HCC. Arginine also affects the antitumor activity of CD8+ T-cells. 107 Arginine deficiency due to the rapid consumption of MDSCs and macrophages results in the protein biosynthesis-mediated cellular exhaustion of T-cells, which causes T-cells to lose their antitumor activity.108–110 Glutamine 111 and tryptophan112,113 reportedly affect the tumor microenvironment. IDO is a crucial rate-limiting enzyme that catalyzes the conversion of tryptophan downstream of the kynurenine pathway 114 and has been reported to recruit immunosuppressive cells. 113 Kakazu et al. 115 reported that amino acid imbalance in the plasma of patients with advanced cirrhosis impaired the production of IFN-γ by peripheral blood mononuclear cells, resulting in the dysfunction of dendritic cells.
Chang et al. 116 reported that anti-PD-1/PD-L1 and anti-CTLA-4 antibodies restore glycolytic capacity in tumors, permitting T-cell glycolysis and IFN-γ production and, therefore, their effector function. Furthermore, they found that blocking PD-L1 directly on tumors reduces glycolysis by inhibiting AKT/mTOR pathway activity.
Cellular Factors Involved in the Immune Suppressive Microenvironment
The cell subsets that make up the tumor microenvironment are diverse and can be categorized as cytotoxic or immunosuppressive. The antitumor immune response induced by immune checkpoint inhibition is mainly mediated by the T-cell lineage, which recognizes and activates antigens presented on the MHC of antigen-presenting cells via T-cell receptors expressed on their surface. There are two subtypes of T-cells: CD8+ T-cells and CD4+ T-cells. Some cytotoxic T-cells cause T-cell exhaustion as their adaptability changes over time. The influx of immunosuppressive cell populations such as Treg cells, MDSCs, and M2 macrophages into the tumor microenvironment can attenuate the effector function of T-cells in patients receiving PD-1 blockade therapy. 117 Treg cells, which are a subtype of CD4 + T-cells, can suppress the activity and proliferation of CD8+ T-cells, causing acquired ICI resistance. 118 Similarly, MDSCs represent a heterogeneous population of immature myeloid cells that can result in acquired resistance to ICIs via the recruitment of immunosuppressive cells, upregulation of immune checkpoints, angiogenesis, and hypoxia.119,120
CD8+ T-cell (CTL/Killer T-cell) and T-cell Exhaustion
TILs can be broadly classified as effector memory T-cells, central memory T-cells, or resident memory T-cells.121–123 TILs often have the phenotype of resident memory T-cells, including CD69 and CD103 expression cells. The expression of phenotypic marker of resident memory T-cells124,125 may be correlated with the prognosis of malignant tumors and the response to anti-PD-1/PD-L1 antibody therapy. On the other hand, a high density of rare cells such as CD8+ FoxP3+ PD-1-low T-cells are closely associated with long-term survival after anti-PD-1 blockade in malignant melanoma. 126
Prolonged antigen exposure and sustained inflammatory stimulations have been considered as potential mechanisms that may induce effector T-cells into an exhausted state. 127 Exhaustion of TILs has also been observed in HCC cases, and is correlated with poor clinical outcomes. 128 Exhausted CD8+ T-cells and Treg cells are enriched and potentially clonally expanded in HCC, and deep single-cell RNA sequencing revealed that one gene of the 11 subsets, laylin, is upregulated on activated CD8+ T-cells and Treg cells and represses the CD8+ T-cell functions in vitro. 129
Multiple repressive immune checkpoint receptors, such as CTLA-4, T-cell immunoglobulin and mucin domain-containing molecule-3 (TIM-3), lymphocyte activation gene-3 (LAG-3), PD-1, and the V-domain Ig suppressor of T-cell activation, are all markers for T-cell exhaustion. These receptors could also be associated with impaired responses to anti-PD-1/PD-L1 antibodies. 130 LAG-3 is structurally homologous to CD4 and binds competitively to MHC class II, which causes reduced efficacy of MHC class II-mediated antigen presentation. Johnson et al. 131 found that patients with malignant melanoma and NSCLC with acquired resistance to anti-PD-1 therapy demonstrated LAG-3 upregulation in their tumors. TIM-3 is an inhibitory receptor expressed on IFN-γ-producing CD4+, Th1, and CD8+ T-cells. PD-1 and TIM-3 coexpression has been reported in severely exhausted T-cells. 132 In immunocompetent mouse models of lung adenocarcinoma, TIM-3 was reportedly upregulated following the administration of anti-PD-1 antibody and anti-TIM-3 antibody, which results in a survival advantage. 56 These studies suggest that a lack of memory T-cells and exhausted T-cells might cause acquired resistance to anti-PD-1 antibodies. 133
To date, the details of the molecular mechanism that leads to the T-cell exhaustion is unknown. Recent studies report that nuclear factor of activated T-cells (NFATs) 134 and their downstream Nr4a receptors135,136 are linked to CD8+ T-cell exhaustion (Fig. 2). Nr4a is a Src family kinase-dependent receptor. 137 Chen et al. 138 found that Nr4a binds to the enhancer region of the PD-1 gene and increases the expression of PD-1 and TIM-3. Nr4a deficiency also causes the downregulation of PD-1 and TIM-3, which are inhibitory receptors. 138 Seo et al.139,140 indicated that Tox and Nr4a transcription factors are critical for the transcriptional program of CD8+ T-cell exhaustion downstream of NFAT.
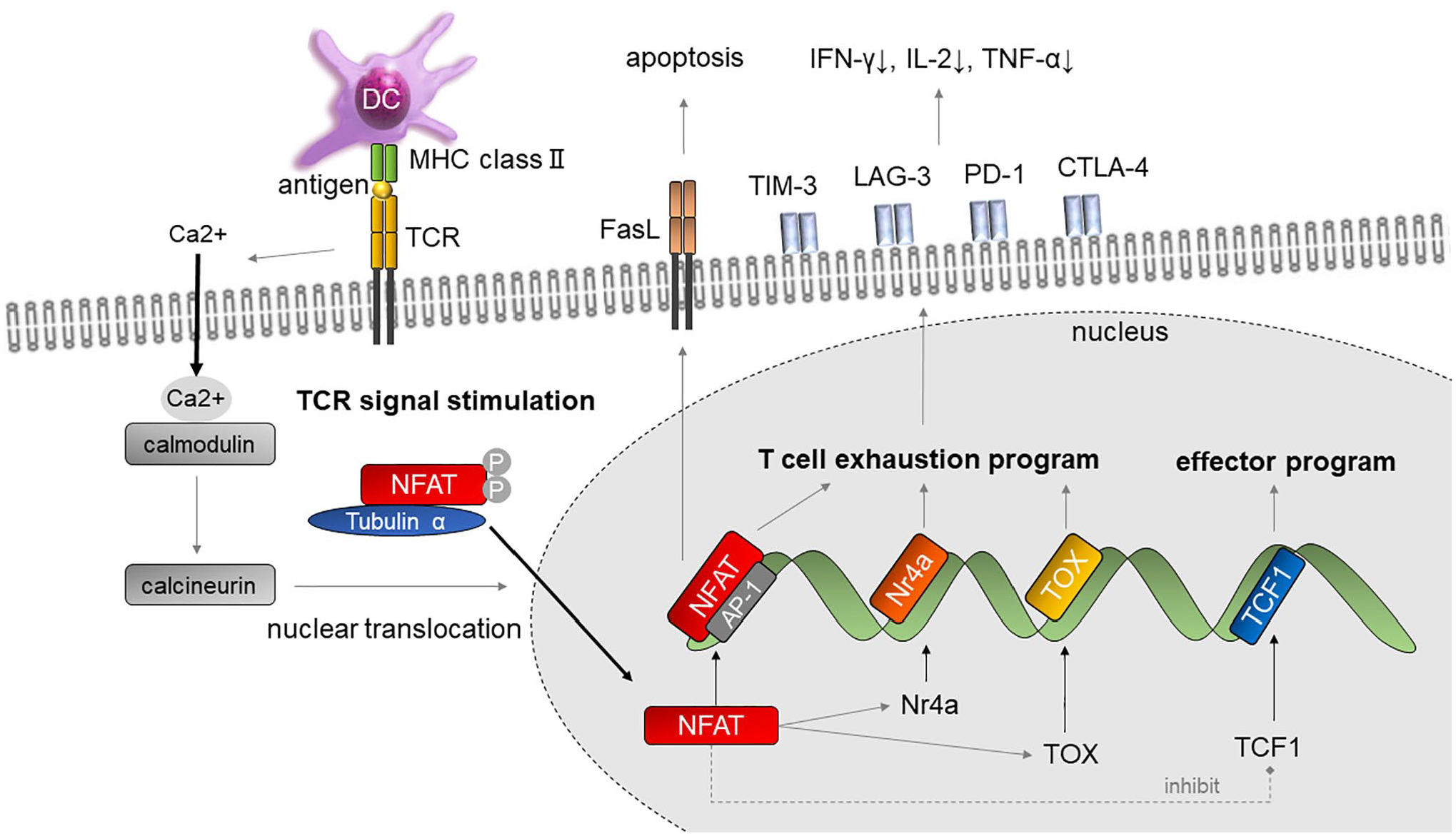
The molecular mechanism of T-cell exhaustion. Persistent antigen exposure due to chronic infection or tumor cells stimulates TCR signaling pathway. Influx of Ca2+ into the cell causes calmodulin to become saturated, which activates calcineurin. Dephosphorylation of NFAT induces nuclear translocation. NFAT binds to the recognition sequence of AP-1 and induces transcription of NFAT, Nr4a, and TOX. Transcription of TCF-1 is repressed. Inhibitory receptors, which are T-cell exhaustion markers, appear and the production of IFN-γ and IL-2 is decreased. T-cell activation also utilizes MAPK activation to promote transcriptional activation through AP-1, which synergizes with NFAT factors to promote inducible gene expression. Abbreviations: PD-1, programmed cell death 1; TIM-3, T-cell immunoglobulin and mucin domain-containing molecule-3; LAG-3, lymphocyte activation gene-3; CTLA-4, cytotoxic T-lymphocyte-associated protein-4; TCR, T-cell receptor; IFN, interferon; IL-2, interleukin 2; TNF-α, tumor necrosis factor alpha; DC, dendritic cell; NFAT, nuclear factor of activated T-cells; AP-1, activator protein-1; MHC, major histocompatibility complex; MAPK, mitogen-activated protein kinase.
Philips et al. 141 reported that T-cell dysfunction markers such as CD38 and CD101 are expressed on TILs in patients who do not respond to anti-PD-1/PD-L1 blockade. Tumor-infiltrating exhausted CD8+ T-cells are characterized by increasing dysfunction which is driven by chromatin remodeling and epigenetic modifications, and eventually reach a state of fixed epigenetic dysfunction in which their chromatin is rendered inaccessible and is resistant to further remodeling and reinvigoration. 141 If the tumor burden remains high, exhausted CD8+ T-cells fail to eradicate the tumor cells, eventually becoming re-exhausted and resistant to reinvigoration through anti-PD-1 therapy. 142
CD4+ T-cells
The mechanism of dysfunction of CD4+ T-cells in the immune microenvironment is not fully understood. Three types of CD4+ helper T (Th) cells, namely Th1, Th2, and Th17 cells, expressed high levels of glucose transporter 1 on their surface and were highly glycolytic; the differentiation and function of CD4+ T-cells may be dependent on the glycolytic system. 143 In addition, the activity of arylamide acetylase 1, a rate-limiting enzyme for fatty acid synthesis, plays an important role in the differentiation of Th17 cells.144,145 These findings indicate that CD4+ Th cells are likely dysfunctional under the specific nutritional condition.
IL-6 has a direct effect on tumor cells through the activation of JAK2/STAT3, Ras/MAPK, and PI3K/Akt signaling pathways, which influence the division and survival of tumor cells, neoangiogenesis, and inflammation.146–148 IL-6 has also reportedly suppressed CD4+ T-cells, inhibiting their differentiation to Th1 cells.98,99 The direct action of IL-6 has been proven by STAT3 activation in CD4+ T-cells and myeloid cells, and by the fact that IL-6 blockade and specific inhibition of soluble interleukin 6 receptor significantly improved the differentiation of CD4+ T-cells into IFN-γ-producing effector Th1 cells in tumor-bearing mice. 99
Some tumor-derived molecules or extracellular vesicles also likely influence the differentiation of CD4+ T-cells. It has been reported that CD4+ T-cells that have come into contact with tumor cell-released autophagosomes directly suppress CD4+ and CD8+ effector T-cells via an antitumor IFN-γ response and promote tumor growth and metastasis. 149 In addition, TGF-β, prostaglandin E2, and tumor-derived exosomes have been proven to act as immunosuppressive molecules for Th cells.150,151
Intrahepatic alpha-fetoprotein-specific CD4+ T-cell responses are rarely detectable in HCC. 152 However, the proportion of alpha-fetoprotein-specific CD4+ Th1 cells in patients with HCC increased significantly during and after embolization of tumor vessels. 153 Thus, the results may provide a rationale for the combining of embolization of tumor vessels with immunotherapy in the treatment for HCC patients. In fact, the combination of ICI and transcatheter arterial chemoembolization154,155 or ablation 156 has been reported to improve the response rate.
Treg Cells
Treg cells can be divided into two groups: “natural Treg cells” which occur spontaneously in the thymus and “induced Treg cells” that originate from peripheral tissues when stimulated by cytokines, such as TGF-β, retinoic acid, and IL-2. Tumor hypoxia promotes the recruitment of Treg cells by inducing the expression of the chemokine CCL28. 157
A master transcription factor called forkhead box P3 (FoxP3) plays an essential role in the induction of differentiation and largely defines the phenotypic and functional characteristics of Treg cells. Nr4a receptors directly activate the gene promoter that encodes the FoxP3. 158 Treg cell function is mediated by an analogous cooperative complex of NFAT with FoxP3. 159 FoxP3 activates the mitochondrial electron transfer system and shifts Treg metabolism toward oxidative phosphorylation (OXPHOS) and fatty acid oxidation (FAO). 160 It has been reported that Treg cells express low levels of Glut1 and have high FAO 143 and OXPHOS rates.161,162 Treg cells may adapt to the tumor microenvironment by suppressing the glycolytic system and increasing the rates of FAO and OXPHOS.
T-cells can be classified into naive Treg (CD45RA+ FoxP3− CD4+ T-cells) and effector Treg cells (CD45RA− FoxP3+ CD4+ T-cells) depending on the presence or absence of CD45RA expression. Effector Tregs suppress antigen recognition using tumor-killing cells, such as CTLs, and inhibit tumor cell clearance by suppressing immune responses through the production of IL-10, TGF-β, and the involvement of CTLA-4.163–165 In fact, increased FoxP3+ Treg cell infiltration into the cancer microenvironment has been observed in a variety of cancers, which allows cancer cells to persist due to the suppression of antitumor immune responses. Increased Treg accumulation in tumors is associated with poor prognosis. 166 On the other hand, numerous animal studies have shown a reduction in tumor size after depleting Treg cells or rendering them functionally defective.167–169
HPD is a severe progression pattern of cancer characterized by an acute acceleration in tumor growth following immunotherapy, and Treg cells reportedly play a pivotal role in HPD.16,170–173 Two mechanisms have been proposed as possible causes of HPD: The first involves the Fc region of anti-PD-1 antibody which causes M2-like differentiation of TAMs, cultivating immunosuppressive conditions in the tumor. 174 The second involves the infiltration of Treg cells into tumors, due to their high level of PD-1 expression, which modulates Treg activity. 175 While blocking the inhibitory signals of PD-1 increases Treg cell activation, Treg cells may still require IL-2 for robust proliferation. 176 One may expect HPD caused by Treg cell expansion to have a shorter time to progression and to be prone to metastasis. The presence of higher Treg levels in the peripheral blood of non-responsive patients suggests that a higher frequency of effector T-cells with PD-1 expression relative to PD-1+ Treg cells in the tumor predicts a positive response to therapy.45,177 Moreover, increased immunosuppression by Treg cells by blocking PD-1 signal has been shown to accelerate tumor development in HPD mouse models. 178 In addition, the growing list of HPD biomarkers can be stratified by the presence of Treg cells in tumors. 179 Although the copy number instability score in cell-free DNA is reportedly useful when predicting the onset of HPD, larger prospective studies are needed. 180
MDSCs
MDSCs are a heterogeneous population of immature and immunosuppressive myeloid cells that regulate immune responses during cancer, infection, chronic inflammation, and traumatic stress. Activating a complex network of signaling pathways induces the differentiation of immature myeloid cells in the bone marrow into MDSCs.181–183 MDSCs are induced in the tumor microenvironment to suppress effector T-cell immune responses by inducing T-cell exhaustion and dysfunction in the tumor microenvironment. 183 It has been reported that STAT3 regulates arginase-I in the MDSCs in cancer tissues. 184
MDSCs have been identified in various cancers, and an increase in the number of MDSCs in the tumor microenvironment has been observed in solid tumors, including those of urothelial carcinoma, glioblastoma, pancreatic adenocarcinoma, and breast cancer. 183 The proportion of CD14+ HLA-DR−MDSCs is higher in patients with HCC, which promotes tumor angiogenesis through VEGF production in HCC tissues. The presence of MDSCs in patients with HCC is correlated with tumor progression and prognosis, but not with the degree of liver fibrosis and inflammation. 185
The nutrient metabolism of MDSCs is dependent on OXPHOS and FAO, and when their metabolism becomes glycolytic-dominant due to the presence of metformin, MDSCs decrease in number and lose their original function. 186 Anti-VEGF inhibitors are thought to reduce the number of MDSCs and Treg cells and IL-1β and IL-6 were identified as biomarkers to evaluate their effects. 187 The number of MDSCs that are present may predict the patient’s response to ICI therapy. 188
TAMs
There is a significant correlation between chronic inflammation and cancer initiation because macrophages play a central role as inflammatory mediators through the production of molecules such as IL-6, TNF-α, and IFN-γ. Deng et al. 189 found that a deficiency of the anti-inflammatory transcription factor, STAT3, in macrophages caused a chronic inflammatory response in the colon, which was sufficient to cause invasive adenocarcinoma. In the tumor microenvironment, macrophages are abundant and present at all stages of tumor progression. Although the origin of macrophages in tumor tissues is still uncertain, MDSCs and Kupffer cells can differentiate into TAMs, most researches on macrophage polarization are simply implemented in vitro and rarely distinguish Kupffer cells from monocyte-derived macrophages. 190 Colony stimulating factor (CSF) 1 is the major lineage regulator of most macrophage populations, which are derived from the yolk sac or bone marrow 191 and high concentrations of CSF1 in tumor tissues are associated with poor prognosis. 192 VEGF-A also recruits macrophage progenitors that differentiate into TAMs due to the influence of IL-4, 193 indicating that VEGF-A and CSF1 can recruit macrophages to tumors independently. 193 Roland et al. 187 demonstrated that the selective inhibition of the binding of VEGF to VEGFR2 can control tumor growth effectively and inhibit the infiltration of suppressive immune cells, such as MDSCs, Treg cells, and macrophages, while increasing the mature dendritic cell fraction.
In primary tumors, macrophages can stimulate angiogenesis and enhance tumor cell invasion and motility. Macrophages infiltrate the premetastatic site and promote tumor cell extravasation, survival, and persistent growth. 194 TAMs express an array of effector molecules that inhibit antitumor immune responses. 194 TAMs are also immunosuppressive, and express the ligand receptors for PD-1 and CTLA-4, which suppress cytotoxic CD8+ T-cells, natural killer T-cells, and natural killer cells upon activation. The single-cell RNA sequencing analysis revealed that TAMs suppress tumor T-cell infiltration in HBV-associated HCC. 195 TAMs secrete IL-10 and TGF-β, which suppress CD4+ and CD8+ effector T-lymphocytes directly or indirectly and induce Treg cells and CCL4, CCL5, CCL20, and CCL22, which recruit natural Treg cells. 196 As a result, infiltration of TAMs is proportional to CD4+ CD25+ FoxP3+ Treg cells and inversely proportional to CD8+ cytotoxic T-cells and natural killer cells in the HCC environment.
Macrophages form a continuous spectrum, including classically activated macrophages, which produce high IL-12 and low IL-10 levels and are associated with killing tumor cells. In contrast, activated M2 macrophages are characterized by the production of high concentrations of IL-10 and tumor progression.197,198 In tumor microenvironment, some agents including CSF1, Wnt/β-catenin, IL8, and IL17 can promote TAMs transform to M2 phenotype. TAMs are also related to the mechanisms of anti-PD-1-induced HPD in that the Fc region of the anti-PD-1 antibody induces M2-like differentiation of TAMs, which cultivates immunosuppressive conditions in the tumor. 174 The HCC microenvironment stimulates M2 polarization, which is a characteristic phenotype of TAMs. It has been reported that MTAs such as lenvatinib modulate cancer immunity in the HCC tumor microenvironment by reducing the number of TAMs and, when combined with PD-1 blockade, exhibit enhanced antitumor activity via the IFN signaling pathway. 199
CAFs
CAFs are essential cellular components of the HCC microenvironment stroma and are characterized by the increased expression of fibroblast activation protein, platelet-derived growth factor receptor b, and prolyl-4-hydroxylase. 103 Although CAFs are derived from hepatic stellate cells (HSCs), their exact origin is unknown. HSCs and CAFs are key players in liver fibrogenesis and hepatocarcinogenesis, respectively. It is important to differentiate between CAFs and HSCs in the premalignant and tumor microenvironments of HCC. This is difficult in practice due to the lack of specific markers to aid the identification of these cells, although fibroblast activation protein, 200 alpha-smooth muscle actin (α-SMA), 201 and fibroblast-specific protein-1202 are considered major markers of CAFs.
CAFs cross talk with cancer cells to influence tumor growth and invasion 203 (Fig. 3). First, CAFs activate myofibroblasts that remodel the ECM by producing ECM components and secreting matrix metalloproteinases (MMPs). 204 In other solid tumors such as breast carcinoma, CAFs reportedly induce further neovascularization and reduce patients’ survival rates 205 due to the recruitment of endothelial progenitor cells, lymphocytes, macrophages, and MDSCs via stromal cell-derived factor 1, which promotes tumor cell proliferation, invasion, and neoangiogenesis.206,207
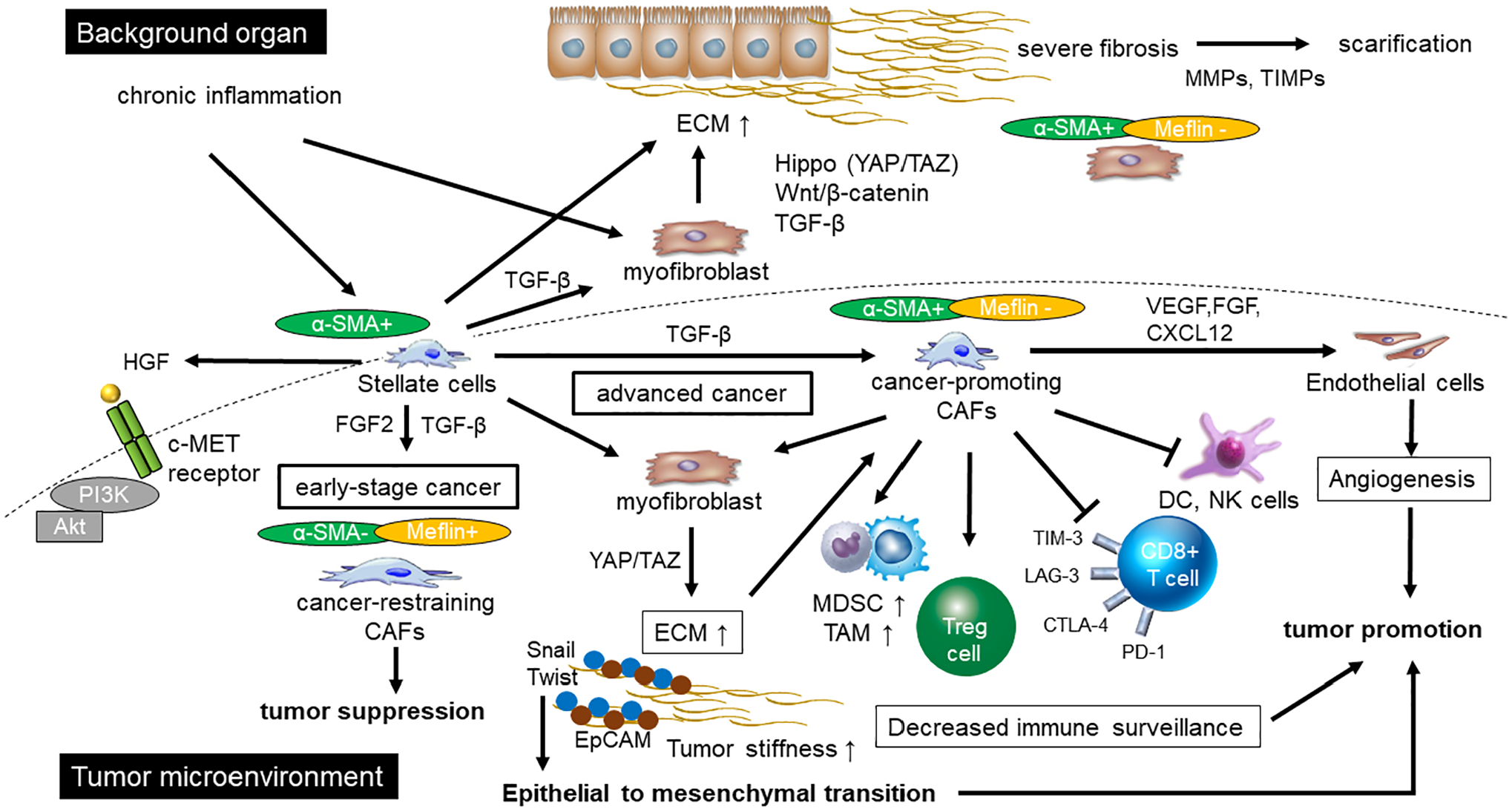
Organ fibrosis and intratumor fibrosis. HSCs, CAFs, and fibroblasts play a central role in fibrogenesis in organs and intratumor. HSCs produce TGF-β, which promotes parenchymal organ fibrosis and CAFs in the tumor environment. Early tumors contain many α-SMA low-negative cancer-restraining CAFs, and TGF-β acts in a tumor suppressive manner. In advanced tumors, the number of cancer-promoting CAFs increases, and TGF-β behaves in a tumor-promoting manner. Cancer-promoting CAFs create a treatment-resistant niche: They promote angiogenesis, suppress immune surveillance, and produce ECM, which hardens the tumor. In scirrhous type HCC, Snail and Twist and stem cell maker are highly expressed around the fibrous stroma and are involved in Epithelial to mesenchymal transition. In the background organs, HSCs and myofibroblast play a central role in promoting fibrosis via TGF-β, Wnt/β-catenin, and YAP/TAZ signaling pathways. α-SMA-positive myofibroblasts lead to more fibrosis and scarring. Abbreviations: PD-1, programmed cell death 1; TIM-3, T-cell immunoglobulin and mucin domain-containing molecule-3; LAG-3, lymphocyte activation gene-3; CTLA-4, cytotoxic T-lymphocyte-associated protein-4; PI3K, phosphatidylinositol 3 kinase; DC, dendritic cell; NK cells, natural killer cells; MDSCs, myeloid-derived suppressor cells; TAM, tumor-associated macrophage; CAF, cancer-associated fibroblast; HSC, hepatic stellate cell; Treg cell, regulatory T-cell; VEGF, vascular endothelial growth factor; FGF, fibroblast growth factor; CXCL12, C-X-C motif chemokine 12; MMP, matrix metalloproteinase; TIMP, tissue inhibitor of matrix metalloproteinase; ECM, extracellular matrix; TGF-β, transforming growth factor beta; α-SMA, α-smooth muscle actin; HGF, hepatocyte growth factor; YAP, yes-associated protein; TAZ, transcriptional coactivator with PDZ-binding motif; EpCAM, epithelial cell adhesion molecule; HCC, hepatocellular carcinoma.
Next, CAFs promote immunosuppression by inhibiting cellular functions. CAFs suppress the activity of natural killer cells by downregulating PVR/CD155, a ligand for activating natural killer receptors in endometrial carcinoma. 208 CAFs derived from HCC inhibit natural killer cell function by secreting the immunosuppressive molecules prostaglandin E2 and IDO, thereby creating favorable conditions for tumor progression. 209 CAFs derived from HCC also have immune regulatory effects on dendritic cells. CAFs derived from HCC recruit regulatory DCs, which are characterized by high expression of CTLA-4 and educate them to acquire a tolerogenic phenotype through IL-6-mediated STAT3 activation. 210 In pancreatic ductal adenocarcinoma, CAFs contribute as important players in immune evasion through the following mechanism: high expression of the PD-L1 and PD-L2 on their surface, promotion of the expression of TIM-3, PD-1, CTLA-4, and LAG-3 in proliferating T-cells, and inhibition of the proliferation of CD8+ T-cells and their function. 211
Recent studies in pancreatic ductal adenocarcinoma models indicate that CAFs are functionally and molecularly heterogeneous. 212 CAFs can act as positive or negative regulators of carcinogenesis; in other words, there are not only cancer-promoting CAFs but also cancer-restraining CAFs, which is partly determined by the presence or absence of Meflin in pancreatic cancers. 213 Another in vitro study with malignant melanoma shows that CAFs induced by fibroblast growth factor 2 and TGF-β from endothelial cells inhibited cancer progression 214 (Fig. 3).
Baglieri et al. 203 suggested that CAFs involved with HCC are positive regulators of cancer. To date, several mechanisms have been proposed that support the mechanism of tumor growth in the liver 203 : CAFs create a stiff microenvironment due to the reorganization and increase in the density of the ECM, which in turn affects tumorigenesis. CAFs secrete inflammatory cytokines and other factors which may promote tumor growth, angiogenesis, and epithelial-to-mesenchymal transition. CAF-derived cytokines/chemokines regulate tumor immune escape and promote tumor growth and metastasis 215 and targeted CAF therapy could restore a tumor immune response.
HSCs
In cases of chronic liver injury, liver fibrosis is involved in a process characterized by the activation of HSCs into myofibroblasts that produce ECM components such as collagen.216–218 HSCs and myofibroblasts are the main producers of ECM components in the liver and play an important role in HCC progression through the release of hepatocyte growth factor (HGF) and TGF-β, and the induction of MDSCs and Treg cells219–221 (Fig. 3).
Activated HSCs can produce cytokines that promote cancer proliferation and migration, such as TGF-β, which is a central regulator in all stages of disease progression from initial liver injury through inflammation and fibrosis to cirrhosis and HCC. Transcription factor SNAI1 (Snail) and Twist is one of the key inducers of epithelial to mesenchymal transition that is a transdifferentiation program associated with tumor metastasis (Fig. 3). It has been reported that Snail and Twist are a target of TGF-β, which is an essential factor in liver cancer formation, 222 and Yang et al. 223 have revealed that overexpression of Snail and/or Twist correlated with the poor prognosis of HCC. In scirrhous type of HCC, Snail is mainly detected in the peripheral cells of tumor nests, next to the fibrous stroma, with coexpression of cytokeratin 19/epithelial cell adhesion molecule, stem cell marker. 224 Overexpression of TGF-β-signaling molecules was significantly higher in scirrhous type of HCCs than in other type of HCCs or intrahepatic cholangiocarcinoma, indicating TGF-β-signaling is a potential key regulator for epithelial to mesenchymal transition induction in scirrhous type of HCC. 224 From all of this, HSCs were recently postulated as a component of the prometastatic liver microenvironment. 225 This is probably due to the induction of nuclear β-catenin accumulation and transcribe number of migration marker genes including MMP-2 and MMP-9 in neoplastic hepatocytes. 226
TGF-β has a bipartite effect on tumors because it acts as a tumor suppressor in the early stages of liver damage and regeneration, whereas it acts as a tumor promoter during cancer progression. 227 This is consistent with previous reports in pancreatic cancer 213 that TGF-β has a tumor suppressive effect in the early stage of tumor progression due to the presence of many cancer-restraining CAFs, and a tumor-promoting effect in the advanced stage due to the increase of cancer-promoting CAFs (Fig. 3).
HSCs also produce HGF, a potent mitogen for epithelial cells. The c-met proto-oncogene encodes the tyrosine kinase receptor for HGF, which stimulates the migration of HCC cells through tyrosine phosphorylation of c-met via the activation of PI3K. 228 Intrahepatic and extrahepatic metastasis is characteristic of HCC, and c-Met is reportedly correlated with tumor differentiation 229 or the propensity to metastasize. 230 Some clinical studies have shown that the expression of HGF and its receptor c-met is elevated in cirrhotic tissues and in 80% of HCC cases. Tyrosine kinase and PI3K inhibitors prevent the migration of HCC cells. 228 A different study showed that HGF secreted from HSCs might reduce HCC sensitization to chemotherapeutic agents by promoting epithelial to mesenchymal transition and cancer stem cell-like properties through the HGF/c-Met pathway. 231
It has been reported that HSCs decrease immune surveillance: The tumor cells of patients with HCC secrete amphiregulin, a pro-oncogenic epidermal growth factor receptor (EGFR) ligand that activates HSCs232,233 and is involved in the induction of Treg cells. 234 HSCs inhibit lymphocyte infiltration in tumors and the spleens of tumor-bearing mice, induce apoptosis of infiltrating mononuclear cells, and increase the number of infiltrating FoxP3+ immunosuppressive Tregs, resulting in reduced immune surveillance.235,236
Immune Suppressive Effect of Liver Fibrosis in the Microenvironment of HCC
Local Fibrosis Within the Tumor and Resistance to ICIs
Pancreatic ductal adenocarcinoma is one of the most difficult cancers to treat with ICIs because of tumors with low expression of PD-L1, low CD8+ T-lymphocyte infiltration, and low expression of tumor-specific neoantigen.14,237 Pancreatic ductal adenocarcinoma is characterized by an abundant desmoplastic environment, which is the accumulation of fibrosis such as collagen in cancerous tissue, and pancreatic stellate cell and CAFs have also been shown to be involved in cancer stromal formation. 238 The accumulation of fibrosis and hypovascular tumor microenvironment contributes to ICI resistance, and CAFs may play an important role in immune evasion in this situation.239,240 CAFs inhibit the proliferation of CD8+ T-cells and their function through the promotion of the expression of immunosuppressive markers. 211 However, studies on pancreatic ductal adenocarcinoma have demonstrated that there are not only cancer-promoting CAFs (α-SMA positive/Meflin low to negative) but also cancer-restraining CAFs (α-SMA low to negative/Meflin positive). 239 Complex signaling pathways are involved in the differentiation into these two opposites (Fig. 3).
Intrahepatic cholangiocarcinoma is similar to pancreatic ductal adenocarcinoma, which is characterized by the presence of abundant and hypovascularized desmoplastic stroma,241,242 in which α-SMA+ CAFs and ECM are abundant. In addition, the stroma contains inflammatory cells, in particular, a high number of macrophages. Recent in vivo and in vitro studies revealed a pivotal role of α-SMA+ CAFs and with the sustained activation of the EGFR in intrahepatic cholangiocarcinoma promotion and resistance to chemotherapy.242–244 This would be similar to a tumor-promoting role of a desmoplastic tumor microenvironment in breast, lung, colon, prostate, and pancreas. 202 CAFs also express tenascin-C, an ECM protein with tumor-promoting roles in many cancers,245,246 and tenascin-C expression correlates with poor prognosis in intrahepatic cholangiocarcinoma patients. 214 It has been reported that increased stiffness of tumor also results in activation of β-catenin signaling during the development of colon cancer 247 and suppression of PTEN expression in breast cancer. 248 Not only physical signals such as stiffness but also receptor-independent signaling pathways such as β-catenin, Hippo, and PTEN are also important contributors to hepatocarcinogenesis.16,134,135
Scirrhous type is a rare variant of HCC (−5%), 249 and Wnt/β-catenin mutations are not observed in scirrhous type of HCC. 250 Scirrhous type of HCC expresses both cholangiocarcinoma-like and stem-cell-like genomic traits. In addition, overexpression of TGF-β signaling and epithelial to mesenchymal transition-related genes, Snail and Twist was observed, which may contribute to the aggressive behavior of scirrhous type of HCC. 224 Further studies are needed to determine the role of local intratumor fibrosis and the relevance of these receptor-independent mechanosensitive signaling pathways such as stiffness for the development and progression of HCC.
Immunosuppressive Effects of Severe Fibrosis of the Background Liver in the Tumor Microenvironment
Organ fibrosis is characterized by the central role of macrophages and activation and nuclear regulation of signaling pathways such as TGF-β, Wnt/β-catenin, and YAP/TAZ, which are located downstream of the Hippo signaling pathway. 251 The loss of Hippo signaling in the liver activates YAP/TAZ, STAT3, Wnt/β-catenin, and Notch signaling, and abnormal activation of these downstream pathways has been shown to lead to liver carcinogenesis. 252 The most characteristic feature of liver fibrosis is the accumulation of collagens, predominantly type I collagen, resulting in a 2- to 5-fold increase of total collagen content in the cirrhotic liver. 253 Specific genomic profiles of HCC appear to influence the activation of the stromal compartment as demonstrated by lower fibrosis in patients with Wnt/β-catenin-mutated HCCs.254,255
Clinical evidence supports an important role of stromal cells in facilitating tumor progression. Patients with abundant desmoplasia and increased expression of α-SMA or periostin in background organs have shorter survival.256–258 In addition, CAFs, HSCs, and myofibroblasts, which promote liver fibrosis, produce immunosuppressive niche through differentiation and recruitment of immunosuppressive cells and dendritic cells.259,260 In HCC, several studies have established that the presence of background liver fibrosis or the presence of either α-SMA+ myofibroblasts or activated HSC signatures is associated with increased recurrence after curative HCC resection.261–264 Ju et al. 263 have reported that a high degree of activated HSCs, macrophage, and Treg cells infiltration around the tumor was contributed to death rates and high recurrence in patients with curative HCC resection.
Another report exists which claims that liver fibrosis may directly affect the cancer immune microenvironment, especially through immune surveillance of T-cells. Guidotti et al. 265 reported that T-cells are trapped by platelets that adhere to sinusoidal hyaluronan via CD44, and liver fibrosis impairs antigen recognition by intravascular CD8+ T-cells. ECM produced by activated HSCs may also provide a physical barrier that blocks the migration of immune cells. 225 Muller et al. 266 stated that fibrous bridging may prevent the spread of clones with harmful mutations to more distant sites in the liver, suggesting that fibrous bridging in the liver also inhibits the free migration of immune cells. On the other hand, activation of Wnt/β-catenin signal, reportedly, induced the gene involved in fibrogenesis in HSC. 267 There is increasing evidence from clinical studies that the presence of liver fibrosis, α-SMA+ myofibroblasts, or activated HSC signatures is associated with a poor prognosis, suggesting a key role of the fibrotic tumor microenvironment in promoting HCC progression.
Interaction of Immunosuppressive Cells, Exhaustion of T-cells, and Advanced Fibrosis in the Tumor Microenvironment
Tumor factors that have been shown to influence primary refractoriness to anti-PD-1/PD-L1 antibody in HCC are low PD-L1 expression, low infiltration of CD8+ T-cells, and the presence of Wnt/β-catenin activating mutation.15,268–270 In clinical trials, ICI resistance has not yet been adequately demonstrated in HCC for the following factors: MSI-high, genetic mutations related to antigen presentation such as β-2 microglobulin or IFN-γ signaling pathway, HLA class I genotype, and gut microbiome (Fig. 1).
Importantly, although activation of Wnt/β-catenin is one of the hallmarks of liver cancer, it is known that inhibition of this signal suppresses the differentiation of myofibroblast in pulmonary fibrosis. 271 It is also reported that inhibition of β-catenin by small inhibitory RNA induced downregulation of fibrogenic genes in HSC, suggesting a role of nuclear β-catenin in the fibrogenic pathway. 267 In addition, activation of Wnt/β-catenin is known to disturb the effector function of CD8+ T-cell and induce their exhaustion in human cancers including HCC. 272 Based on this evidence, it is conceivable that the activation of the Wnt/β-catenin induced by Wnt ligand may contribute to the fibrosis as well as the establishment of immune suppressive microenvironments in cancer including HCC.
Based on previous reports, we speculate that the following factors involved in acquired refractoriness to anti-PD-1/PD-L1 antibody may be particularly important in constituting an immunosuppressive tumor microenvironment (Fig. 4).
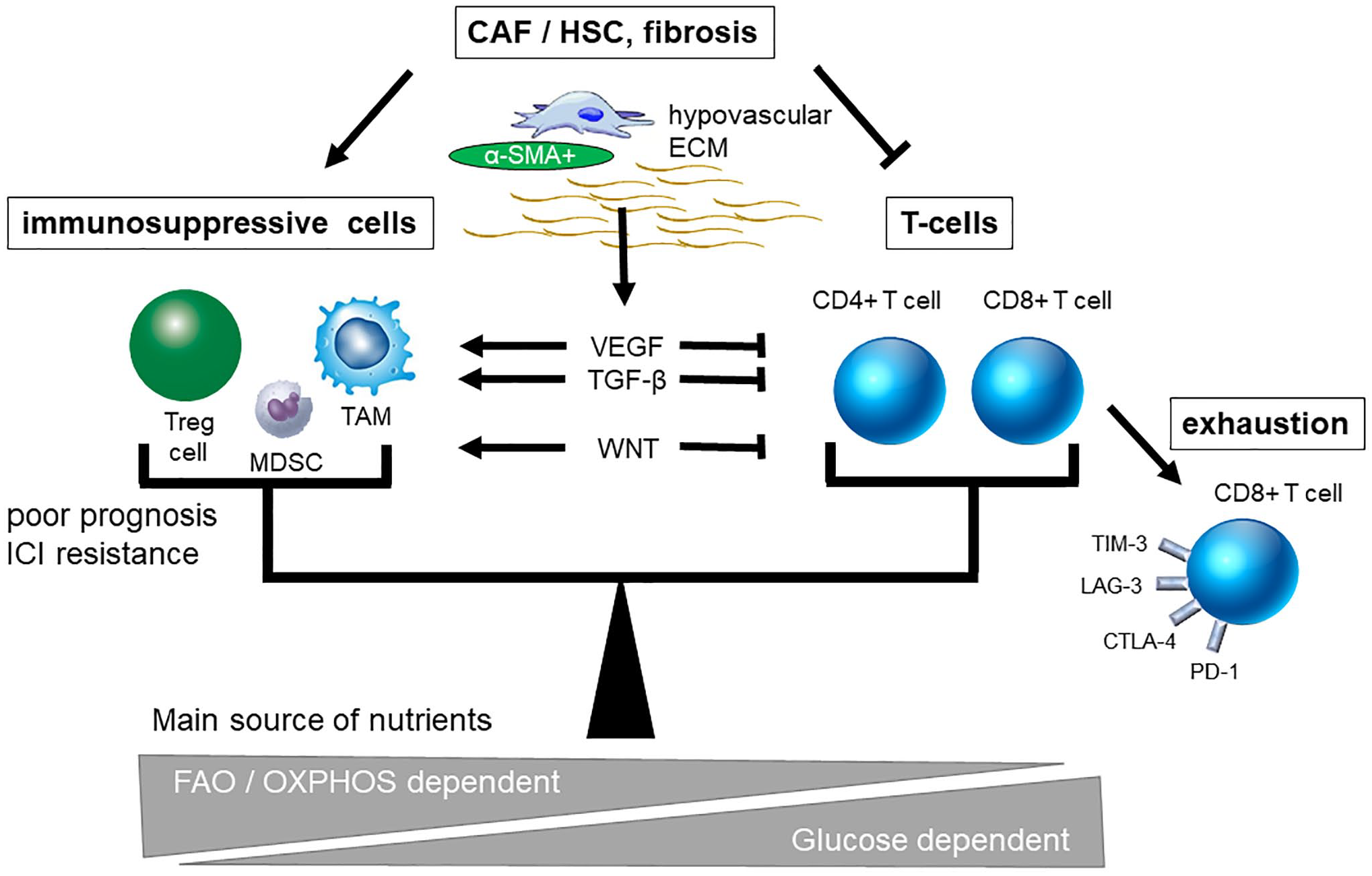
Triangle of immunosuppressive cells, exhausted T-cells, and advanced fibrosis in the tumor microenvironment. Immunosuppressive cellular factors including Treg cells, MDSCs, and TAMs play an important role in immunosuppressive tumor microenvironment. T-cell exhaustion is observed due to chronic liver inflammation and cirrhosis-associated immune dysfunction. In addition, the presence of fibrosis, which is induced by CAF, HSC, and myofibroblast, inside and outside the tumor has also been reported to accelerate the progression of immunosuppressive condition. These factors are closely intertwined with each other to disturb antitumor immunity. In the microenvironment where tumors utilize more glucose, nutritional competition occurs and shifts the niche toward more immunosuppressive. Abbreviations: PD-1, programmed cell death 1; Treg cell, regulatory T-cell; MDSCs, myeloid-derived suppressor cells; TAM, tumor-associated macrophage; CAF, cancer-associated fibroblast; HSC, hepatic stellate cell; ECM, extracellular matrix; VEGF, vascular endothelial growth factor; TGF-β, transforming growth factor beta; α-SMA, α-smooth muscle actin; OXPHOS, oxidative phosphorylation; FAO, fatty acid oxidation; ICI, immune checkpoint inhibitor; TIM-3, T-cell immunoglobulin and mucin domain-containing molecule-3; LAG-3, lymphocyte activation gene-3; CTLA-4, cytotoxic T-lymphocyte-associated protein-4.
Immunosuppressive cells, including Treg cells, MDSCs, and TAMs, have been demonstrated in many solid tumors, and such immunosuppressive cells contribute to ICI resistance in HCC as well. The importance of infiltration of PD-1+ Treg cells and TAMs has been specifically noted because they cause HPD during ICI treatment and worse outcome than without treatment. The frequency of HPD in HCC was 12.7% 16 ; we have to encourage careful monitoring of patients to prevent clinical deterioration induced by anti-PD-1/PD-L1 antibody.
T-cell exhaustion is also commonly observed in patients with chronic liver disease before liver carcinogenesis. In cases of chronic viral infection, PD-1 inhibitory receptors are expressed on CD8+ T-cells when they are in a state of dysfunction.19–21 Pfister et al. 273 reported that exhausted CD8+ T-cells express PD-1 receptors in non-alcoholic steatohepatitis-related HCC. Lebosse et al. 22 reported that TIM-3, CTLA-4, and PD-1 levels were highly expressed on CD8+ T-cells in patients with cirrhosis, and that cirrhosis-associated immune dysfunction is caused by IFN-γ-mediated T-cell dysfunction. Data defining the clinical relationship between T-cell exhaustion marker expression and the acquisition of ICI resistance in HCC are currently insufficient and require further study.
In addition, progression of fibrosis during chronic liver damage also offers niche for developing HCC. Few HCCs have a fibrous component in the tumor, but many cases have severe fibrosis in the background liver. Although some gene expression profiles of cholangiocarcinoma-associated fibroblasts were different from non-tumorigenic liver fibroblasts, 258 further investigation must be needed whether this fact clearly separates the origins and functions of localized fibrosis within the tumor and fibrosis in background organs. It is intriguing to speculate that activation of Wnt/β-catenin plays a role for developing immune suppressive microenvironment as well as induction of liver fibrosis because activation of Wnt/β-catenin pathway is frequently observed in HCC, especially in the early stage of tumor.
On the other hand, Finki et al. 274 showed that development of microniche could recruit tumor progenitor cells of HCC. They described the appearance of the niche was associated with the development of HCC and progression of liver fibrosis. Therefore, niche that can be a hotbed of HCC may appear in the early stage of hepatocarcinogenesis and the several players including HSC and myofibroblast may participate there. The immune cells, immune checkpoint molecules, and other mesenchymal cells including CAF and HSC are closely intertwined to create an immunosuppressive microenvironment in solid tumors. In addition, in an environment where tumors utilize more glucose, nutritional competition occurs, biasing the microenvironment to be more immunosuppressive. Tissue hypoxia, poor nutrition, and vascular disorganization are typically observed in patients with chronic liver injury. Hypoxia inhibits liver regeneration and promotes angiogenesis, fibrogenesis, and hepatocarcinogenesis. 275 Although it is possible that nutritional competition between tumor cells, hepatocytes, and the presence of immune cells in the liver causes T-cell exhaustion, the role of nutritional competition in HCC associated with liver cirrhosis has not yet been fully explored.
The advent of ICIs caused a major paradigm shift in treatment for HCC and other solid tumors. ICI resistance involves a complex niche created by a combination of specific genetic mutations such as Wnt/β-catenin, acquisition of epigenetic events, and infiltration of immunosuppressive microenvironment such as Treg cells, TAMs, HSCs, and CAFs. Because multiple heterogeneous intrahepatic HCC nodules exist at the same time due to multicentric carcinogenesis, heterogeneous gene mutations may cause complex treatment resistance in patients with liver cirrhosis. ICI resistance factors without genetic mutation include intratumor infiltration of Treg cells and other immunosuppressive cells, T-cell exhaustion due to chronic inflammation and cirrhosis-associated immune dysfunction, and the presence of fibrosis inside and outside the tumor. Severe liver fibrosis may inhibit T-cell migration and antigen presentation directly. Because the primary tolerance of HCC is only partially understood and there is insufficient evidence to explain the immunosuppressive microenvironment of HCC, further research on tumor immune microenvironments of HCC is needed, especially considering severe fibrosis of the background liver.
Footnotes
Competing Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
Conceptualization, TA; methodology, TA; writing—original draft preparation, TA and NN; writing—review and editing, TA and NN; supervision, MK; project administration, NN and MK.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported in part by a Grant-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (KAKENHI: 16K 382, N. Nishida, and 18K07922, M Kudo) and a grant from Smoking Research Foundation (N. Nishida).