
Abstract
Brain tumors in adults may be infrequent when compared with other cancer etiologies, but they remain one of the deadliest with bleak survival rates. Current treatment modalities encompass surgical resection, chemotherapy, and radiotherapy. However, increasing resistance rates are being witnessed, and this has been attributed, in part, to cancer stem cells (CSCs). CSCs are a subpopulation of cancer cells that reside within the tumor bulk and have the capacity for self-renewal and can differentiate and proliferate into multiple cell lineages. Studying those CSCs enables an increasing understanding of carcinogenesis, and targeting CSCs may overcome existing treatment resistance. One approach to weaponize new drugs is to target these CSCs through drug repurposing which entails using drugs, which are Food and Drug Administration–approved and safe for one defined disease, for a new indication. This approach serves to save both time and money that would otherwise be spent in designing a totally new therapy. In this review, we will illustrate drug repurposing strategies that have been used in brain tumors and then further elaborate on how these approaches, specifically those that target the resident CSCs, can help take the field of drug repurposing to a new level.
Keywords
Introduction
Tumors of the brain are rare but deadly entities whose treatment remains challenging. 1 Glioblastoma (GBM; World Health Organization [WHO] grade IV glioma) is the most frequent and aggressive primary brain tumor in adults. Life expectancy remains bleak at around 15 to 19 months. 2 The challenges associated with treatment of brain malignancies are extensive, ranging from inability to use current cancer therapies to limited interest/diminished funding from pharmaceutical industries. 3 Due to the brain’s complexity and frailty, many tumors are unable to be completely excised, which hinders surgical treatment and worsens prognosis. 3 Furthermore, the highly selective blood–brain barrier (BBB) hinders chemotherapeutic agents from reaching the site of carcinogenesis. 4 Our limited understanding of the biology of these widely variant tumors and their microenvironment poses a grand challenge to brain cancer treatment. 4
Cancer stem cells (CSCs) are a subpopulation of cancer cells that resemble normal human stem cells, having the capacity for self-renewal, differentiation, and proliferation into multiple cell lineages through symmetric and asymmetric cell division. 5 The study and investigation of CSCs (or “tumor-initiating cells”) have allowed us to further understand their role in metastasis, which accounts for 90% of cancer deaths in the world. Overall, the use of conventional therapies such as surgical resection, chemotherapy, and radiation is limited and often does not remove or target the CSCs, which are the cells responsible for tumor recurrence and metastasis, especially in the brain. Thus, targeted therapies, such as those that use cell surface markers, as well as various tumorigenic proteins found in CSCs, could provide for better patient outcomes (Fig. 1).
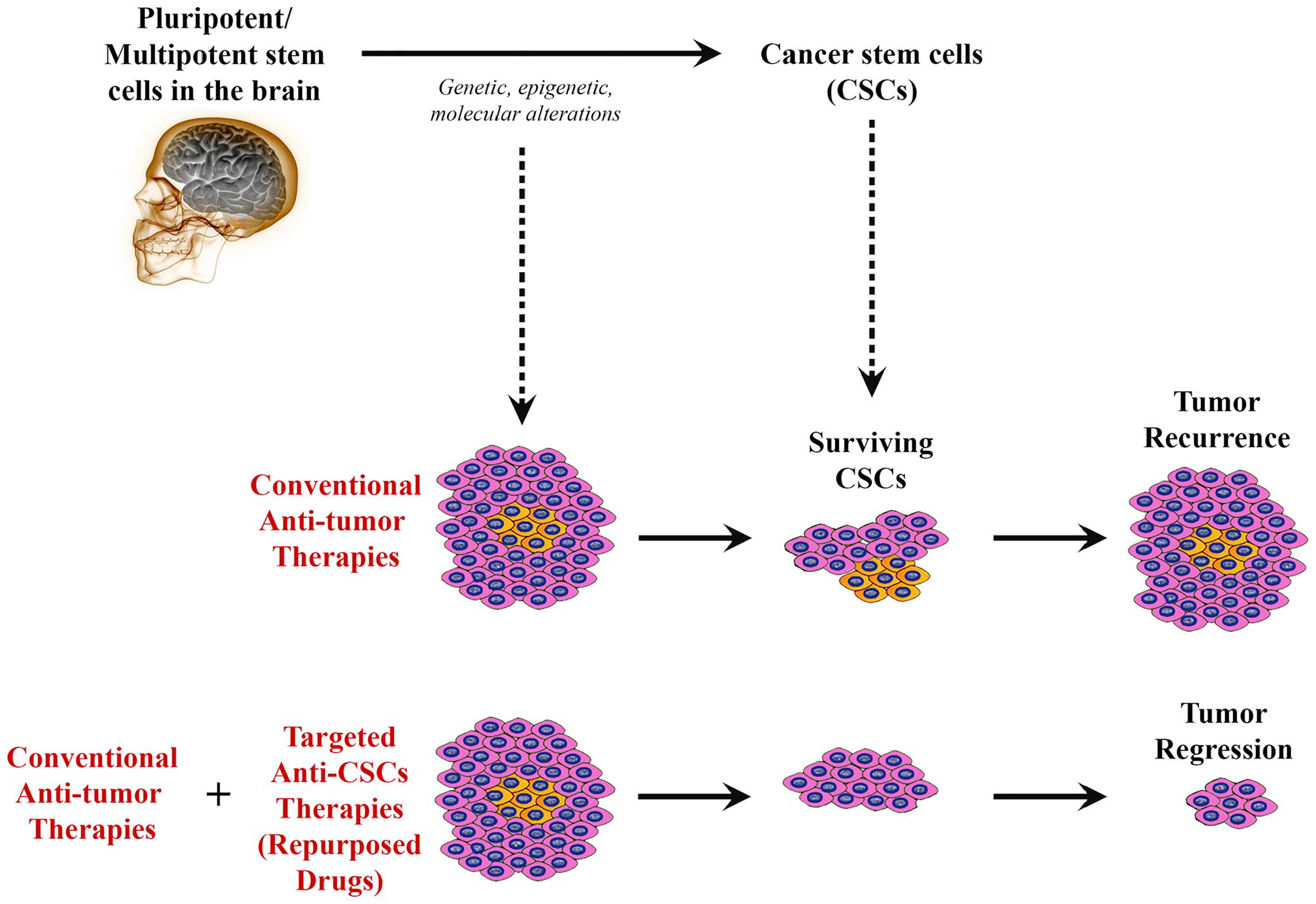
Schematic diagram illustrating how drug repurposing to target cancer stem cells (CSCs) in brain tumors can promote tumor regression. Despite that cancer cells within a tumor might be mostly affected by conventional therapies leading to shrinkage of the tumor, surviving CSCs will remain to resist to such therapies, leading to cancer recurrence with time. However, combining traditional anticancer therapies with novel targeted therapies directed against CSCs (“repurposed drugs”) could potentially elicit tumor regression and prevent recurrence.
The field of drug repurposing (aka drug repositioning) has recently become more attractive to many researchers. It is defined as the use of preexisting therapeutics—that have been previously deemed to be safe and efficacious in a defined medical condition—for purposes different from the one that they were initially studied for. 6 The advantage of studying preexisting drugs rather than de novo drug studies can be summarized in two words: time and cost.6,7 In this review, we will consider how drug repurposing can be leveraged to provide better outcomes for patients with brain cancer.
Cancer Stem Cells in Brain Tumors
CSCs have the capacity for self-renewal, differentiation, and proliferation into multiple cell lineages through symmetric and asymmetric cell division. 5 First identified in hematological malignancies, specifically acute myeloid leukemia (AML), and then subsequently in solid malignancies, CSCs have modified our understanding of cancer initiation, progression, and metastasis. 8 The first study on CSCs was done in 1997, where Bonnet and Dick demonstrated that despite having the morphological phenotype of leukemic blasts, only a subpopulation of CD34+/CD38− leukemic blast cells, and not CD34+/CD38+ cells, were able to repopulate the entire tumor and initiate human AML in mice with non-obese diabetic/severe combined immunodeficiency disease (NOD/SCID mice). 9 Subsequently, CSCs were identified and isolated in solid tumors such as breast cancers, 10 brain tumors, 11 prostate cancers, 12 lung cancers, 13 colon cancers, 14 head and neck squamous cell carcinomas (HNSCCs), 15 and pancreatic 16 and liver cancers. 17 Increasing studies have demonstrated that CSCs are responsible for tumor heterogeneity, invasion, metastasis, and tumor recurrence via epithelial-to-mesenchymal (EMT) transformation and multidrug resistance. Therefore, identification of CSCs is crucial as it may present a key therapeutic target on which novel therapeutic agents can act.
In vivo studies have demonstrated that when a subpopulation of the original malignant cells is transplanted into immunodeficient mice, the ability to reform the parental tumor is observed, indicating the existence of CSCs.11,18–20 Based on analyses, including those that examined differential allele expression of the X-linked gene G6PD in chronic myeloid leukemia (CML) and AML and those that have shown the activation of self-renewal via the β-catenin pathway, among others, CSCs can be derived from either normal stem cell lineages or early progenitor cells that have acquired certain mutations that confer stem-like cell properties on these originally non-self-renewing cells.21,22 Of note, if the CSCs do arise from these mutated early progenitor cells, they depend on inactivation of normal inhibitory pathways of self-renewal and activation of normal stem cell pathways.23–27 The tumor microenvironment (TME) has also been demonstrated to have the capacity to generate CSCs via activation mediated through extracellular cues. 28
It is believed that CSCs are responsible for the tumorgenicity and high proliferative capacity of multiple cancers, a concept further elaborated on when comparing CSCs with other cancer cells because CSCs are able to regenerate a tumor and cause metastasis, whereas other cancer cells have limited proliferative and regenerative capacity.29,30 With this in mind, the heterogeneity observed in many cancers can be attributed to the mutagenesis that occurs within CSCs, producing different phenotypic cancer cells described. 30 In addition to CSCs being multipotent, they share several other features with normal stem cells, such as enhanced DNA damage response mechanisms, relative quiescence, decreased apoptosis, and increased immune system evasion, which amplifies the stem cell component of tumors. 29 Despite these other features, a more critical topic of concern is the drug resistance that arises from the CSCs believed to be the cause of the cancer relapse seen in several cancers. This is in part attributed to their ability to self-renew and promote greater heterogeneity as a response to the therapeutic agents, as well as the interactions between the agents and the TME.29,31–33 Relatedly, although CSCs are typically quiescent, they can also induce a cell cycle arrested state to enhance their ability to be resistant to various therapies.24,34–38
When considering the importance of targeting these cells in combating tumor relapse, it is important to first identify the unique biomarkers associated with them. However, due to the heterogeneous and plastic nature of CSCs and their inward location within tumors, molecular components of CSCs have been difficult to define. 32 There are promising cell surface markers consistently seen on CSCs that can be used as a means of targeting CSCs more readily. CSC-specific cell markers include, but are not limited to, CD24, CD26, CD29, CD34, CD44, CD133, CD166, aldehyde dehydrogenase (ALDH), and Ep-CAM (CD326).32,39,40 Marker expression on CSCs differs from one cancer to another due to their origin.41–43 This offers a point of interest to target CSCs via these stem cell markers as it may be a more precise mechanism of identifying CSCs. However, it has been noted that although normal stem cells or progenitor cells may share similar cell surface markers and enzymes, there does appear to be differences in glycosylation that would require analysis to effectively target the markers. 40 The markers’ validity requires examination as well because although the molecular component may play a role in the function of the cells, it may not be unique to the CSC and thus sufficient for identification and targeting, as seen with Lgr5 in intestinal stem cells. 41 Overall, the particular marker may assist in identifying the CSC for one type of cancer, but not another. For example, A2B5 appears to be a unique marker for CSCs in GBM, whereas CD44 is more widespread after having been observed in various cancer CSCs, including those in the breast, bladder, stomach, ovary, and GBM, among others.39,44–47
Methodologies for the Identification of CSC
CSCs can be identified by various approaches, namely, (1) cell surface markers, (2) side population study, (3) ALDH activity, and (4) molecular signatures exemplified by the expression of stemness genes and transcription factors.
Cell Surface Markers
A variety of cell surface markers/antigens have been identified in several cancers that may serve as CSC biomarkers. These include CD20, CD24, CD34, CD44, CD90, CD117, and CD133. Although a universal biomarker for CSCs does not exist, CD44 and CD133 have gained much attention for both epithelial and mesenchymal malignancies.
CD44 is a transmembrane glycoprotein that is implicated in cell adhesion, migration, differentiation, proliferation, and survival. It functions as a receptor for hyaluronan, as well as other extracellular matrix components, growth factors, and cytokines. CD44 was first described as a CSC marker in breast cancer in 2003. 10 Increasing evidence has demonstrated that CD44 serves as a CSC marker and plays an important role in regulating the functions and properties of CSCs, including self-renewal, tumorigenesis, chemoradiation resistance, and metastasis.48,49
CD133 (Prominin 1) is a transmembrane glycoprotein expressed in neural progenitor cells, epithelial cells, and non-epithelial cells. It has also been identified in stem cells in both normal and cancer cells. 50 CD133 interacts with the Wnt/β-catenin and PI3K–Akt signaling pathways, which in turn promotes angiogenesis and cancer cell growth. 50 Different CSC subtypes that are known to express CD133 include breast, 51 colon, 52 gastric, 53 pancreatic, 54 hepatocellular, 55 lungs, 56 prostate, 57 melanoma, 58 and GBM. 59 Although CD133 has been used reliably as a CSC biomarker in various cancers and correlates with worse prognosis, it is not entirely specific.
Side Population Assay
The side population (SP) assay is a flow cytometry method used to identify stem cells based on the dye efflux properties of the ATP-binding cassette (ABC) transporters. 60 Cells that have the capability to express the stemness genes (ABC multidrug transporters) also can self-renew, differentiate into multiple lineages, and therefore display stem cell properties. Similar to the CSC biomarkers mentioned above, the SP assay has been used to identify both normal cells and CSCs such as in HNSCC, 61 bladder cancer, 62 colon cancer, 63 endometrial cancer, 64 ovarian cancer, 65 hepatocellular carcinoma, 66 lung cancer, 67 osteosarcomas, 68 medulloblastoma (MB), 69 and GBM. 70
ALDH Activity
ALDHs are a group of enzymes that catalyze the oxidation of aldehyde substrates to their corresponding carboxylic acids and play an important role in cellular detoxification. 71 ALDH1A1 is one of the key ALDH isozymes that is expressed not only in normal hematopoietic progenitor cells 72 but also in various CSCs, making it a useful CSC biomarker. 73 ALDH1A1 overexpression and increased enzyme activity have been associated with chemoresistance and radioresistance and correlates with poor prognosis across different cancers such as non–small cell lung cancer, 74 stomach cancer, 75 and ductal carcinoma of the breast. 76
Molecular
Signatures Exemplified by the Expression of Stemness Genes and Transcription Factors
Increasing evidence has demonstrated that stem cell–associated molecular features are biologically important in cancer. 77 In addition, the expression of stemness genes across multiple cancers has shown association with suppressed immune response, higher intratumoral heterogeneity, and dramatically worse outcomes. 78 In a study in breast cancer, the expression of stemness genes such as OCT3, SOX2, KLF4, MYC, NOTCH1, and NANOG in tumor cells is associated with metastasis and the acquisition of non-CSC to CSC plasticity with the formation of EpCam+/CD44high/CD24low cells. 79 The expression of stemness genes, particularly OCT4, SOX2, and NANOG, is the most analyzed transcription factor in CSCs. Increased expression of these genes has been shown in gliomas, 80 lung carcinoma, 81 prostate carcinoma, 82 and bone sarcoma. 83 Other transcriptional factors to be considered include Bmi-1, Snail, and Twist. Bmi-1 was identified as a protooncogene that promoted tumorigenesis, and its overexpression confers self-renewal potential and chemoresistance and radioresistance associated with increased incidence of metastasis. 84 Snail and Twist are essential in maintaining CSCs, promoting EMT, and mediating invasiveness and metastasis in various tumors. 85 Overexpression of Twist has also been observed in breast cancer, 86 lung cancer, 87 and gastric cancer. 88
Identification of CSCs in Various Brain Tumors
Although CSCs have been identified within multiple hematological and solid tumors, malignancies of the brain require increased consideration due to the challenges faced by treatments and therapeutics used to target these areas, including limited access for surgical resection, the inability of certain treatments to cross the BBB, and their unique genetic and epigenetic characteristics allowing for resistance to those agents able to cross the BBB.3,89–91
CSCs commonly found in brain tumors like gliomas have been investigated, and studies assessing their ability to self-renew have demonstrated that when these cells are isolated from the rest of the tumor, they are able to form neurospheres—indicative of their renewal potential.11,92 This supports the notion that these cells are required for the establishment of the tumor in vivo. 11 Furthermore, the proximity of CSCs to other cells within the TME in the brain has demonstrated potentially unique environmental characteristics, leading to the success of the tumor in this location. 93 For instance, in GBM, CSCs have been noted to be in close contact with endothelial cells (ECs), suggesting that the soluble secreted materials from the ECs may maintain the self-renewal capacity and differentiation of CSCs. When CSCs and ECs have been cultured in vitro, there is enhancement of CSC function, whereas when CSCs have been transplanted into models undergoing antivascular endothelial growth factor A (VEGFA) therapy, the CSC population was reduced, emphasizing the role of this vascular niche in the brain. 93 Alongside this vascular niche, a hypoxic niche has also been observed in the brain, where hypoxia has induced the activation of hypoxia-induced factor 1α resulting in CSC expansion.94–96 Similarly, CSCs have a dynamic relationship with their respective niche, allowing for active regulation to promote niche formation and maintenance. 97
In addition, the evaluation of various tumorigenic proteins has been completed and reported in brain tumors, such as GBMs, which promote the ability of the tumor to self-renew, promote angiogenesis, resist therapeutic agents, and facilitate transcription, proliferation, differentiation, and metastasis.89,98 For instance, transforming growth factor-beta (TGF-β), PI3K/Akt, Wnt/Beta-catenin pathway, Shh signaling, and mitogen-activated protein kinase (MAPK) have been documented in brain CSCs to promote the self-renewal of the tumor.99–101 Also, the enhanced expression of forkhead box protein M1 (FOXM1) within CSCs has been shown to be metastatic inducers and one of the mechanisms used by the tumor to resist therapy. 102 The upregulation of VEGF and its respective receptor demonstrated increased angiogenic potential, alongside the use of the histone chaperone complex known as FACT to facilitate transcription of the CSC markers, enabling multiple functions including interaction with the TME. 98 Relatedly, brain CSCs have been shown to adapt to nutrient restrictions, possibly another mechanism of evading treatment, via upregulation of the high-affinity neuronal glucose transporter GLIT3. 103 Of note, pediatric brain tumors, such as MB, use signaling pathways similar to those seen in adult brain CSCs, including Shh, Notch and Wnt, which govern specification, proliferation, and survival, indicating potential widespread targeting across these tumors.104,105 Studies have also shown that particular genetic alterations have been associated with the existence of the tumor, including mutations affecting EGFR and HDM2 (gain of function), PTEN, and NF1 (loss of function), among others.106,107 Cell surface markers noted in certain brain CSCs like those seen in GBM include CD133, nestin, and A2B5, representing potential specific target sites. Noteworthy mentioning are also the stromal-derived factor-1α (SDF-1α) and its receptor C-X-C chemokine receptor type 4 (CXCR4), where SDF-1α plays a role in homing of therapy-resistant CXCR4+ GBM CSCs 108 that are shown to reside in protective niches around arterioles recapitulating hematopoietic stem cell (HSC) niches in the bone marrow. 109 Interestingly, both markers are targets of AMD3100 (plerixafor), a drug that is used to mobilize HSCs out of their niches to improve the yield of stem cell transplantation. Plerixafor is currently being investigated in a repurposing effort in GBM as an adjuvant inhibitor of vasculogenesis, 110 forcing slowly dividing CSCs out of their niches. 111 Results from a phase I/II trial using plerixafor in GBM showed that the drug was well tolerated and improves local control of tumor recurrences (ClinicalTrials.gov; NCT01977677). 112
Among the primary brain tumors, GBM is a highly aggressive and lethal cancer with frequent tumor recurrence. Accumulating evidence supports the concept that GBM harbors CSCs, which confer resistance to chemotherapy and radiotherapy, cancer recurrence, and poor prognosis. 113 Therefore, targeting CSCs emerges as a promising method to eradicate this devastating disease. To effectively identify and target CSCs, various common surface markers such as CD133/prominin, Nestin, Musashi-1, Sox2, HMG box, CD44, L1CAM (CD171), CD15 (SSEA-1), and A2B5 were studied. 114 Notably, novel therapeutic modalities specifically targeting GBM CSCs bearing CD133 and CD44 have demonstrated encouraging results both in vitro and in vivo, expanding their role as theranostic biomarkers.115,116
Repurposing of Approved Drugs in Brain Tumors
Repurposing of Approved Drugs in Cancer
The field of drug repurposing (aka drug repositioning) has been recently attractive to many researchers. It is defined as the use of preexisting therapeutics—that have been previously studied to be safe and efficacious in a defined medical condition—for purposes different from the one that they were initially indicated for. 6 Repurposed drugs comprise four categories. The first includes drugs that were discontinued due to observed toxicities but later found to be effective in certain diseases. The second group includes drugs that were studied for a particular indication but were found to have an additional disease-fighting property (off-label). The third group includes drugs that are efficacious in several diseases but only used in the treatment of one disease. The fourth category includes drugs that exhibit synergistic action with the conventional pharmacological agents used to treat a particular pathology. 117
Advantages of Drug Repurposing and Obstacles to Drug Repurposing
The approach of studying preexisting drugs carries benefits over de novo drug studies. These advantages can be summarized in two words: time and cost. A study by Kaitin et al. published in 2010 demonstrated that it takes an average of 8.3 years for an antineoplastic drug to evolve from the time of filing of the investigational drug application to the first New Drug Application/Biologic License Application submission, compared with 3 to 4 years for a drug to be studied in a drug repurposing trial.6,7 Hence, when a researcher/clinician is to follow the strategy of drug repurposing and study a particular drug, there is a wealth of published data demonstrating the drug pharmacokinetics and pharmacodynamics, reducing the time and cost needed to complete, to a certain extent, phases I and II of a clinical trial. 118 Moreover, and from an economic perspective, drug repurposing trials study drugs in a cost-effective manner, for example, bringing a repurposed drug to market with an estimated cost of $300 million compared with $2–$3 billion as an estimated cost to study a new chemical entity. 119
On the contrary, the repositioning process of a particular candidate drug might face some hurdles. The due diligence is a challenging step in the repositioning process as a repositioning candidate should have a competitive profile. 6 In addition, redirecting a preexisting drug for a new use is considered as a high-risk investment as the repurposed drug may fail to achieve a benefit–risk profile sufficient to support its use for new indications. Adding to that, the drug of interest will need to go through phases to determine the appropriate dose for the new indication and in some cases will need to repeat early phases of the study to determine the efficacy and safety, all of which can cause an increase in cost and time required for the drug to be approved.118,120
Approaches to Drug Repurposing
The challenges associated with the treatment of brain malignancies are extensive, ranging from the inability to use current cancer therapies to diminished funding from pharmaceutical entities. 3 Due to the brain’s complexity and frailty, many tumors are unable to be completely excised, which hinders surgical treatment and worsens prognosis. 3 Furthermore, the highly selective BBB hinders chemotherapy agents from reaching the site of carcinogenesis. 4 Along with our limited understanding of the biology of the widely variant tumors and their microenvironment, all these factors pose a great challenge to brain cancer treatment. 4 Interestingly, although brain malignancies are deadly, they are generally infrequent compared with other forms of cancers. As a result, the demand and interest for novel therapies from pharmaceutical industries are diminished. 3 Moreover, the discovery and development of novel drugs is an arduous process which demands high costs and extensive time for these agents to be tried and approved. 121
Within the field of drug repurposing, various techniques have been developed, which contribute to the discovery of compounds of interest to treat various brain malignancies (Fig. 2). The exploration of links between brain tumor advancement and medications use is facilitated by large databases containing patients’ information, such as the Clinical Practice Research Datalink. 122 In addition, these relationships can also be explored by investigating clinical trial databases and even organizations’ specific data that have already been filed. 122 However, malignancy regression cannot be attributed to drug use unless experimental analysis is first performed. Approaches in silico can be used to select drugs of interest for repurposing by using computational algorithms to recognize interactions between drugs and targets of interest. 123 The databases used vary in the drug–target interactions they explore, ranging from genomics, 124 proteomics, 125 and disease-specific and even molecular targets, among others. The idea is to identify drugs that target a certain disease mechanism, protein, or pathway that is relevant to the malignancy in question. Although this approach requires data on the disease phenotype and the targets, it has proven to be time- and labor-efficient compared with other more traditional methods. 126 A different technique to new drug recognition is activity-based repurposing. This approach uses drug targets for different diseases that have been screened previously; this is all done in the absence of concurrent structural information. 89 The library of Integrated Network–based cellular signature (LINCS) is a program that analyzes how cells respond to certain stimuli, from chemical and genetic mutations to responses to different agents such as drugs. 127 Understanding the basis of cell signaling and regulation in disease states is crucial to determining functional drugs and possible therapeutic agents. 128 Hence, activity-based drug repurposing presents a powerful tool where knowing the molecular basis of disease can serve as a way to target therapy by using drugs that are known to be effective for a certain type of molecular phenomenon and apply them to another tumor type. 129 An example of a drug group that has been repurposed in this way is the ALK inhibitors for the treatment of various brain tumors such as large cell lymphoma and neuroblastoma (NB) where the ALK kinase is overactivated. 130
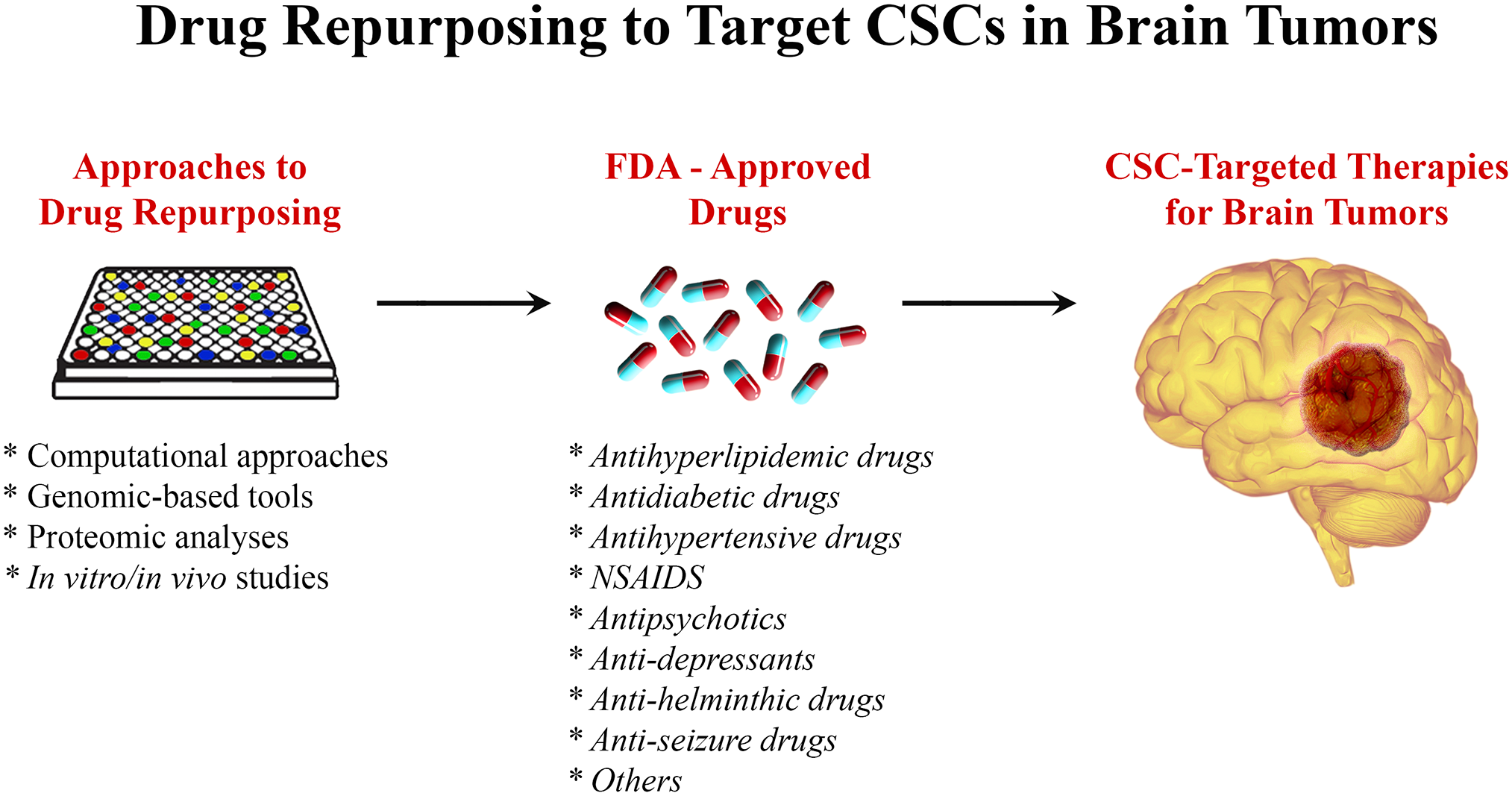
Drug repurposing to target CSCs in brain tumors. Using different approaches including genomics- and proteomics-based tools, computational approaches, and in vitro/in vivo studies, FDA-approved drugs could be repurposed to target CSCs in brain tumors. Abbreviation: CSC, cancer stem cell; FDA, Food and Drug Administration; NSAIDs, nonsteroidal anti-inflammatory drugs.
Targeting Cancer Stem Cells in the Treatment of Meningioma
Meningiomas are neoplasms thought to be derived from meningothelial cell arachnoid cap cells and are among the most common primary central nervous system (CNS) tumors, representing approximately 37% of all primary CNS neoplasms. 131 Current management guidelines indicate gross total resection as the primary treatment for intracranial meningiomas. 132 Clinically, the major factor in recurrence is the extent of resection, which may be complicated by adhesion or encasement of vital structures such as cranial nerves, cerebral arteries, or the brainstem.131,132 Several studies have reported the presence of CSCs in meningioma,133,134 which can be targeted using targeted therapies. Given the high cost, long development time, and technical difficulty of new drug discovery, a promising strategy to identify new therapeutic options is the repurposing of drugs, 118 which can be implicated in meningiomas.
Antiseizure Drugs
Several drugs have been proposed as candidates for repurposing in the treatment of meningiomas. Chiou et al. 135 tested the effect of valproic acid (VPA) on cultured meningioma stem cells. VPA is a drug primarily used in the treatment of seizure disorders. In the study by Chiou et al., authors demonstrated that VPA reduced the viability of meningioma CSCs and increased their sensitivity to radiation. In addition, VPA was found to reduce the expression of Oct4 and anchorage-independent growth, indicating a differentiation-promoting effect of VPA on stem cells that was more pronounced when combined with radiation. Thus, combined treatment of VPA and radiation could be proposed as a possible strategy in the treatment of meningiomas by targeting their CSC population. 135
Anthelminthic Drugs
Mebendazole (MBZ) is a wide-spectrum anthelminthic that works by inhibiting microtubule polymerization. 136 Its role as a possible agent for the treatment of CSCs is also under study. In a review of the literature by Guerini et al., 137 multiple preclinical studies showed MBZ to be an effective inhibitor of multiple mechanisms that facilitate cancer cell viability. Larsen et al. 138 described inhibition of the hedgehog (Hh) signaling pathway and Hh-dependent survival in cultured cell lines. In a study by Bai et al., 139 MBZ inhibited tumor angiogenesis in MB animal models by blocking the activity of VEGF receptor 2. Sasaki et al. also reported that MBZ caused mitotic arrest via depolymerization of tubulin and abnormal spindle cell formation. In murine models, the authors reported a significant suppression of tumor growth after treatment with MBZ. 140 In addition, Skibinski et al. 141 found a proapoptotic effect of MBZ on human meningioma cell lines and an increase in median survival in animal models treated with MBZ. They also described a synergistic effect when combining MBZ therapy with radiation.
Antidiabetic Drugs
Metformin is a biguanide antihyperglycemic agent widely used for the treatment of diabetes mellitus type 2. In vitro and in vivo studies using different CSC models, including breast,142,143 colon, 144 meningioma, 145 cholangiocarcinoma, 146 pancreatic adenocarcinoma, 142 GBM, and NB, 147 have shown inhibition of growth and proliferation of CSCs. Multiple mechanisms of action have been attributed to metformin; its major effects are largely exerted through the activation of AMP-activated protein kinase and the inhibition of mitochondrial respiratory chain (complex I).148,149 The capacity of metformin to regulate several cell signaling pathways is suggested to be the mechanism behind its anticancer properties. 142 In a study by Guo et al., 145 metformin inhibited the mammalian target of rapamycin (mTOR) signaling pathway, and significant inhibition of cell growth was seen in a concentration-dependent manner in vitro and in vivo. Metformin has also been shown to increase the efficacy of classical anticancer drugs. In meningioma and cholangiocarcinoma cell models, the anticancer effect of cisplatin was significantly enhanced when the cells were co-treated with metformin.145,146 In a study by Hirsch et al., 143 and in a similar fashion, metformin increased the therapeutic power of doxorubicin where the combination killed CSCs and non-CSCs in culture, and reduced tumor mass and prolonged remission much more effectively than either drug alone in a xenograft mouse model.
Antihypertensive Drugs
The role of the renin–angiotensin system (RAS) as a target for the treatment of CSCs is also of interest. Better known for its role in blood pressure regulation and electrolyte hemostasis, components of the RAS also mediate angiogenesis, cellular proliferation, and apoptosis. Angiotensin II (ATII) is the main effector of the RAS, via its two receptors ATII type 1 receptor (ATIIR1) and ATII type 2 receptor (ATIIR2). ATIIR1 induces angiogenesis and cellular proliferation. 150 Pro-renin, through its receptor pattern recognition receptor (PRR), is another important component of the RAS implicated in carcinogenesis through its involvement in the Wnt/β-catenin signaling pathway. 151 Cells in numerous cancer types, including brain tumors such as GBM, have been found to express both CSC markers and components of the RAS. 152 In two similar studies, Shivapathasundram et al. 150 reported the expression of PRR, ACE, ATIIR1, and ATIIR2 and Rahman et al. 153 reported the expression of cathepsin B, cathepsin D, and, to a lesser extent, cathepsin G in a putative stem cell population of low-grade meningioma. Given that the RAS is overexpressed in a wide range of tumors, numerous studies have assessed the effect of RAS inhibitors in vitro and on tumor models in vivo. These drugs, across different classes (β-blockers, angiotensin- converting enzyme inhibitors, and angiotensin receptor blockers), have been reported to reduce tumor cell growth, migration, invasion, and metastasis in numerous cancer types. 152 To the best of our knowledge, the effect of RAS inhibitors in meningioma CSCs has not been studied. The overexpression of different components of RAS suggests a potential utility for the repurposing of these drugs for the treatment of meningioma, and more research is needed to explore this possibility.
Another group of antihypertensive drugs, calcium channel blocker (CCB) is a candidate for repurposing. Cytosolic Ca2+ plays an important role in intracellular signal transduction and several processes necessary for cell proliferation and survival, making this ion a possible target in the treatment of cancer. 154 Previous studies have reported the overexpression of Ca2+ channels in various tumor types155,156 In meningioma, Jensen et al. reported a dose-dependent decrease in growth stimulation when cultured human meningioma cells exposed to serum-containing media or different growth factors were treated with verapamil, nifedipine, or diltiazem (voltage-dependent calcium channel–blocking agents).157,158 In a similar study, diltiazem and verapamil were found to decrease the growth of meningioma tumor models in mice. 158 These drugs were also found to potentiate the effects of hydroxyurea on inhibition of growth of meningioma cell models in a study by Ragel et al. 159 In CSCs, the blockade of Ca2+ channels has been reported to inhibit the proliferation, survival, and stemness features of stem-like cells in GBM and ovarian cell models. It also potentiated the effect of chemotherapeutic agents.160–162 The effect of CCB on meningioma CSCs has not been characterized to the best of our knowledge, but these observations suggest another possible treatment option worth studying.
Antihyperlipidemic Drugs
The repurposing of hydroxyl-methyl-glutaryl CoA (HMG-CoA) reductase inhibitors for the treatment of cancer has also been studied. These drugs, classically used for the treatment of hypercholesterolemia, block the conversion of HMG-CoA to mevalonate, the rate-limiting step in cholesterol synthesis. Mevalonate is necessary for normal and neoplastic cells to enter the cell cycle. 163 Several studies have found that drugs in this group inhibit cell proliferation by downregulating several intracellular signaling pathways.163,164 Wu et al. 164 reported simvastatin (SMV)-mediated downregulation of the PI3K/Akt pathway, leading to inhibition of GBM cell proliferation and induction of apoptosis via increase in caspase-3 activation in vitro and in vivo. In meningioma cells, Johnson et al. evaluated the effects of lovastatin (LVS) on meningioma cell proliferation and its influence on the activation of the MEK-1–MAPK/ERK pathway. In their study, cultured meningioma cells were exposed to three different mitogens with and without LVS, and the levels of phosphorylated MAPK kinase (MEK-1/2) and MAPK were compared. LVS significantly reduced the levels of both and inhibited cell proliferation after stimulation, suggesting inhibition of MAPK/ERK activity as one of the mechanisms that explain the effect of LVS on cell proliferation. 163 In a study by Ghering et al., cultured meningioma cell lines were treated with different combinations of statins and thiazolidinediones (TDZ). They compared the antiproliferative and cytotoxic effect of SMV, LVS, atorvastatin, pravastatin, SMV, and two TDZ, pioglitazone (PGZ) and rosiglitazone, and their combinations on various human meningioma cell lines. Among the statins, SMV was the most effective drug in the group. From the TDZ group, PGZ exhibited a significant effect when used as a single agent, and out of all the drug combinations, SMV in combination with PGZ turned out to be the most effective treatment. 165 In CSCs, SMV has been reported to inhibit proliferation and induce apoptosis in breast cancer cell models. 166 LVS has been found to have similar effects in nasopharyngeal carcinoma CSCs. 167 These findings suggest the utility of the repurposing of lipid-lowering drugs for the treatment of meningiomas.
Targeting Cancer Stem Cells in the Treatment of Glioblastoma
GBM (WHO grade IV glioma) is the most frequent and aggressive primary brain tumor in adults. Life expectancy remains bleak at around 15 to 19 months. 2 The majority of GBMs, around 90%, are primary and develop rapidly in older individuals without any proof of a precursor lesion. 168 A minority are secondary, which signifies that they arise from lower WHO grade gliomas such as II or III. 169 Incidence is on the rise but varies drastically between countries and can range between 0.59 and 5 cases per 100,000 individuals.170,171 The direct causes behind this increasing incidence have not been elucidated as of yet. However, it has been postulated that this may be due to better diagnostic modalities and an increasingly aging population as a whole. 172 Surgical resection, if possible, remains the standard of care. It may be followed by radiotherapy 173 or supplemented with chemotherapy such as temozolomide (TMZ), which is of particular benefit in patients with a methylated MGMT promoter. 174
Tumorigenic CSCs are self-renewing cells that are present in the tumor bulk of GBM. 175 They contribute to both tumor initiation and therapy resistance, including conventional chemotherapy 38 and radiotherapy. 176 Indeed, a study by Murat et al. 177 provided the first clinical evidence for the presence of a “glioma stem cell” or “self-renewal” phenotype in treatment resistance of GBM. The study analyzed the gene expression profiles of 80 GBMs and associated them with resistance to therapy, showing that the expression signature dominated by HOX genes, which comprises Prominin-1 (CD133), could serve as a predictor for poor survival in patients treated with concomitant chemoradiotherapy. 177 Another study by Pallini et al. 178 demonstrated that analyzing CSCs may predict the survival of GBM patients where in vitro CSC generation and the presence of ≥2% CD133+ cells in tumor lesions negatively correlate with overall and progression-free survival of patients. Therefore, one of the most challenging aspects of treating GBM is destroying the resident CSC population. 179 Also, it is apparent that developing novel treatment strategies that target CSCs may help advance the entire field of GBM treatment. 180 Novel treatments need to be safe, efficacious, able to penetrate the BBB, and Food and Drug Administration (FDA)-approved 181 (Table 1).
Examples of Some Repurposed Drugs Currently Being Clinically Investigated for the Treatment of Glioblastoma (ClinicalTrials.gov).
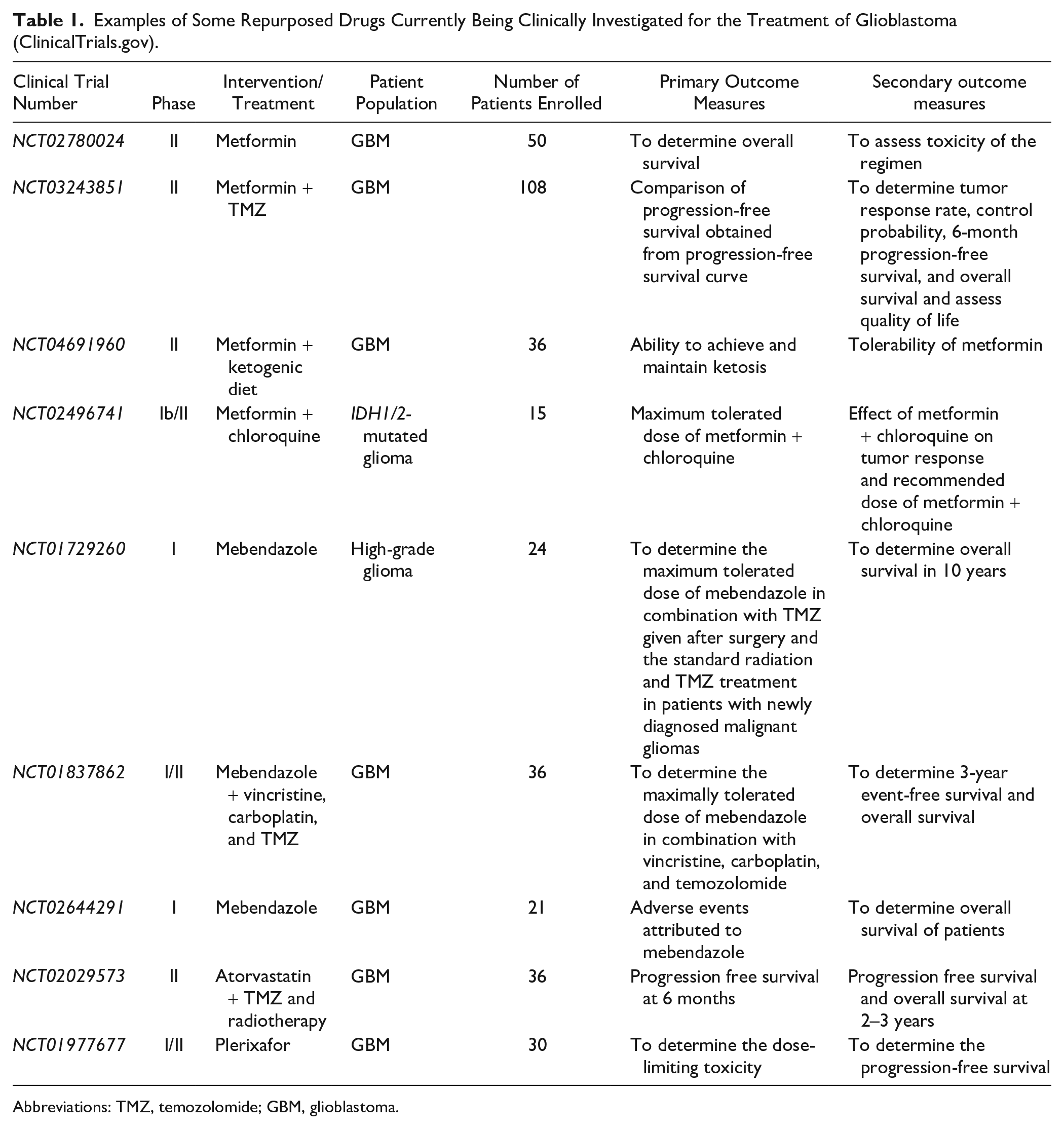
Abbreviations: TMZ, temozolomide; GBM, glioblastoma.
Antidiabetic Drugs
Metformin is currently in phase II of National Institutes of Health (NIH)-approved clinical trial NCT02780024 for its use as an adjuvant component to TMZ and radiotherapy (ClinicalTrials.gov; NCT02780024). In addition, a dose-finding phase Ib/II clinical trial was done on patients with IDH1/2-mutated glioma treated with a combination of metformin and chloroquine (ClinicalTrials.gov; NCT02496741), revealing promising results and opening novel treatment avenue for those patients.182,183 In mouse xenograft neoplastic models, metformin was able to preferentially target and kill CSCs and prolong remission. 184 The mode of action is postulated to be through attenuation of the inflammatory feedback pathway, particularly on transcription factor NF-κB and interleukin 6 (IL-6). 185 Metformin and phenformin were shown to inhibit cell migration of LN229 glioma cells in vitro and in vivo. In addition, both drugs were associated with increased reactive oxygen species (ROS) production within the cell mitochondrion. 186 Phenformin is superior to metformin in terms of chemical potency and was shown to induce apoptosis of GBM CSCs and inhibit their self-renewal. Phenformin decreases stemness and mesenchymal marker expression while increasing miR-124, miR-137, and let-7 expression. 187 Silencing let-7, which normally acts as a tumor suppressor microRNA by targeting HMGA2, attenuates the effects of phenformin on inhibiting CSCs. 188 Dichloroacetate, which acts as an inhibitor of the glycolytic enzyme pyruvate dehydrogenase kinase that in turn decreases lactic acid production induced by biguanides, has been shown to potentiate the anti-CSC effect of phenformin. 187
Antihyperlipidemic Drugs
Statins are of interest in GBM therapy because they have been proposed to induce apoptosis in glioma cells. 187 SMV’s antineoplastic action is via proliferation reduction and apoptosis induction of the C6 glioma cell line. This is achieved by way of increasing phosphorylation of c-jun through activating the JNK pathway. 189 Atorvastatin, on its own and via MT1-MMP microglial decreased expression, was able to suppress both the invasive and migratory potential of glioma cells. 190 Furthermore, LVS had a synergistic effect with tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL)-inducing apoptosis in 50% of cells in three human GBM malignant cells lines: M059K, M59J, and A172 in contrast to the failure of TRAIL to do so when used alone. 191 Statins could be used as an adjunct to TMZ to increase the efficacy of chemotherapy and decrease resistance. 192
Antihypertensive Drugs
The RAS system has been implicated in the hallmarks of cancer, 193 which is one explanation to as why this class of drugs may be helpful in glioma treatment. 194 ATII and ATIIR1 interaction aids in tumor angiogenesis via stimulating vascular smooth muscle cells and ECs to produce VEGF.195,196 Furthermore, the interplay of the Ang-II/AT1R system has been shown to be a contributing factor toward neoplastic immunosuppression, which may hinder the productive utilization of immunotherapy. 197
GSK3-β Inhibitors
Glycogen synthesis kinase 3-β (GSK3-β) has been implicated in many tumors including GBM, which paves the way for potential therapeutic strategies.198–200 GSK3-β is a serine/threonine kinase 201 linked to several pathways involved in tumorigenesis, such as Wnt/β-catenin 202 and PI3K/PTEN. 203 One study showed that, in vivo, infrared (IR)-induced inhibition of GSK3-β reduced the tumor load, thus extending survival. GSK3-β, at high dosages, increased GBM cell sensitivity to IR via 53BP1 phosphorylation at serine 166. Consequently, GSK3-β inhibitors such as SB216763 may become a valuable asset in treating radiotherapy-resistant GB. 204 Another study demonstrated the in vitro effects of BIO (an indirubin) and CHIR9902, both GSK3-β inhibitors, on glioma cells. Specifically, BIO decreases migration and invasion in spheroid assays, whereas CHIR9902 renders ECs more permeable, hence making the BBB more permissible to drugs. 205
In C6 glioma cells that have the isodehydrogenase 2 (IDH2) mutant, lithium chloride resulted in a decrease in proliferation and migration via inhibition of GSK3-β activity. At the same time, β-catenin accumulated less in these cells. These results illustrate a potential use of lithium chloride as a treatment modality. 206
Another promising treatment avenue is kenpaullone, a GSK3-β and cyclin-dependent kinase inhibitor. 207 Kenpaullone exhibits TMZ-enhancing activity against GBM CSCs possibly via suppressing SOX2 that attenuates stemness. This activity contrasts with the case when kenpaullone was administered alone possibly due to the increased BBB penetrance of the combined therapy. 208 An additional GSK3-β inhibitor is tideglusib which suppresses GBM tumorigeneses via downregulating KDM1A. 209
Nonsteroidal Anti-inflammatory Drugs
Nonsteroidal anti-inflammatory drugs (NSAIDs) are a drug group with anti-inflammatory, analgesic and antipyretic characteristics that act through inhibition of cyclooxygenase (COX). 210 Inflammation is a key component of tumorigenesis explaining the role of NSAIDs in anticancer therapy, specifically via decreasing tumor burden and angiogenic and metastatic potentials. 211 IL-6, an inflammatory cytokine, promotes the development of non-cancerous cells into CSCs specifically via upregulation of Oct4 gene expression by activating IL-2R/JAK/STAT3 pathway. 212 Certain prostaglandins, such as PGE2, have been shown to be implicated in cancers such as colon carcinoma. 213 This could explain the role of inflammation in persistence of the tumor burden via the resident CSCs and hence resistance to traditional GBM therapy routes. Two NSAIDs, ibuprofen and diclofenac, have been shown to potently inhibit glioma cells in vitro, albeit via distinct mechanisms; elucidating their role as potential adjuvants to GBM therapy. Ibuprofen mainly acts via a COX- and lactate-independent mechanism. On the contrary, diclofenac acts primarily via decreased STAT-3 signaling and downstream modulation of glycolysis in a more lactate-dependent manner. 214 The role of lactate is to induce TGF-β2 via its activating protein, thrombospondin 1 (THBS-1). This serves an important role in GBM pathogenesis via regulating cell migration. 215
Antipsychotic Drugs
Antipsychotics have displayed potential for utilization as anticancer therapies particularly against GBM. 216 As a general rule, antipsychotics have adequate BBB penetrance, a property that is of significant use in malignant brain gliomas. 217 Another interesting fact is that patients with schizophrenia have a lower risk of getting cancer than those without, at a rate of 1.93% to 2.97%, respectively. 218 Chlorpromazine, an FDA-approved psychotropic drug, was shown to efficaciously inhibit proliferating glioma cells which were resistant to chemotherapy. Chlorpromazine specifically induced arrest in the proliferation of cells that expressed COX4-1. 219 Thioridazine (THD), a drug belonging to the class of phenothiazine used to treat psychosis and schizophrenia, exhibits anti-GBM properties. THD, through the Wnt/β-catenin pathway, enhances p-62 mediated apoptosis in glioma cells. 220 THD also acts by upregulating AMPK, leading to apoptosis. 221 Traditionally used for its dopamine receptor antagonistic properties in schizophrenia, THD has shown a role in sensitizing GBM cells to TMZ in vivo and hence reducing tumor growth and increasing survival in mouse xenografts. The antineoplastic role is mediated by impairing autophagy, a survival mechanism for glioma cells, independent of its action on dopamine receptors. 222 Triflupherazine (TFP), another dopamine receptor antagonist in the class of phenothiazines, could be repurposed for GB treatment when combined with radiotherapy. The combination of TFP and radiotherapy targets GB CSCs and prevents the radiotherapy-induced phenotypic switch of GB into a more aggressive variant. 223
Antidepressants
The quest for finding new drugs to overcome GB resistance has also included antidepressant drugs, one of which is fluoxetine. Fluoxetine is a selective serotonin reuptake inhibitor (SSRI) that is used in the treatment of major depression. 224 In glioma cells, fluoxetine was shown to bind to α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor, causing a calcium influx into the cell which consequently results in an increase in mitochondrial calcium, thus triggering apoptosis. In vivo studies showed that fluoxetine’s action was comparable with that of TMZ in suppressing glioma growth. 225
One tumorigenesis theory proposes that cancer is a “mitochondrionopathy,” meaning that mitochondria in cancer cells are both structurally and functionally diseased as opposed to those in healthy cells. 226 Those who support the “metabolic theory” propose that if one can activate the malfunctioning mitochondria in the neoplastic GB cells, cell death may progress normally and facilitate chemotherapy. 227 Tricyclic antidepressants, such as imipramine and amitriptyline, may be able to restore GB mitochondrial function and respiratory function and thus serve as potential therapeutic adjuvants. 228 In terms of mechanism of action, both drugs served to suppress p56 NF-κB, which is strongly activated in many tumors.228,229 Impramine and amitriptyline also suppressed lactate release, a potent oncogenic metabolite that induces inflammation, from GB cells.228,230 Another antidepressant drug, fluvoxamine, exhibited potential anti-GB therapeutic potential via inhibiting actin polymerization and subsequent glioma cell migration in mice models with human GB CSCs. 231
Anthelminthic Drugs
In a study by Bai et al., fenbendazole was incidentally found to decrease engraftment of brain tumors in nude mice that were being treated for a pinworm infection. This discovery led the group to explore the anticancerous effects of other benzimidazoles. They also found that MBZ was cytotoxic at IC50 concentrations ranging between 0.1 and 0.3 µM. MBZ was also shown to increase survival by 68% in mouse glioma models. 232 Other than disrupting tubulin polymerization, benzimidazoles were postulated to act via inhibiting the expression of VEGF 233 and HIF-1α. 234 MBZ-induced apoptosis may potentially occur through phosphorylation and consequent inactivation of Bcl-2. 235
“The Reverse swing-M” trial is a phase I going to phase II study conducted by Patil et al. to determine the maximal tolerable dose (MTD) of MBZ in conjunction with other treatment modalities in fighting recurrent high-grade glioma. It was shown that the MTD of MBZ is 1600 mg TDS when combined with TMZ or TMZ radiation. When using MBZ with single-agent lomustine (CCNU), the MTD was a dosage of 800 mg TDS. 236 Another NIH-approved clinical trial NCT01729260 is a phase I study whose goal is to determine the MTD of MBZ in patients who have been newly diagnosed with GB and are on TMZ (ClinicalTrials.gov; NCT01729260).
Targeting Cancer Stem Cells in the Treatment of Pediatric Brain Tumors
Brain tumors are the second most prevalent malignancies in the pediatric population after leukemia and account for the majority of the solid tumors in children. They are also counted as the leading cause of tumor-related childhood mortality. 237 In 2007, WHO initially classified brain tumors based on the histopathologic appearance. 131 With advances of biomedical research, this classification expanded in 2016 to involve molecular parameters along with histopathologic patterns to classify brain malignancies. 238
Despite the presence of a wide therapeutic armamentarium to treat brain tumors, the presence of the CSC subpopulation within the tumor bulk contributed to cancer resistance to the conventional therapy and consequent recurrence of the tumor.98,239,240 To overcome this resistance, researchers have been exploring various molecular pathways in CSCs and identifying therapeutic strategies to target this subpopulation of cells.
NB and GB cell lines demonstrated chemoresistant and radioresistant activity, respectively, of CD133+ cells with Wnt overexpression and that inhibition of this pathway decreased the viability of these cells.241,242 Moreover, targeting Wnt signaling promoted NB cell line differentiation and enhanced their sensitivity to chemotherapy. 243 GSK-3β is a serine/threonine kinase that represents a downstream switch to multiple pathways in CSCs.199,244 GSK-3β inhibition has been shown to induce apoptosis in NB cell lines via activating the Wnt signaling pathway in a p53-independent manner.199,245
MB is the most common primary brain tumor diagnosed in childhood. Studies were able to isolate CD133+ CSCs within the perivascular niche of MBs. 93 Like many other tumors as well as GB, highly aggressive MB cells have shown to upregulate CSC markers such as CD133 and Nestin. 240 Although the functional role of CD133 is not well delineated, overexpression of CD133 is associated with chemoresistance 246 and high tumor-initiating capacity. 247 Similarly, CD133 cell surface marker expression has been used to identify CSCs in NB cell lines which display more resistance to anticancer drugs. 248
Future Directions
To conclude, drug resistance is a cardinal feature of CSCs in all cancers. Currently, conventional therapies target the bulk of the tumor, mainly the non-CSC component, and patients typically experience recurrence and metastasis due to the CSC enrichment which was not tackled. 41 Some of these resistance mechanisms include dormancy, the TME, expression of multidrug resistance proteins, such as ALDH, and EMT, 249 among others, which emphasize the need to contemplate these mechanisms when considering therapies against CSCs. 250 In addition, when we are concerned with brain tumors and their respective CSCs, there are unique challenges facing treatments and therapeutics that need to be kept in mind, as discussed previously.3,89,91 The utilization of conventional therapies like chemotherapy, radiation, and surgical resection is limited and often does not remove or target the CSCs, which are the cells responsible for tumor recurrence and metastasis, especially in the brain. Therefore, innovative solutions are required to tackle the problems presently faced by CSCs in the brain: drug repurposing is a promising approach. This idea—the use of drugs in ways other than their original FDA-approved indications—could offer novel avenues to bypass the chemoresistance and recurrence seen with conventional therapy and treatment. 89 Indeed, targeted therapies, such as those that use cell surface markers, as well as various tumorigenic proteins, seen in CSCs could pave the way for better patient outcomes. Using the markers that are specific to CSCs in general, along with the markers specifically observed in brain tumor CSCs, bioinformatic and computational methods could be used to match different drug types previously indicated for different diseases to be used as treatment for CSCs.
Footnotes
Acknowledgements
The authors thank all members of the Arkadi M. Rywlin M.D. Department of Pathology and Laboratory Medicine, Mount Sinai Medical Center (Miami Beach, FL) and the Herbert Wertheim College of Medicine, Florida International University (Miami, FL) for their help with this work.
Competing Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
HFB was involved in conceptualization, project administration, supervision, investigation, methodology, writing—original draft preparation, writing—reviewing and editing, visualization, and validation. DD was involved in investigation, methodology, writing—original draft preparation, writing—reviewing and Editing, visualization, and validation. AAA, ME, KSO, JCAM, RD, RSu, AZ, and RA were involved in methodology, writing–original draft preparation, and validation. RSa was involved in supervision, writing—reviewing and editing, visualization, and validation. RP was involved in project administration, supervision, writing—reviewing and editing, visualization, and validation. All authors have read and approved the final manuscript.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.