
Abstract
Cancer stem cells (CSCs) drive metastasis, treatment resistance, and tumor recurrence. CSCs reside within a niche, an anatomically distinct site within the tumor microenvironment (TME) that consists of malignant and non-malignant cells, including immune cells. The renin–angiotensin system (RAS), a critical regulator of stem cells and key developmental processes, plays a vital role in the TME. Non-malignant cells within the CSC niche and stem cell signaling pathways such as the Wnt, Hedgehog, and Notch pathways influence CSCs. Components of the RAS and cathepsins B and D that constitute bypass loops of the RAS are expressed on CSCs in many cancer types. There is extensive in vitro and in vivo evidence showing that RAS inhibition reduces tumor growth, cell proliferation, invasion, and metastasis. However, there is inconsistent epidemiological data on the effect of RAS inhibitors on cancer incidence and survival outcomes, attributed to different patient characteristics and methodologies used between studies. Further mechanistic studies are warranted to investigate the precise effects of the RAS on CSCs directly and/or the CSC niche. Targeting the RAS, its bypass loops, and convergent signaling pathways participating in the TME and other key stem cell pathways that regulate CSCs may be a novel approach to cancer treatment:
Keywords
Introduction
Cancer is the leading cause of death globally across all income levels, with its incidence predicted to rapidly rise. This is largely attributed to aging populations, and to a lesser extent, adoption of certain lifestyles. 1 The growing burden of cancer and the escalating cost of treatment 2 underscore an urgent need for more effective and affordable treatment.
There are two concepts guiding cancer research: (1) the stochastic model, also known as the clonal expansion model, and (2) the cancer stem cell (CSC) model, also known as the hierarchical model of cancer. 3 The stochastic model proposes that cancer cells result from normal cells acquiring genetic mutations and oncogenic properties in a stepwise fashion. The high mutation rate increases the probability of cell clones being formed that are adapted to the selection pressures of a given tumor site. 3 However, the CSC model proposes CSCs as the origin of cancer and the driver of treatment resistance, 4 cancer metastasis, and recurrence. 5 CSCs divide asymmetrically giving rise to non-tumorigenic cancer cells which form the bulk of the tumor, and identical highly tumorigenic CSCs that sit atop the cellular hierarchy in an undifferentiated state. 6 CSCs have several proposed origins, including non-malignant stem cells and/or progenitor cells, 5 mature cells, and from the fusion of stem cells and mutant cells. 7
The stochastic model and the hierarchical model of cancer are not mutually exclusive, as the concept of cellular plasticity bridges the apparent dichotomy of the two concepts. 8 Cancer cells are plastic and possess the ability to interconvert between differentiated and stem-like phenotypes, depending on a continuum of cell fates and the surrounding microenvironmental niche. 8 It has also been observed that hierarchical cell populations contain transitory cells that change their position within the cellular hierarchy 9 and that stochastic cell populations can generate hierarchically organized cell populations. 10 Furthermore, cancer cells can dedifferentiate and enter the CSC pool to gain tumor repopulation capability. This may be inherited, as proposed in the hierarchical concept by which cancer cells are endowed with intrinsic stem cell properties, rather than acquiring them in a stepwise fashion. Alternatively, this capability may be acquired by gene mutations, as proposed by the stochastic model. 8
Metastasis is the primary cause of cancer death. 11 Metastasis, resistance to chemotherapy, radiotherapy, 12 and immunotherapy 13 have been attributed to the presence of CSCs, which have been identified in many cancer types14–17 using stemness-associated markers and functional investigations. 17 Several strategies to target CSCs have emerged and they been investigated in clinical trials, including (1) monoclonal antibodies against CSC-specific surface biomarkers; CSC-associated signaling pathways such as the Notch (by various methods), Hedgehog, Wnt, and mitochondrial oxidative phosphorylation pathways, (2) inhibiting C-X-C chemokine receptor type 4 (CXCR4), part of the CSC microenvironment, (3) CSC-directed immunotherapies, such as chimeric antigen receptor T-cell therapy, and (4) immune checkpoint blockers targeting the receptors programmed cell death protein 1 and cytotoxic T-lymphocyte-associated protein 4. 18 The activity of human epidermal growth factor receptor 2–targeted therapies and CDK4/6 inhibitors against CSCs has been recently realized.19,20
The tumor microenvironment (TME) is an important regulator of CSCs. 21 The TME contains immune cells such as tumor-associated macrophages (TAMs), tumor blood vessels and lymphatics, cancer-associated fibroblasts (CAFs), pericytes, and adipocytes. 22 Intercellular communication within the TME is achieved with cytokines, growth factors, chemokines, inflammatory mediators, and remodeling enzymes in the tissue. 22 Targeting non-malignant cells and the mediators of intercellular communication, and other TME components, has been proposed as a treatment strategy for cancer. 22
Understanding the interactions between CSCs and the TME is crucial, as CSCs are a key mediator of treatment resistance, tumor metastasis, and recurrence. 5 Key interactions between CSCs and immune cells result in evasion of immune detection and elimination and can create an anti-inflammatory and/or protumorigenic microenvironment milieu. 5 CSCs also alter the balance between protumorigenic and antitumor immune cell activity within the TME. 5 CSCs achieve successful immune evasion through reduction of antigenicity by downregulating components involved in antigen presentation and processing, such as transport associated with antigen processing, and/or the major histocompatibility complex. 23 The downregulation of these components facilitates immune evasion of CSCs from the adaptive immune system, and thus their survival. 5
The renin–angiotensin system (RAS) plays an important role in the TME through its effects on tumor cells, non-malignant cells, hypoxia, angiogenesis, and the inflammatory response. 21 This endocrine system regulates blood pressure, blood volume, and electrolyte homeostasis 24 and functions at both systemic and local levels. 25 Physiologically, angiotensinogen is synthesized and released by the liver and is converted to angiotensin I (ATI) by renin, released by the juxtaglomerular apparatus of the kidney. ATI is converted to angiotensin II (ATII) by angiotensin-converting enzyme (ACE). ATII is the main effector for the RAS that binds to ATII receptor 1 (AT1R) and ATII receptor 2 (AT2R) to exert its physiological effects. 25 Angiotensin 1–7 (Ang1–7) is a cleavage product of ATII that maintains cardiovascular homeostasis by binding to the G protein–coupled receptor MAS. 26 ATII is converted to angiotensin III by aminopeptidase A. Bypass loops which include the proteases cathepsins B, D, and G, and chymase provide redundancies to this system 26 (Fig. 1).
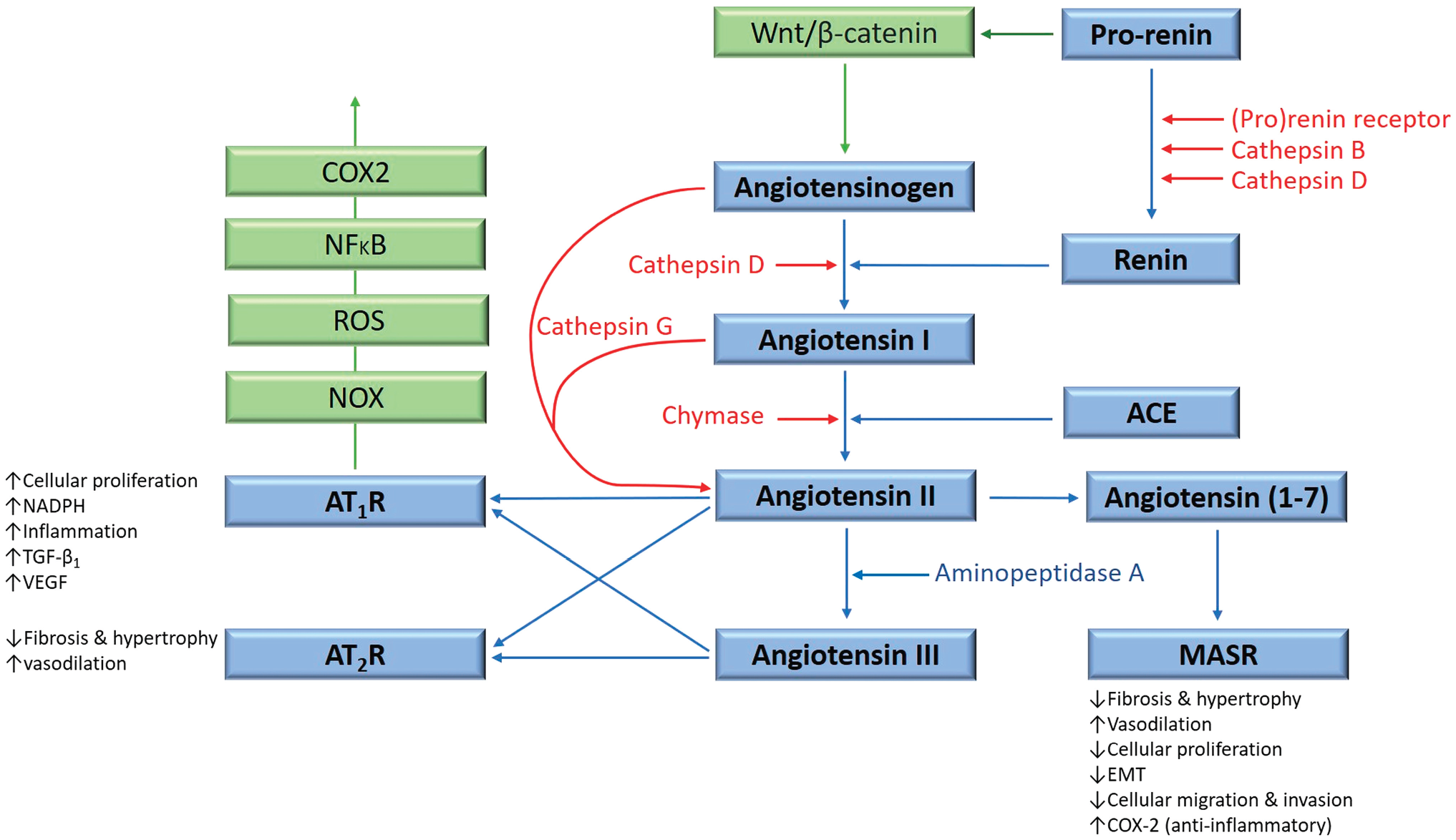
The renin–angiotensin system (RAS), its bypass loops, and converging signaling pathways. In the classical RAS (blue), angiotensinogen is converted to angiotensin I (ATI) by renin. ATI is converted to angiotensin II (ATII) by angiotensin-converting enzyme (ACE). ATII is the main effector for the RAS that binds to ATII receptor 1 (AT1R) and ATII receptor 2 (AT2R) to exert its physiological effects. Angiotensin 1–7 (Ang1–7) is a cleavage product of ATII which binds to the G protein–coupled receptor MAS receptor (MASR), which in turn exerts physiological effects. ATII is converted to angiotensin III (ATIII) by aminopeptidase A. ATII and ATIII bind to both AT1R and AT2R. Proteases including cathepsins B, D, and G, and chymase constitute bypass loops of the RAS (red), providing redundancies to this system. Other signaling pathways converge on the RAS (green) with the Wnt/β-catenin signaling pathway being upstream of the RAS, activated by the prorenin receptor. Inflammatory signaling via the NOX-ROS-NFκB-COX2 axis, downstream of the RAS, is induced by activation of AT1R. Abbreviations: COX2, cyclooxygenase 2; ROS, reactive oxygen species; VEGF, vascular endothelial growth factor; TGF-β1, transforming growth factor β1; EMT, epithelial-to-mesenchymal transition; NOX, NADPH oxidase; NFκB, nuclear factor kappa B.
The RAS is implicated in cancer, 27 fibrotic conditions,28,29 cardiovascular, 30 renal, 31 and lung 32 diseases. Components of the RAS33–35 and cathepsins B, D, and G that constitute bypass loops of the RAS36–38 are expressed by CSCs in many cancer types. Targeting CSCs by inhibiting the RAS, its bypass loops, and converging signaling pathways has been proposed as a potential treatment strategy for cancer by repurposing existing low-cost medications.25,26
CSCs are also regulated by crucial stem cell pathways, including the Notch, Wnt, Hedgehog, and Hippo 5 pathways. In clinical trials, some of these CSC-associated pathways have been targeted to regulate and eliminate CSCs, which sustain and drive tumor development. Given the RAS is also an important stem cell pathway, it may also play a role in directly regulating CSCs. 39
This article discusses the interaction between CSCs and the TME, and the role of the constituents of the TME, including immune cells and the RAS, in regulating the CSC niche. We propose a system-wide approach to targeting CSCs by exploiting the TME using inhibitors of the RAS, its bypass loops, and convergent signaling pathways.
Tumor Microenvironmental Niche and CSCs
CSCs reside in a niche, an anatomically distinct microenvironment within the overall TME. Cells within the niche produce factors that induce CSC self-renewal and angiogenesis. They also attract immune cells and other stromal cells that release growth factors, which promote tumor invasion and metastasis.8,40 Tumor development is influenced by a balance of protumorigenic factors and antitumor immune cell activity within the TME. CSCs also influence this balance within the TME to drive and sustain tumorigenesis. CSCs and the TME are highly relevant clinically, as treatment resistance caused by CSCs12,13 ultimately leads to treatment failure and cancer death. Treatment resistance is partly attributed to quiescence by which CSCs are able to arrest the G0 phase of the cell cylce41,42; upregulation of drug efflux pumps such as ATP-binding cassette (ABC) transporters, for example, ABCB1, ABCC1, and ABCG2 43 ; metabolic mediators 44 ; and activation of DNA repair mechanisms. 45
Interactions between CSCs and their microenvironmental niche also contribute to treatment resistance. 46 TME-induced conversion of non-CSCs into CSCs is one such interaction, underscoring the plasticity of non-CSCs that may contribute to therapy resistance. 8 This highlights the capacity of cancer cells to acquire CSC characteristics under the influence of TME-related factors, for example, in ovarian cancer, in which cancer cells acquire a CSC-like phenotype following cisplatin administration in vitro. 47 CSCs are sustained by several crucial stem cell signaling pathways, including the Notch, Wnt, Hedgehog, and Hippo pathways, which are often altered in CSCs, 48 supporting CSC traits such as maintenance and survival. 48 These pathways interact with oncogenic signaling pathways that function within the TME, including the nuclear factor kappa B (NFκB), mitogen-activated protein kinase (MAPK), phosphatidylinositol-3 kinase, and epidermal growth factor receptor (EGFR) cascades, 5 which are treatment targets for some cancers. 48 In addition, cathepsin-related signaling pathways operating within the TME may contribute to cancer growth and invasiveness. Cathepsins B, X, and K are involved in treatment resistance and tumor cell apoptosis, 49 while other cathepsins cause tumor suppression. 50 In particular, cathepsin K has been demonstrated in glioblastoma CSC niches. 51 It is tightly regulated by cathepsin K activating proteases and other components of the TME, supporting a functional role of cathepsin K in the glioblastoma CSC niche. 51 Cathepsin B is also highly active within glioblastoma CSC niches, which affects the interactions between glioblastoma cells and endothelial cells, and endothelial cell proliferation. 51 Despite these findings, the precise effects of the functionally diverse cathepsins demonstrated in CSC niches, as in glioblastoma, remain unclear.
The Immune System and the TME
The ability of CSCs to evade immune detection and induce a protumorigenic environment 52 is attributed to their interactions with immune cells within their niche. For example, TAMs induce proliferation of CSCs in hepatocellular carcinoma via interleukin 6 (IL-6)-induced activation of STAT3, which causes cytokine release, and a positive feedback loop that contributes to CSC self-renewal5,53 (Fig. 2). In breast cancer, the epithelial-to-mesenchymal transition (EMT) process upregulates EphA4 and the stem cell marker CD90, which mediate interactions between TAMs and CSCs by binding with their respective receptors. 54 Activation of the EphA4 receptor causes activation of Src and NFκB. 54 NFκB in CSCs induces the release of cytokines that sustain CSC properties.54,55 Myeloid-derived suppressor cells (MDSCs) are another immune cell type that support CSCs. In ovarian cancer, MDSCs promote CSC characteristics by causing microRNA-101 expression, inducing expression of stemness genes. 56
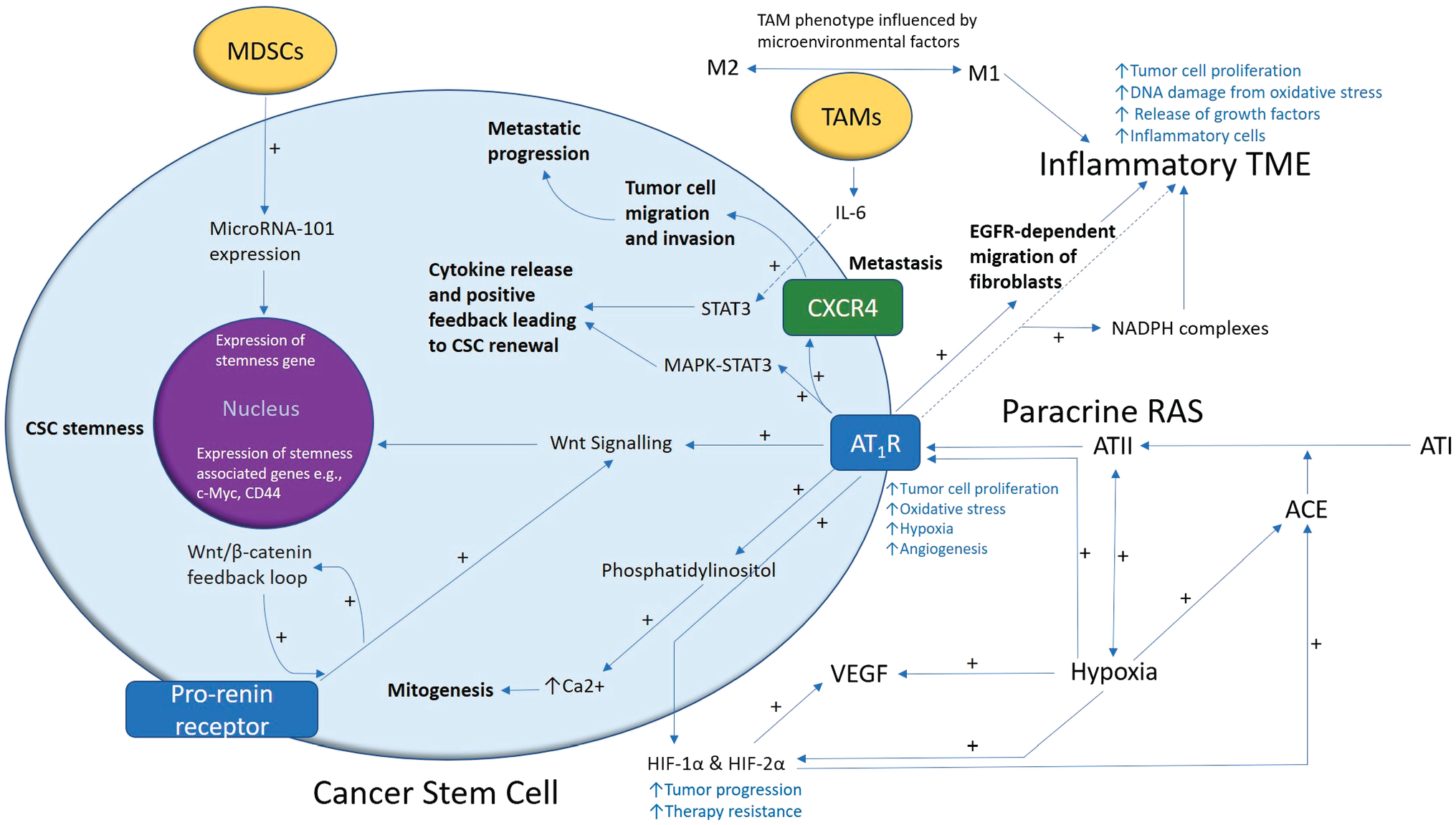
A proposed model demonstrating the role of the renin–angiotensin system (RAS) in cancer stem cell (CSC) niche. A CSC (with the cytoplasm in light blue and the nucleus in purple) residing within the tumor microenvironment (TME). Angiotensin II (ATII), the physiologically active end-product of the paracrine RAS, activates ATII receptor 1 (AT1R) leading to increased tumor cell proliferation, oxidative stress, hypoxia and angiogenesis, and inflammation — the hallmarks of cancer. This contributes to an inflammatory TME by increasing the number of inflammatory cells, partly by increasing the number of NADPH complexes, leading to tumor cell proliferation, DNA damage from oxidative stress, and release of growth factors. AT1R also activates phosphatidylinositol signaling which increases cytosolic Ca2+ to promote mitogenesis. Hypoxia increases paracrine RAS activity by upregulating angiotensin-converting enzyme (ACE) and the expression of hypoxia-inducible factor 1α (HIF-1α) and HIF-2α, which increase tumor progression and treatment resistance. HIF-1α, HIF-2α, and hypoxia increase the expression of vascular endothelial growth factor (VEGF). VEGF increases angiogenesis. Binding of AT1R to C-X-C chemokine receptor type 4 (CXCR4) promotes tumor cell migration and invasion, leading to metastatic progression. AT1R, via MAPK-STAT3 signaling, contributes to a cytokine release that leads to CSC renewal. AT1R signaling also contributes to the migration of fibroblasts in an epidermal growth factor receptor (EGFR)-dependent fashion. AT1R signaling and the prorenin receptor, which act in a feedback loop with Wnt/β-catenin, increase Wnt signaling which promotes CSC stemness by upregulating stemness-associated markers. Myeloid-derived suppressor cells (MDSCs) promote CSC characteristics by increasing microRNA-101 expression that induces expression of stemness-related genes in CSCs. Under the influence of the TME, polarization of tumor-associated macrophages (TAMs) — immune cells that are located within the TME — changes from the M1 to M2 phenotype. M2 TAMs induce the proliferation of CSCs via interleukin 6 (IL-6)-induced activation of STAT3, leading to cytokine release and positive feedback contributing to CSC renewal. Abbreviations: ATI, angiotensin I; AT2R, ATII receptor 2; Ang1–7, angiotensin 1–7; ATIII, angiotensin III; MAPK, mitogen-activated protein kinase.
Glioma CSCs promote macrophage polarization toward the M2 phenotype, which is immunosuppressive and protumorigenic, by producing cytokines such as transforming growth factor β and macrophage inhibitory cytokines. 57 Interestingly, CSCs can attenuate the anticancer properties of TAMs and dendritic cells. 5 CSCs in malignant melanoma and glioblastoma inhibit proliferative T-cell responses and IL-2 production in vitro,58,59 and CSCs in head and neck squamous cell carcinoma cause an immunosuppressive and procancer effect through induced proliferation of regulatory T cells. 58 These interactions underscore the complex relationships between CSCs and their surrounding TME, and the many potential therapeutic targets that exist.
The RAS and the TME
The RAS participates in the TME to facilitate hallmarks of cancer, such as angiogenesis, tumor cell proliferation, and hypoxia. 60 This is largely mediated by binding of ATII to AT1R, which increases cellular proliferation and vascular endothelial growth factor (VEGF) production, causing angiogenesis, inflammation, and oxidative stress. 21 The local (paracrine) RAS can act independently of or synergistically with the endocrine RAS. Administration of ATII increases the ability of cells with a CSC phenotype to form spheroids, and growth in a 3D culture Matrigel-based assay in vitro. 61 ATII also improves cell survival via an anchorage-independent process, and their expression of the stemness-associated marker CD133 is significantly upregulated. 61 This shows the RAS is not only a critical regulator of stem cells, but also increases the stemness of cancer cells via the action of ATII. 61 This and the fact that CSCs in multiple cancer types express components of the RAS and its bypass loops 60 ,62–65 suggest the RAS and its convergent signaling pathways play an important role in regulating CSCs within the TME. 26
ATII causes cellular proliferation by activating MAPK-STAT3 66 and phosphatidylinositol signaling via AT1R, to increase cytosolic Ca2+ which in turn affects mitogenesis 67 (Fig. 2). ATII also affects cell growth by modulating tyrosine kinases 68 and stimulates migration of fibroblasts via activation of AT1R in an EGFR activation–dependent process.21,69 In a breast cancer model, AT1R signaling promotes tumor cell contraction, migration, and invasion, by enhancing the expression of CXCR4—a chemokine receptor involved in the attraction of tumor cells to lymph nodes 70 (Fig. 2). AT1R signaling also increases expression of focal adhesion kinase (FAK)/RhoA signaling molecules and their downstream effectors ROCK1, ROCK2, and MLC, which cause cell contraction and pseudopodia formation, allowing cell migration and invasion. This is relevant as FAK/RhoA signaling molecules are key promoters of directional cell movement. 70 CXCR4 signaling is crucial for AT1R-induced lymph node metastasis in breast cancer. 70 These observations support the use of RAS modulating agents to mitigate cancer and metastasis.
The TME is often hypoxic, which promotes tumor progression and treatment resistance, largely via the effects of hypoxia-inducible factor 1α (HIF-1α) and HIF-2α 21 (Fig. 2). RAS activation in the TME contributes to hypoxia by ATII-mediated production of reactive oxygen species (ROS) 52 which create a proinflammatory environment containing proangiogenic cytokines.27,52 Hypoxia upregulates ATII, ACE, and AT1R.71,72 ATII activates HIF-1α via AT1R activation, leading to upregulated expression of both VEGF and ACE. This increases ATII formation that binds to and increases AT1R signaling.69,73 The effect of the RAS in increasing the expression of VEGFa is supported by the reduction of VEGF-A mRNA and protein, and reduced tumor growth in lung tumors following ACE inhibitor (ACEI) administration in vivo. 74 Furthermore, administration of an AT1R antagonist prevents an ATII-mediated increase in VEGF-A mRNA and protein in vivo. 74
Inflammation contributes to the TME through various mechanisms, notably the higher number of inflammatory cells, increased ROS, DNA damage, growth factors, and other inflammatory milieu. 21 The RAS influences inflammation 75 by activation of AT1R by ATII. This causes oxidant signaling via NADPH oxidase (NOX) complexes that play a role in inflammation.27,76 Components of the RAS are expressed by CSCs and non-CSCs, and stromal cells such as TAMs and CAFs. The expression of the RAS by TAMs is relevant, as these cells can either increase or decrease inflammation depending on their polarization. M1 macrophages produce a large amount of proinflammatory cytokines. 77 The TME can polarize M1 macrophages to form M2 macrophages, which reduce inflammation, and increase angiogenesis and tissue repair. 77 The RAS may also contribute to an immunosuppressive TME by reducing infiltration of TAMs. 78
EMT is a critical process in carcinogenesis and metastasis. 79 The involvement of the RAS in EMT and cellular differentiation is demonstrated in kidney disease and metastatic colorectal cancer to the liver. 80 Furthermore, high levels of ATII induce EMT in tubular cells toward a myofibroblastic phenotype in chronic nephropathy; this causes renal fibrosis, a process prevented by ACE inhibitor (ACEI) and angiotensin receptor blocker (ARB) administration. 81 Treatment of human colorectal cancer with ARBs decreases the EMT markers ZEB1, vimentin, and MM-9. 80 The effects of RAS inhibition in tumor models and cell lines have been demonstrated in many cancer types, for example, inhibition of tumor cell growth, proliferation and migration, metastasis, and prolonged survival in tumor-bearing animals. 26 Administration of losartan, an AT1R inhibitor, improves delivery of chemotherapy agents to breast cancer and pancreatic cancer xenografts, via a tumor stroma-mediated reduction in matrix stiffness and decompression of tumor microvessels. 82 The finding of reduced tumor growth upon transplantation of melanoma cells into Agtr1−/− mice further supports the critical role of the RAS in the TME. 83 Candesartan, an ARB, significantly reduces angiogenesis and metastases in a mouse lung cancer model 84 and prostate 85 and ovarian 86 cancer xenograft models. Reduction in tumor growth rates by losartan has also been observed in multiple cancer types.82,87 Irbesartan, another ARB, reduces tumor growth and angiogenesis in metastases of colorectal cancer to the liver in an in vivo model. 88
The RAS has multiple effects on the TME and cancer cells, although it is not clear whether this critical endocrine cascade also directly affects the stemness of CSCs. The RAS is important for the maintenance and differentiation of stem cells 26 and has been shown to drive differentiation of mesenchymal stem cells into adipocytes. 89 ACE is required for hemangioblast expansion and modulation of AT1R and AT2R signaling which directly affects differentiation of the blasts toward a hematopoietic or endothelial lineage. 90 The RAS also plays an important role in other developmental processes involving stem cells91–93 including vasculogenesis, myeloid differentiation, and erythropoiesis.91–94 The RAS acts in a feedback loop with the Wnt/β-catenin pathway, as prorenin can induce Wnt/β-catenin (Fig. 2). This is important for embryonic development, as it induces differentiation of pluripotent stem cells. 95 This is relevant, as CSCs express stemness-associated markers such as the Yamanaka factors 96 and because components of the RAS are targets of Wnt/β-catenin. Wnt signaling is implicated in cancer, and its end targets include the stemness-associated markers CD44 and c-MYC. 97 It could be that CSCs require activation of Wnt signaling, as seen in leukemia stem cells in acute myeloid leukemia. 98 This suggests that RAS modulators could be used to inhibit Wnt signaling and its downstream effects on CD44 and c-MYC, and possibly other stemness-associated markers, expressed by CSCs.26,99,100 Animals treated with losartan develop tumors containing more differentiated cells compared with untreated tumors which contain less differentiated cells. 101 This supports the possible influence of the RAS on cancer cell differentiation. 101 Furthermore, 20% of animals in the losartan treatment group are tumor free, compared with control animals, by the end of the study at 100 days. 101 This suggests that AT1R signaling is important for tumor progression, the loss of differentiation in tumor cells, and possibly the stemness of CSCs. Further research is required to determine the precise role of the Wnt/β-catenin pathway within the RAS, in regulating CSCs within the CSC niche. These findings call for further investigation of RAS inhibition as a potential therapeutic option for treating cancer, with clinical trials.
RAS Inhibitors and Cancer
Hypertension is common in cancer patients, and given the widespread use of RAS modulators for cardiovascular and renal diseases, their effects on cancer incidence and outcomes have been investigated in many observational studies. 94 Numerous studies have shown that RAS inhibitors (RASIs), especially ACEIs and ARBs, used for the treatment of hypertension, improve not only cardiovascular disease outcomes but also survival outcomes for cancer patients (Table 1). For example, RASIs have been associated with improved survival of patients with non-small cell lung cancer in some studies107,108 although not in others. 109
Studies Investigating the Association Between RASIs and Cancer Incidence and Outcomes.
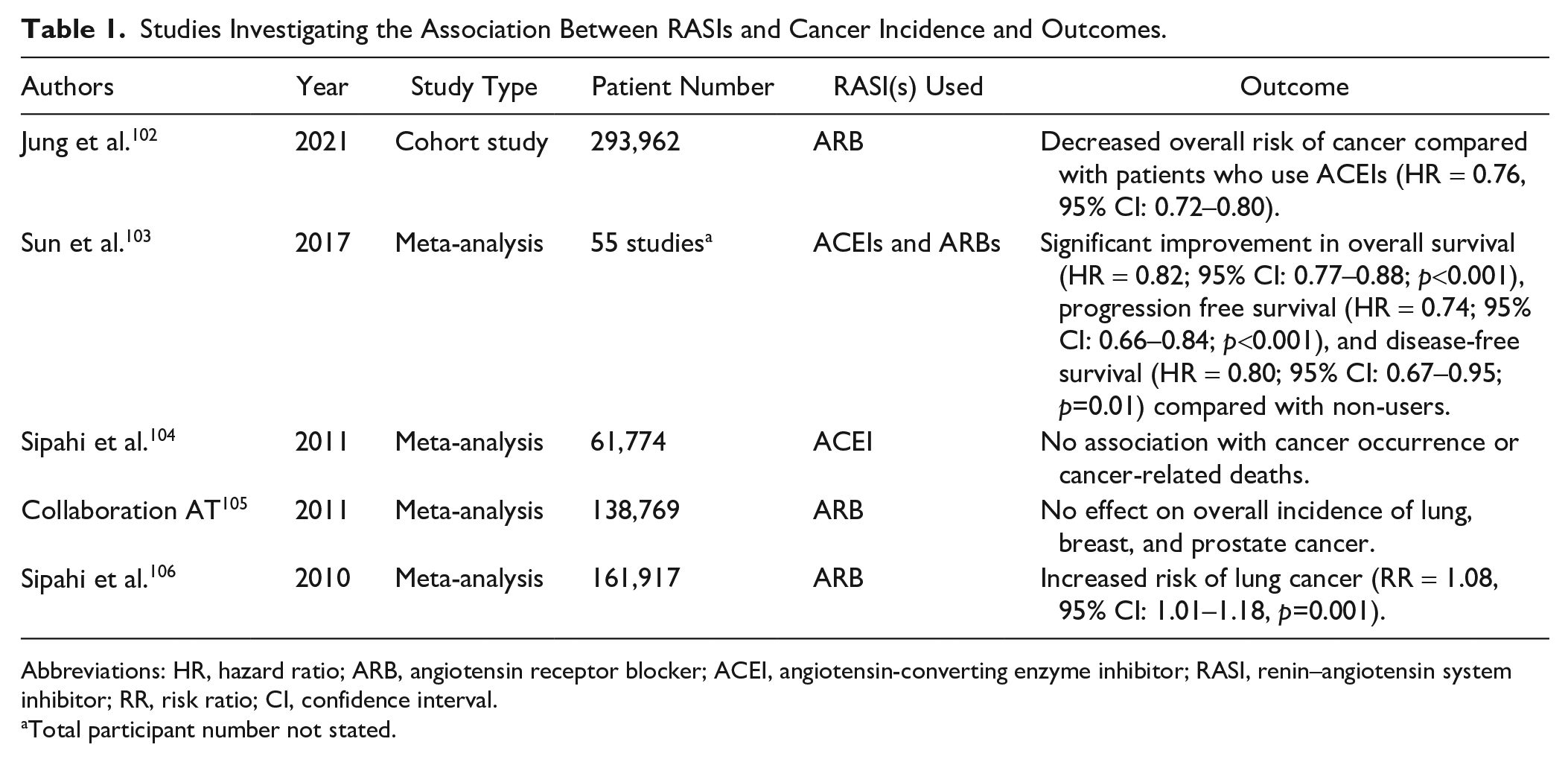
Abbreviations: HR, hazard ratio; ARB, angiotensin receptor blocker; ACEI, angiotensin-converting enzyme inhibitor; RASI, renin–angiotensin system inhibitor; RR, risk ratio; CI, confidence interval.
Total participant number not stated.
ACEI administration has not been shown to change the incidence of cancer in several studies.104,106,110 A meta-analysis of 161,917 patients from 13 studies reports an increased risk of lung cancer in patients who were administered ARB. 106 Another meta-analysis of 61,774 patients from 14 studies concludes that ACEI therapy does not affect cancer occurrence or cancer-related deaths. 104 The incidence of certain cancers increases in organ transplant recipients, with immunosuppressive therapy being a major risk factor. For example, cutaneous squamous and basal cell carcinomas are common in renal transplantation patients. ACEI or ARB use has been associated with an approximate 2-fold risk reduction of skin cancers in renal transplantation patients compared with non-users. 111 This association is supported by functional data, which show ATII blockade in an SCID-beige mouse pulmonary metastasis model effectively blocks cyclosporin-associated tumor progression in vivo. 112 A cohort study demonstrates ARB use is associated with a decreased overall risk of cancer compared with patients who use ACEI [hazard ratio (HR) = 0.76, 95% confidence interval (CI): 0.72–0.80] with a gradual dose–response relationship also being observed, and the cancer risk decreasing in those on prolonged ARB regimes compared with ACEI use. 102
Other studies show no improvement of outcomes of cancer patients administered with RASIs (Table 1). For example, a meta-analysis of 138,769 patients concludes that ARB administration does not affect the overall incidence of cancer including lung, breast, and prostate cancer or overall cancer deaths in those treated with ARBs. 105 In contrast, a recent meta-analysis of 55 studies demonstrates a significant improvement in overall survival (HR = 0.82; 95% CI: 0.77–0.88; p<0.001), progression-free survival (HR = 0.74; 95% CI: 0.66–0.84; p<0.001), as well as disease-free survival (HR = 0.80; 95% CI: 0.67–0.95; p=0.01) for cancer patients using RASIs compared with non-users. This study also shows the effect of RASIs is dependent on the cancer type, with differences demonstrated between hepatocellular carcinoma, renal cell carcinoma, gastric cancer, and bladder cancer. 103 These conflicting findings from observational studies highlight the need for further investigation with well-designed studies, including double-blinded randomized controlled trials to determine the effect of RASIs on the incidence and outcomes of cancer patients.
There is, however, substantial evidence demonstrating that RASIs decrease tumor growth and progression, angiogenesis, and metastasis.26,94 Existing in vitro and in vivo functional data consistently show an anticancer effect of RASIs, and the overall epidemiological data suggest RASIs could improve the survival of cancer patients with the degree depending on the cancer type and the type of RASI used. Further functional work using more sophisticated model systems, such as organoid systems, may advance our understanding of the mechanisms underpinning the relationship between CSCs and the TME, and particularly the RAS. Randomized controlled trials are warranted to establish the precise role of RASIs on cancer incidence and outcomes.
Therapeutic Targeting of the CSCs: Future Directions
Despite the emergence of new targeted therapies for cancer such as small-molecule inhibitors, immunotherapy, and biochemotherapy (combination of chemotherapy and immunotherapy), treatment resistance and treatment failure remain an unsolved clinical problem. For example, treatment of BRAF V600E mutant malignant melanoma with a BRAF inhibitor, or in combination with an MEK inhibitor, results mostly in an initial partial treatment response due to genetic and epigenetic mechanisms determined by the TME, as well as immunogenic and transcriptomic alterations.113,114 These changes may contribute to reactivation of MAPK signaling and subsequent treatment resistance. 115 Furthermore, targeted therapy is not always successful due to the high intratumor genetic and epigenetic heterogeneity, and cellular plasticity. This increases the possibility of switching a cancer cell into a slow-cycle state, which causes treatment resistance. Non-coding RNAs also contribute to treatment resistance by becoming resistant to anti-BRAF treatment, influenced by dynamic deregulation of microRNA. In turn, this leads to alteration of cell survival pathways and enhancement of proinflammatory and proangiogenic cues. 116 These resistance mechanisms underscore the need for a holistic system-wide approach that affects the tumor-forming capacity and stemness of CSCs, the proposed origin of cancer and metastasis.
The RAS plays a critical role in the CSC niche that regulates CSCs. In addition to the presence of bypass loops that provide redundancies, there are several inflammatory and developmental signaling pathways that converge on the RAS. For example, prorenin receptor induces Wnt/β-catenin signaling within a feedback loop. AT1R signaling can also cause inflammatory signaling via the NOX-ROS-NFκB-COX2 (cyclooxygenase 2) axis 26 (Fig. 1). Each primary step of this axis could be targeted using NOX inhibitors, ROS inhibitors, metformin, and nonsteroidal anti-inflammatory drugs, respectively (Fig. 1). Wnt/β-catenin can also be targeted by its respective inhibitor. 26 These axes and many steps of the RAS and its bypass loops can be inhibited using existing, low-cost medications. For example, renin secretion can be inhibited by β-blockers such as propranolol, 117 ACE can be targeted with ACEIs, and AT1R with ARBs. Enzymes that constitute bypass loops of the RAS such as cathepsins B, D, and G, and chymase can be blocked by their respective inhibitors. The MAS receptor has been demonstrated to have anticancer effects, making it a potential treatment target. Aminopeptidase A may be targeted with aminopeptidase A inhibitors.
This holistic system-wide approach of targeting the RAS, its bypass loops expressed on CSCs, and convergent signaling pathways, to regulate the TME that affects the tumor-forming capacity and stemness of CSCs, warrants further functional work with in vitro and in vivo models such as organoid systems. This treatment approach is currently being tested in a phase I/II clinical trial on glioblastoma and metastatic melanoma. 25
Footnotes
Competing Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: S.T. is an inventor of the patents Cancer Diagnosis and Therapy (PCT/NZ2015/050108), Cancer Therapeutic (PCT/NZ2018/050006), Novel Pharmaceutical Compositions for Cancer Therapy (US/62/711709), and Cancer diagnosis and therapy (United States Patent No. 10281472).
Author Contributions
EJK drafted the manuscript and STT critically reviewed and commented on the manuscript. The authors approved the final manuscript.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.