
Abstract
Proteolytic activity is perturbed in tumors and their microenvironment, and proteases also affect cancer stem cells (CSCs). CSCs are the therapy-resistant subpopulation of cancer cells with tumor-initiating capacity that reside in specialized tumor microenvironment niches. In this review, we briefly summarize the significance of proteases in regulating CSC activities with a focus on brain tumor glioblastoma. A plethora of proteases and their inhibitors participate in CSC invasiveness and affect intercellular interactions, enhancing CSC immune, irradiation, and chemotherapy resilience. Apart from their role in degrading the extracellular matrix enabling CSC migration in and out of their niches, we review the ability of proteases to modulate CSC properties, which prevents their elimination. When designing protease-oriented therapies, the multifaceted roles of proteases should be thoroughly investigated.
Keywords
Introduction: Proteases and the Proteolytic System
To date, 884 known and putative protease genes have been annotated in the human genome (https://www.ebi.ac.uk/merops/). 1 According to the chemical mechanisms of catalysis, proteases are grouped into five major classes: serine proteases, cysteine proteases, threonine proteases, aspartic proteases, and metalloproteases. In addition, there are proteases of mixed and unknown catalytic types. Based on similarities in their primary amino acid sequences, proteases are grouped into families that are further combined into structural clans. 1
Metalloproteases are characterized by the presence of a catalytic metal ion in the active site of the enzyme and represent the largest protease group in humans. The family includes matrix metalloproteases (MMPs),2 –4 a disintegrin and metalloproteases (ADAMs), 5 and ADAMs with a thrombospondin motif (ADAMTSs).6 –8 In humans, 24 MMPs have been characterized, some of which are secreted, whereas others are anchored in the cell membrane. MMPs are best known for their role in remodeling the extracellular matrix (ECM).3,4 They degrade structural components of the ECM, including different types of native collagens, fibronectin, elastin, laminin, and proteoglycans, and proteolytically activate pro-MMPs. Their activity also results in the release and activation of matrix-bound growth factors and/or their precursors, for example, transforming growth factor-β (TGF-β) and vascular endothelial growth factor (VEGF). 9 Based on their protein structure and in relation to their substrate specificity, MMPs are divided into six groups: collagenases (MMP1, MMP8, MMP13), gelatinases (MMP2, MMP9), stromelysins (MMP3, MMP10, MMP11), matrylysins (MMP7, MMP26), membrane-type MMPs (MMP14, MMP15, MMP16, MMP17, MMP24), and other non-classified MMPs.10,11
Half of the 22 known ADAMs possess enzymatic activity. These transmembrane proteins shed the ectodomains of other transmembrane proteins, including growth factor and cytokine precursors, their receptors, and adhesion proteins.5,12 On the contrary, all 19 ADAMTSs are secreted enzymes involved in proteolytic processing of ECM proteoglycans, procollagens, and other substrates.6 –8
Another important group of proteases are cathepsins, that is, lysosomal proteases primarily responsible for intracellular protein catabolism.13,14 However, under certain conditions, cathepsins are released from lysosomes or even out of cells, where they perform other functions.14,15 According to the catalytic amino acid residue, cathepsins are divided into cysteine proteases (cathepsins B, C, F, H, K, L, O, S, V, W, and X), aspartic proteases (cathepsins D and E), and serine proteases (cathepsins A and G). 14
Proteases, their substrates, and endogenous protein inhibitors form a complex network called the degradome.16 –18 Proteolytic processing is tightly regulated and both directly and indirectly involved in virtually all biological processes, including cell proliferation, differentiation, death, and migration, and, importantly, in tissue remodeling.16,17,19,20
Among the numerous functions of proteases, there is evidence on their role in specialized anatomically delineated tissue microenvironments called niches that host and sustain certain cell types, such as stem cells (SCs).21 –23 Different cell types within SC niches provide a cocktail of cell-bound and secreted factors that, in concert with the niche ECM, shape the fate of resident SCs, either maintaining their quiescent state or inducing their proliferation and differentiation. 24 Among the best characterized SC niches is the bone marrow hematopoietic stem cell (HSC) niche,24,25 where proteolysis by MMPs23,26,27 and cathepsins27,28 has been demonstrated. Proteases regulate SC functions by reshaping the extracellular scaffold of the niche, degrading cell surface adhesion molecules, and/or regulating the bioavailability and activity of cytokines and growth factors such as stromal cell–derived factor 1α (SDF-1α), SC factor, TGF-β, and VEGF (reviewed in Tay et al., 24 Saw et al., 26 and Maurer et al. 27 ). SCs in the niches are mostly in a quiescent/dormant cellular state. 29 However, upon various endogenous cues, or cues from the microenvironment, several types of proteases are induced to release SCs from their niches. Secreted by SCs or from various niche-resident cells, such as neutrophil granulocytes, osteoblasts, and osteoclasts in the HSC niche, MMP8 30 and MMP931,32 have been reported to mobilize HSCs via cleavage of the cytokine, SDF-1α, thus preventing its binding to C-X-C chemokine receptor type 4 (CXCR4). The SDF-1α–CXCR4 axis and mobilization of HSCs are also affected by several cathepsins, including cathepsin X28,33 and cathepsin K. 34
Role of Proteases in Cancer
To maintain the fine tuning of complex proteolytic networks, proteolytic activity is tightly regulated at the levels of protease expression, activation, posttranslational modifications, and intercellular and intracellular trafficking. Furthermore, protease regulation is enhanced by the presence of selective endogenous protease inhibitors.19,35 –37 Dysregulated proteolysis underlies numerous pathologies, including cancer,38,39 where proteases are most commonly upregulated and act as protumorigenic factors.40 –42 However, some proteases are also involved in tumor suppression and are downregulated in cancer.43 –45 By taking part in various proteolytic cascades and networks, proteases are involved in several processes of tumor development and progression,39,46 –49 as summarized in Table 1. The primary function of proteases derived from either cancer or non-cancerous cells in the tumor microenvironment (TME)38,39,60 is not only ECM degradation, as originally anticipated, but also the modulation of other pericellular events, in particular the activity and bioavailability of growth factors, cytokines, cell surface receptors, and adhesion molecules, as well as the modulation of intracellular signaling pathways.39,49,50 Some proteases or their domains also possess non-enzymatic functions, 50 such as the inactive proform of cathepsin X 74 and several MMPs whose hemopexin domain is involved in protein–protein interactions. 23
Roles of Proteases in Cancer Development and Progression.

Abbreviations: MMPs, matrix metalloproteases; EMT, epithelial-to-mesenchymal-like transition; ECM, extracellular matrix; ADAMs, a disintegrin and metalloproteases; ADAMTSs, ADAMs with a thrombospondin motif.
Proteases in Cancer SCs and Their Niches
Cancer cells within a tumor are heterogeneous and occur in a variety of hierarchically organized and functionally distinct cell states. The modern concept of cancer stem cells (CSCs), which reside at the top of the tumor hierarchy or, alternatively, at the “bottom” of cancer evolution, was first introduced in leukemia 75 and later specified by the Eaves group. 76 The concept of a pool of CSCs with stemness characteristics has since been confirmed in many other malignancies, 77 for example, in breast, 78 colon,79,80 pancreatic, 81 and brain cancers, 82 including glioblastoma 83 and medulloblastoma. 84 CSCs, also called tumor-initiating cells, are a small subpopulation of cells within tumors that exhibit characteristics reminiscent of normal adult SCs. The key common characteristic of normal SCs and CSCs is asymmetric division (i.e., the ability to self-renew, yet differentiate via generation of progenies to mature, functionally differentiated cell types). In cancer, these mature cells comprise the bulk of the tumor mass, being initiated and maintained by CSCs, enabling fast tumor growth. 85 CSCs, like normal SCs, are mostly present in a so-called dormant state. In the TME, CSCs may undergo epithelial-to-mesenchymal-like transition (EMT) that allows their migration and, through a plethora of proteases, also invasion into the surrounding parenchyma and metastasis. 86
Apart from sharing the characteristic “stemness,” both normal SCs and CSCs reside in distinct microenvironments called niches. The niche milieu contains different cell types, including endothelial cells, immune cells, cancer-associated fibroblasts, and mesenchymal SCs.87 –89 In different cancer types, different types of niches have been identified. 88 They are best characterized in glioblastoma where perivascular, hypoxic, and invasive niches have been described. 90 A link between CSCs and the perivascular niche has also been reported in other brain tumors 91 and in several other cancers,92,93 for example, in colorectal cancer, 94 skin squamous cell carcinoma, 95 melanoma, 96 and head and neck squamous cell carcinoma. 97
Intercellular crosstalk and physical components of a niche either maintain the quiescent state of CSCs or induce their proliferation and differentiation, influence their metastatic potential, and play a role in protecting CSCs against the immune system.88 –90,98 Microenvi-ronmental conditions in the niche may also protect CSCs against therapeutic intervention. In medulloblastoma, CSCs display features imitating those of neural progenitor cells and SCs 84 ; they reside in perivascular niches and exhibit enhanced radioresistance compared with cells of the tumor bulk. 99 Similarly, slow-cycling perivascular tumor cells were shown to be therapy-resistant in a mouse model of glioma. 100 Due to their importance for disease progression and modulation of cancer-related processes such as inflammation and angiogenesis, stromal factors were proposed as molecular markers for diagnosis, prognosis, and treatment selection in prostate cancer. 101
Proteases play an important role within the CSC niches. Expressed and/or released either by cancer (stem) cells or by other cells of the niche, some proteases, such as cathepsins, are activated by the hypoxic and acidified TME.102 –106 In the following sections, we briefly summarize the ways in which proteases affect CSCs and their niches (Fig. 1). Specifically, we focus on primary brain tumor glioblastoma and glioblastoma stem cells (GSCs) that are commonly identified by the expression of a selected set of biomarkers [CD133, CD44, CD15 (SSEA-1), CD70 (CD27 L), Nestin, Olig2, SOX2, ALDH1A3, S100A4, Nanog, and OCT4].107,108 We have localized GSCs in hypoxic periarteriolar niches109,110 that closely resemble bone marrow HSC niches. 111
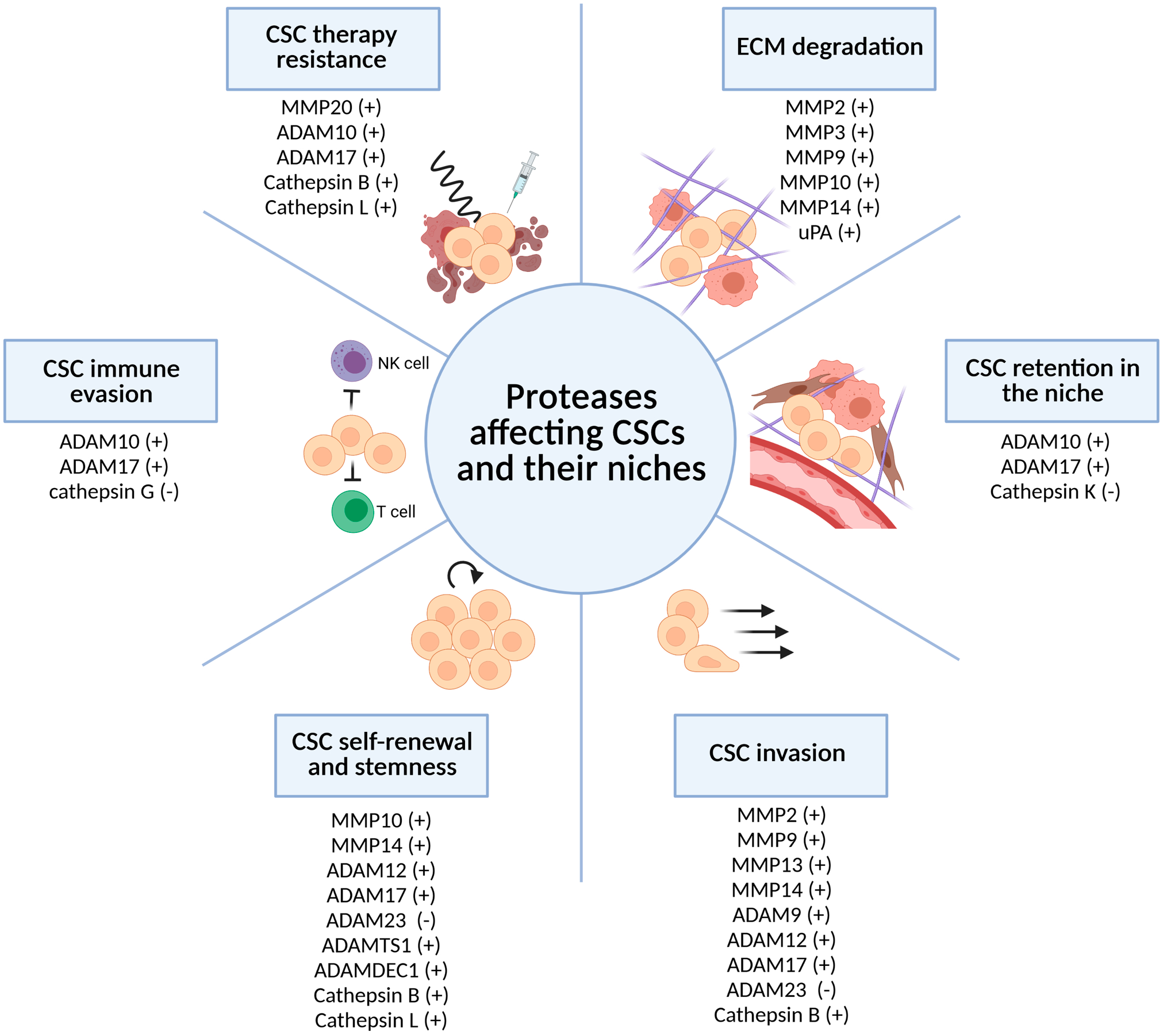
Proteases affect CSC functions and their niches. Proteases degrade niche ECM and influence several CSC properties, including their retention in the niche, invasion, self-renewal, stemness, immune evasion, and therapy resistance. Proteases either support (+) or inhibit (−) CSC-related processes. Created with BioRender.com. Abbreviations: CSC, cancer stem cell; ECM, extracellular matrix.
Proteases and the ECM
As mentioned previously, proteases were first investigated as key enzymes remodeling the ECM, which is increasingly recognized as a functional physical and biochemically active component of the niche, also with direct impact on the regulation of CSCs.112,113 Increased expression of several proteases in breast CSCs contributes to degradation of the ECM and invasion into their surroundings.114 –116 On the contrary, ECM components provide physical support and CSC anchorage, assuring their self-renewal.112,113,117 Furthermore, the ECM represents a reservoir of growth factor and cytokine precursors that are activated and released upon proteolysis and affect CSCs.112,113 Among these, TGF-β, 118 VEGF, 119 and insulin-like growth factors 120 can be highly protumorigenic in neoplasia. In addition, partial proteolytic degradation of certain ECM components generates bioactive peptide fragments (so-called matrikines) that are able to regulate cellular activities.121,122 Thus, besides their well-established roles in ECM degradation and direct promotion of cancer cell invasion, proteases also influence CSC self-renewal, EMT, chemoresistance, and immune evasion.116,123,124
Proteases and CSC Migration Out of Niches
Components of the ECM serve as anchoring sites for CSC adherence. CSCs interact with the ECM through several cell surface receptors, including integrins, discoidin domain receptors, and CD44, which maintain CSCs by inducing intracellular stem and proliferative signaling pathways. 112 Cathepsin K is an atypical papain-clan cysteine cathepsin, originally reported as a collagenolytic protease produced by osteoclasts, 125 which appears to be overexpressed in several cancers, particularly glioblastoma. 126 In addition to various types of native collagens, cathepsin K can also hydrolyze various other fibrillar proteins and proteoglycans. 125 In glioblastoma, cathepsin K, along with cathepsins B and X, is colocalized with GSC markers CD133 and nestin, and GSC niche markers osteopontin and SDF-1α and its receptor CXCR4.105,127 It was suggested that cathepsin K could induce GSCs to migrate out of their niches,104,109 and this notion was later supported by Hira and coworkers who demonstrated the ability of cathepsin K to cleave and inactivate SDF-1α in vitro. 128 This mechanism was first observed in the HSC niche where interactions between SDF-1α, secreted by osteoblasts and endothelial cells, and CXCR4, expressed by HSCs, retain HSCs,33,34 highlighting the similarity of the GSC niche and the physiological HSC niche.110,111 In addition, Siney et al. 129 suggested a possible role of ADAM17 and ADAM10 in the retention of GSCs in the tumorigenic niche, as inhibition of both proteases increased CSC migration and differentiation.
Proteases and CSC Invasion
In response to signals from the TME, CSCs can undergo the EMT and acquire migratory capacity to initiate the first steps in metastasis.86,130 CSCs also contribute to tumor spread via enhanced angiogenesis, that is, the formation of alternative vascular structures by vasculogenic mimicry, which is analogous to the mimicry of embryonic vasculogenesis by tumor cells first described by the Hendrix group in melanoma 131 and later in other cancers. 132 Vasculogenic mimicry requires activation of the transmembrane metalloproteases, MMP14 and MMP9, which support the proteolytic cascade in melanoma CSC invasion. 131 In breast cancer, CSCs line vasculogenic mimicry channels and synergize their formation in the perivascular niche. 133
Westhoff et al. 134 showed that invasion of GSCs is enhanced by activation of MMPs involved in fibronectin processing, thus forming routes facilitating GSC migration. The invasiveness of GSCs was shown to be supported by several metalloproteases, such as MMP2, MMP9, 135 and MMP13, 136 as well as ADAM9 and ADAM17.137,138 Invasion of GSCs is also directly supported by stromal cells proteases. Ye and coworkers have shown that tumor-associated microglia and macrophages enhance the invasiveness of GSCs via the release and activation of the TGF-β1 signaling pathway, leading to upregulation of MMP9, 139 whereas microglia-derived MMP14 activates GSC-derived MMP2. 140 The roles of urokinase-type plasminogen activator receptor (uPAR) and cathepsin B in GSCs were investigated by Alapati et al. 141 who showed that simultaneous downregulation of both proteins inhibited irradiation-induced integrin signaling to the cytoskeleton and to the nucleus via protein kinase C. In addition, the complex of mitogen-activated protein kinase kinase 1 and phospho-c-jun N-terminal kinase was translocated from the cytosol to the nucleus, resulting in migratory arrest of GSCs. 142
In breast CSCs, MMP14 is one of the most upregulated proteases, especially under hypoxic conditions that trigger its relocalization to the cell surface.143,144 This enzyme mediates the conversion of stationary CSCs into invasive CSCs—the mechanism thought to drive CSC metastasis. 144 MMP2,145 –147 MMP9,148,149 MMP14, 148 ADAM12, 150 and ADAM17 151 have also been shown to govern CSC invasion in other cancers. However, as emphasized above, proteases can also act as tumor suppressors. For example, in lung adenocarcinoma, ADAM23 suppresses migration and metastasis of CSCs by inhibiting integrin αvβ3 function. 152
Proteases and CSC Self-renewal and Stemness
CSC characteristics are maintained by a plethora of microenvironmental cues, including autocrine and paracrine growth factors (TGF-β, fibroblast growth factor, epidermal growth factor, VEGF), cytokines (interleukin [IL]-1β, IL-6, IL-8, tumor necrosis factor-α), and specific ligands that promote Hedgehog, Wnt, and Notch signaling pathways, leading to activation of NF-κB, JAK-STAT, GLI, β-catenin, LEF/TC, and NIC-CSL families of transcription factors.153 –156 Proteases play a crucial role as part of the signaling cascades in CSCs that sustain their self-renewal and stemness.
Metalloproteases
The metalloprotease ADAM17 assists in processing of Notch1 receptors and solubilization of the Notch ligands, Jagged-1 and Jagged-2. 157 In glioblastoma, ADAM17 has been identified as a mediator of stemness 158 and invasion of U87 GSCs 137 ; similar effects were proposed in colorectal CSCs 157 and in colorectal carcinoma CSC crosstalk with endothelial cells. 94 ADAM17-mediated activation of Notch1 was also detected in liver CSCs where the enzyme was activated by overexpression of inducible nitric oxide synthase. 159 Another protease secreted by GSCs, a disintegrin and metalloproteinase domain–like protein decysin 1, has been linked recently to the maintenance of GSCs, primarily via activation of an autocrine fibroblast growth factor signaling loop. 160 In addition, the claudin-low breast CSC phenotype was promoted by ADAM12, a protease induced during the EMT. 150 ADAMTS1 was recently associated with induction of stemness in uveal melanoma. 161 However, the tumor suppressor protease, ADAM23 (see above), was downregulated in the lung adenocarcinoma CSC subpopulation, which likely contributes to the stemness phenotype. 152
The maintenance of stemness of lung CSCs 162 and epithelial ovarian CSCs 163 was associated with MMP10, which inhibited the non-canonical Wnt signaling ligand, Wnt5a, and activated the canonical Wnt signaling pathway. MMP14 promoted the EMT and induced CSC properties in an oral squamous cell carcinoma cell line. 164 Consistent with these results, knockdown of MMP14 strongly affected CSC properties in breast carcinoma SCs. 143 In conclusion, metalloproteases of different classes affect multiple intracellular signaling pathways that are central to the maintenance of CSCs.
Cysteine and Serine Proteases
Cathepsin B and uPAR play important roles in regulating symmetric GSC division and self-renewal. Their expression correlates with the expression of the Hedgehog signaling components, SOX2 and BMI1, which are regulated by GLI factors, although the mechanisms have not been fully explained. 165 Wang et al. 166 demonstrated downregulation of the GSC marker, CD133, after knockdown of cathepsin L. We and others found that, due to excessive mRNA splicing in cancer cells, N-terminally truncated cathepsin L diffuses into the nucleus167,168 to proteolytically modify histones, thus affecting antiapoptotic gene expression and differentiation in cancer cells. 169
Proteases and SC Immune Evasion
The microenvironment around CSCs contains a broad spectrum of immune cells such as macrophages, myeloid-derived suppressor cells, natural killer (NK) cells, regulatory T-cells, cytotoxic T-lymphocytes (CTLs), and T-helper cells. 170 However, CSCs have evolved several ways to avoid their recognition and destruction by the immune system and to shape the TME into an immunosuppressive landscape. For example, CSCs can transform infiltrating macrophages from the M1 phenotype into the tumor-supportive M2 phenotype or tumor-associated macrophages.170,171 Among other factors, proteases and their endogenous inhibitors are involved in crosstalk between CSCs and immune cells in the TME.
An interesting example is NK cells, cytotoxic lymphocytes characterized by their ability to specifically recognize and eliminate target cells that lack the expression of major histocompatibility complex (MHC) class I molecules normally involved in cell surface presentation of antigenic peptides to CTLs. 172 GSCs have been shown to lack active serine cathepsin G, resulting in impaired cleavage of MHC class I molecules on these cells, thereby escaping recognition by NK cells.173,174 On the contrary, cysteine cathepsins normally contribute to NK cell cytotoxicity by proteolytically activating effector granzymes and perforin that are released by activated NK cells to induce target cell death. 175 In NK cells cocultured with oral squamous carcinoma CSCs, decreased levels of mature cathepsins C and H, and an increased level of their inhibitor, cystatin F, were observed, promoting the anergic state of NK cells.176,177 It has been proposed that CSCs or other stromal cells secrete cystatin F, which when internalized lowers the cytotoxic potential of NK and other cytotoxic cells in the TME. 123 Consistent with this suggestion, uptake of extracellular cystatin F by CTLs resulted in decreased activities of cathepsins C, H, and L, leading to impaired activation of granzymes A and B, and consequently lowered T-cell cytotoxicity. 178 High levels of proteinase inhibitor 9, a potent inhibitor of granzyme B, were detected in breast CSCs, most likely providing another means of CSC immune escape. 179 CSCs have also evolved mechanisms to evade γδ T-cells, a distinct subpopulation of T-cells that differ from the more commonly considered αβ T-cells in terms of their antigen recognition, activation, and effector functions. 180 In CSCs, increased ADAM10 and ADAM17 expression has been associated with increased shedding of the cell surface MHC class I polypeptide-related sequence A, which has been proposed as the main mechanism underlying CSC resistance to γδ T-cell cytotoxicity. 181
Proteases and CSC Therapy Resistance
Aside from residing in a quiescent state, being by itself protective against drugs that target exposed DNA during mitosis, CSCs also exhibit high expression of multidrug resistance proteins and enzymatic DNA damage repair mechanisms. Safeguarded in the shelter of their niche, these cells often represent the main reason for treatment failure and cancer recurrence.182 –184
As mentioned previously, active cathepsin B has been detected in GSC niches. 105 Using fluorogenic metabolic mapping, we detected cathepsin B activity in both non-irradiated and irradiated GSC NCH644 cells (Fig. 2). Expression of this enzyme was upregulated upon irradiation of glioblastoma U87 stem-like cells. 187 Apart from the role of cathepsin B and uPAR in maintaining the stemness characteristics of GSCs described above, 165 highly expressed cathepsin B and uPAR also protected these cells from irradiation-induced DNA damage, and this effect was reversed by silencing of the two genes. 187 Similarly, radiation resistance mediated by cathepsin L has been shown. 166 The well-known sheddase, ADAM10, marks CSCs with active Notch signaling that mediates chemoresistance. Targeted inhibition of active ADAM10 inhibited Notch activity and tumor growth in mouse models, particularly regrowth following chemotherapy. This suggests targeted inhibition of active ADAM10 is a potential therapy for ADAM10-dependent tumor development and drug resistance. 188 Colorectal CSC chemoresistance has been linked to another sheddase, ADAM17. 157 In oral carcinoma CSCs, overexpression of MMP20 supported stemness and was proposed to reduce the sensitivity of this cell population to chemotherapeutic agents. 189 Overall, these studies confirm the involvement of proteases in the therapeutic resistance of CSCs and support their therapeutic potential.
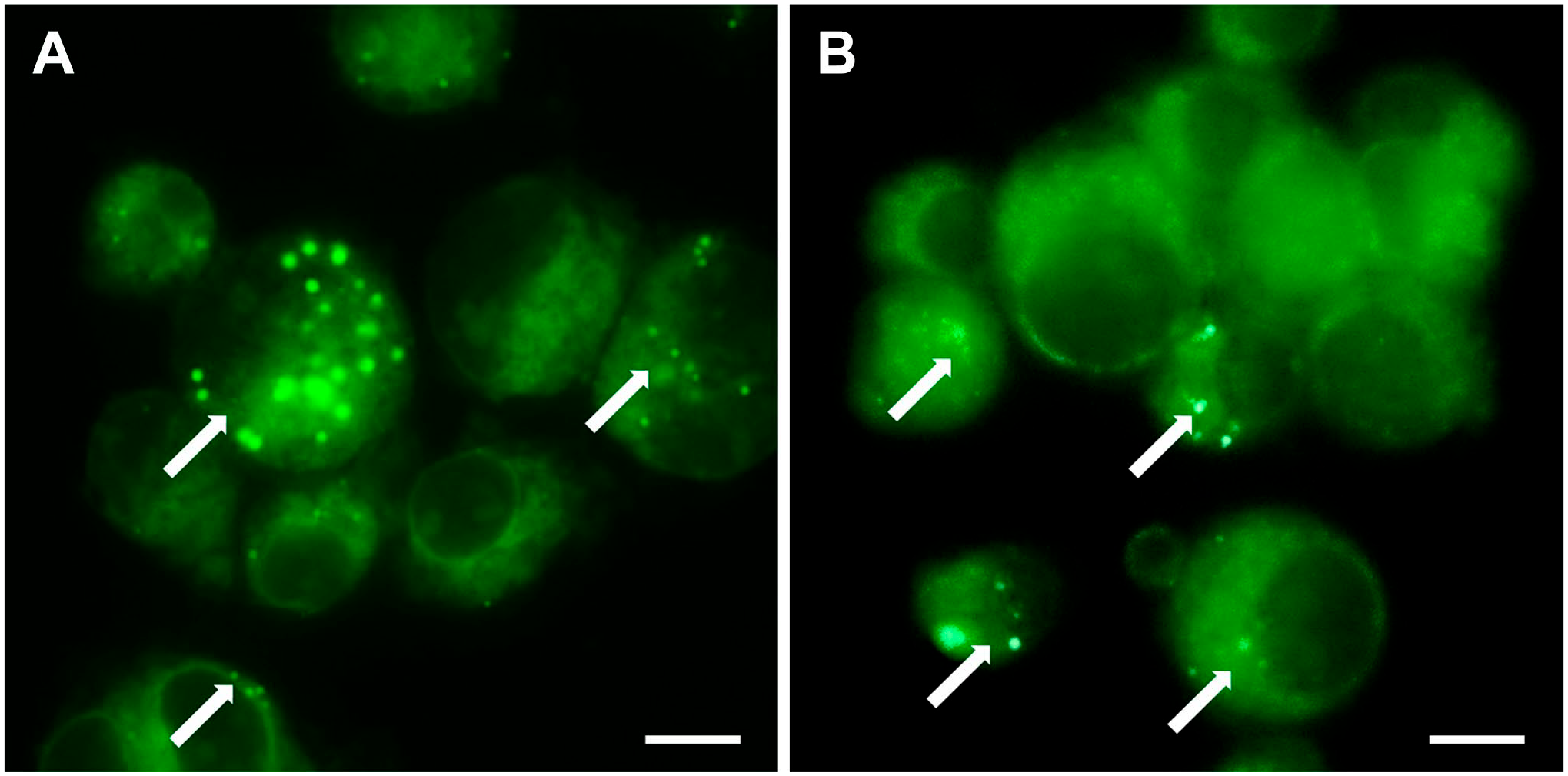
Metabolic mapping of cathepsin B activity. Mapping was done in non-irradiated (A) and irradiated (2 Gy) (B) NCH644 cells, a glioblastoma stem cell line. Enzymatic activity was detected as green fluorescent dots (marked by white arrows). The method is based on the coupling of NSA with 4MbNA, which is cleaved from a protease-specific substrate. Enzymatic release of 4MbNA and its coupling with NSA result in the formation of a fluorescent green product. Background fluorescence occurs due to the nonspecific binding of NSA to protein NH2 groups. The method was adapted from Van Noorden et al.
185
Cells were grown as floating spheres in supplemented NB medium as described by Podergajs et al.
186
Intact spheroids were irradiated with 2 Gy. Four hours after irradiation, the spheroids were washed with Ca2+- and Mg2+-free Hank’s balanced salt solution and with 100 mM sodium phosphate buffer (pH 6.0). Subsequently, spheroids were incubated in 100 mM phosphate buffer (pH 6.0) containing 1 mM dithiothreitol, 1.3 mM EDTA, 2.7 mM
Proteases as Therapeutic Targets in Cancer
In anticancer therapies, targeting proteases has long been a promising therapeutic tool to counteract invasiveness of cancer cells. Selective targeting of CSCs in their specific niches has recently attracted much attention in the field of cancer therapy,190,191 and the inclusion of proteases as therapeutic targets seems to be an option. However, several attempts at protease inhibition have failed to deliver the desired outcomes. To date, only three proteasome inhibitors have been approved in cancer therapy for the treatment of multiple myeloma (Table 2).15,35,212 Reasons for the treatment failures with other protease inhibitors might, first, be due to inappropriate clinical trial designs, for example, including cancer patients with advanced disease where a hypermutated state was able to overcome single target therapy. Second, the lack of sufficient selectivity and specificity of the protease inhibitors may have led to undesired off-target effects by disturbing the normal physiological functions of these proteases in adjacent tissue or systemically. Finally, due to protease redundancy, the efficiency of selective inhibitors may hinder their effectiveness.15,40,212,213
Protease Inhibitors and Protease-cleavable Conjugates in Cancer Clinical Trials.
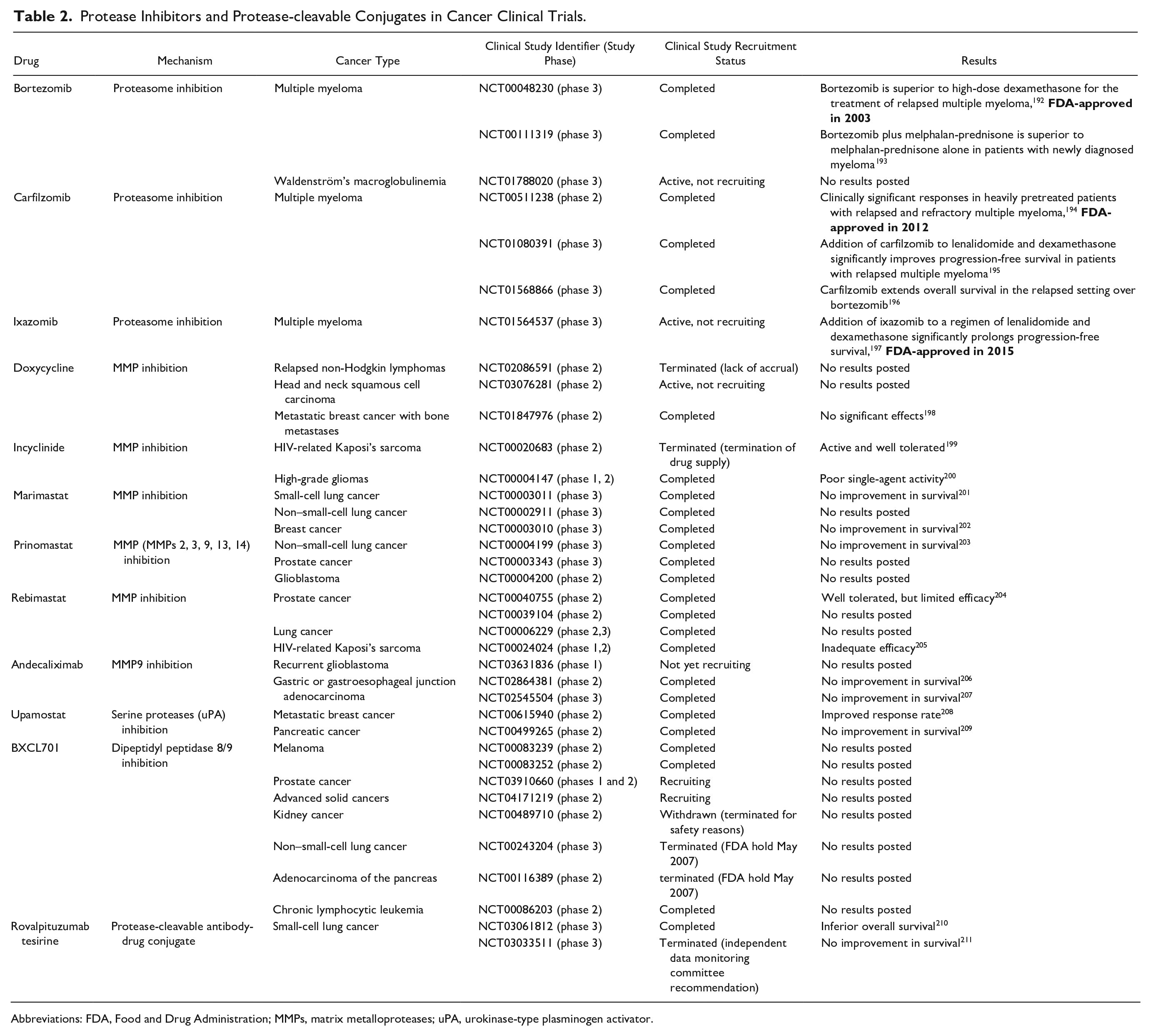
Abbreviations: FDA, Food and Drug Administration; MMPs, matrix metalloproteases; uPA, urokinase-type plasminogen activator.
Despite the initial clinical disappointments, novel approaches have emerged in which proteases are not used as treatment targets, but instead as therapeutic triggers (Table 2). These technologies enable the delivery of anticancer therapeutics directly to the tumor site where their activation or release is mediated by proteases (e.g., MMPs, cathepsins, or uPA) within the TME.213 –215
Based on current knowledge in the field, targeting or exploiting proteases in the CSC niches may contribute to the elimination of quiescent CSCs. This, arguably, represents the bottleneck in successful tumor eradication. Several examples of such approaches have already shown promising results,190,216,217 providing hope for future cancer treatments. Nevertheless, deliberate target selection may be the key to success. On one hand, proteases bolster CSC proliferation and invasion, whereas, on the other hand, they can also induce CSC detachment and mobilization out of the niche by modulating cytokines.104,109,128 This may lead to their differentiation and higher therapeutic sensitivity. However, CSC mobilization may not necessarily present the optimal therapeutic intervention because it may lead to enhanced aggressiveness of CSCs expressing hybrid phenotypes during the EMT.130,218 Hence, continued efforts are needed to better characterize the heterogeneous and phenotypically distinct CSC pool, and the interplay of these cells with the TME in the niches. When designing CSC niche protease-oriented therapeutic applications, the multifaceted roles of the proteases should be thoroughly examined and modeled in experimental animals in terms of the complex and dynamic TME, before human studies.
Footnotes
Competing Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
BB and AH conceptualized the research. AH, MN, BM, and BB wrote the original draft of the manuscript. AH, TLT, and BB revised and edited the manuscript. All authors have read and approved the final version of the manuscript.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Slovenian Research Agency (grant program P1-0245, grant project J3-2526, post-doctoral project Z3-1870) and by the European Program of Cross-Border Cooperation for Slovenia-Italy Interreg TRANS-GLIOMA.