
Abstract
Both heparanase and syndecan-1 are known to be present and active in disease pathobiology. An important feature of syndecan-1 related to its role in pathologies is that it can be shed from the surface of cells as an intact ectodomain composed of the extracellular core protein and attached heparan sulfate and chondroitin sulfate chains. Shed syndecan-1 remains functional and impacts cell behavior both locally and distally from its cell of origin. Shedding of syndecan-1 is initiated by a variety of stimuli and accomplished predominantly by the action of matrix metalloproteinases. The accessibility of these proteases to the core protein of syndecan-1 is enhanced, and shedding facilitated, when the heparan sulfate chains of syndecan-1 have been shortened by the enzymatic activity of heparanase. Interestingly, heparanase also enhances shedding by upregulating the expression of matrix metalloproteinases. Recent studies have revealed that heparanase-induced syndecan-1 shedding contributes to the pathogenesis and progression of cancer and viral infection, as well as other septic and non-septic inflammatory states. This review discusses the heparanase/shed syndecan-1 axis in disease pathogenesis and progression, the potential of targeting this axis therapeutically, and the possibility that this axis is widespread and of influence in many diseases.
Keywords
Introduction
Work on heparan sulfate proteoglycans intensified in the 1980s as it became clear that this category of molecules had important functional capacities. Merton Bernfield’s Laboratory at Stanford University performed pioneering studies on what was then referred to as the epithelial cell surface proteoglycan, a glycosaminoglycan containing molecule that was present on a normal murine mammary gland epithelial cell line designated NMuMG. 1 A series of publications from the Bernfield Laboratory subsequently revealed a number of key features of this proteoglycan, much of which was enabled by the development of monoclonal antibody 281-2 that recognized the core protein of the proteoglycan. 2 The core protein consisted of a transmembrane domain flanked by a cytoplasmic domain that interacted with the cytoskeleton and an extracellular domain that bore both chondroitin sulfate and heparan sulfate glycosaminoglycan chains, the latter of which could bind to the extracellular matrix (ECM).3–5 Immunohistochemistry and electron microscopy using monoclonal antibody 281-2 revealed widespread localization on the surface of most epithelial cells and surprisingly also on plasma cells. 6 Another important feature and key discovery was that this proteoglycan could be proteolytically shed from the cell surface as an intact ectodomain containing the extracellular core protein domain with attached glycosaminoglycan chains. 7 With the advent of expression cloning techniques, a milestone in the proteoglycan field occurred in 1989 with publication of the cloning and sequencing of the epithelial cell surface proteoglycan. That proteoglycan was given the name syndecan, and later designated as syndecan-1. 8
Subsequent studies over the next decade from a number of labs established that there were two major families of cell surface proteoglycans, the transmembrane syndecans (syndecan 1–4) and the glycosylphosphoinositide-linked glypicans (glypican 1–6). Biological studies revealed that these proteoglycans function primarily to bind ligands and regulate their interaction with signaling complexes at the cell surface, thereby fine tuning cell signaling. 9 As the field expanded, an even wider array of heparan sulfate proteoglycan functions were discovered, including regulation of metabolism, transport, information transfer, and even recently the revelation that syndecans can regulate multiple cellular functions via control of calcium flux through cell surface membrane channels.10,11
Along with these advances in understanding heparan sulfate proteoglycan function came the increasing awareness that shedding of these proteoglycans through the action of metalloproteinases (syndecan sheddases) or phospholipases (glypican sheddases) was an important means of regulating proteoglycan function and subsequent cell behaviors that could participate in disease pathogenesis.12–14 In most cases, shedding results in the release of an intact ectodomain containing fully functional glycosaminoglycan chains capable of acting in either autocrine or paracrine fashion. In addition, shedding removes the proteoglycan from the cell surface, rendering it less likely to participate in signaling events at the cell surface, particularly those events where membrane-bound syndecans are involved in forming signaling complexes. Evaluation of syndecan-1 in various cell types and under different conditions revealed that shedding can be stimulated by an array of agonists that initiate the shedding process through upregulation of protein sheddases. 12
In parallel to the development of the heparan sulfate proteoglycan field, research on heparanase, the sole mammalian heparan sulfate–degrading enzyme, was also progressing and significantly advanced following its cloning in 1999.15–17 Pioneering work in the laboratory of Israel Vlodavsky, among others, demonstrated that heparanase had a major impact on tumor progression by enhancing tumor growth, metastasis, and angiogenesis.18–20 Heparanase enzymatic activity trims heparan sulfate chains on the surface of cells in the ECM. Through its action on heparan sulfate, it can regulate cell signaling and remodel the ECM. Heparanase can also translocate to the nucleus where it regulates the expression of multiple genes (e.g., vascular endothelial growth factor [VEGF], hepatocyte growth factor [HGF]), thereby greatly expanding the importance of this unique enzyme.21–23 Although much of the early work on heparanase focused on cancer, more recently it has become clear that this enzyme plays important roles in the pathogenesis of many diseases.24,25
While studying heparanase, our lab made the surprising discovery that this enzyme dramatically enhances shedding of syndecan-1 from the surface of myeloma and breast cancer cells. 26 This enhanced effect on shedding was seen when cells were transfected with the gene for human heparanase or when enzymatically active recombinant heparanase was added to cells in the culture. Subsequent studies using human myeloma cells demonstrated that heparanase-induced shedding of syndecan-1 was the result of two distinct mechanisms. Heparanase stimulated extracellular signal–regulated kinase (ERK) signaling in the tumor cells, leading to upregulated expression of the syndecan-1 sheddase matrix metalloproteinase (MMP)-9. 27 In addition, heparanase via its enzyme activity shortens the heparan sulfate chains of syndecan-1, thereby rendering the core protein increasingly susceptible to cleavage by MMP-9. 28 Since our discovery of the unique link between heparanase and syndecan-1 shedding and its role in regulating tumor progression, other researchers have confirmed the importance of the heparanase/shed syndecan-1 axis in a variety of pathological states (Fig. 1). This review summarizes several examples of those studies and underscores the growing realization that this axis exerts a broad impact on disease pathogenesis.
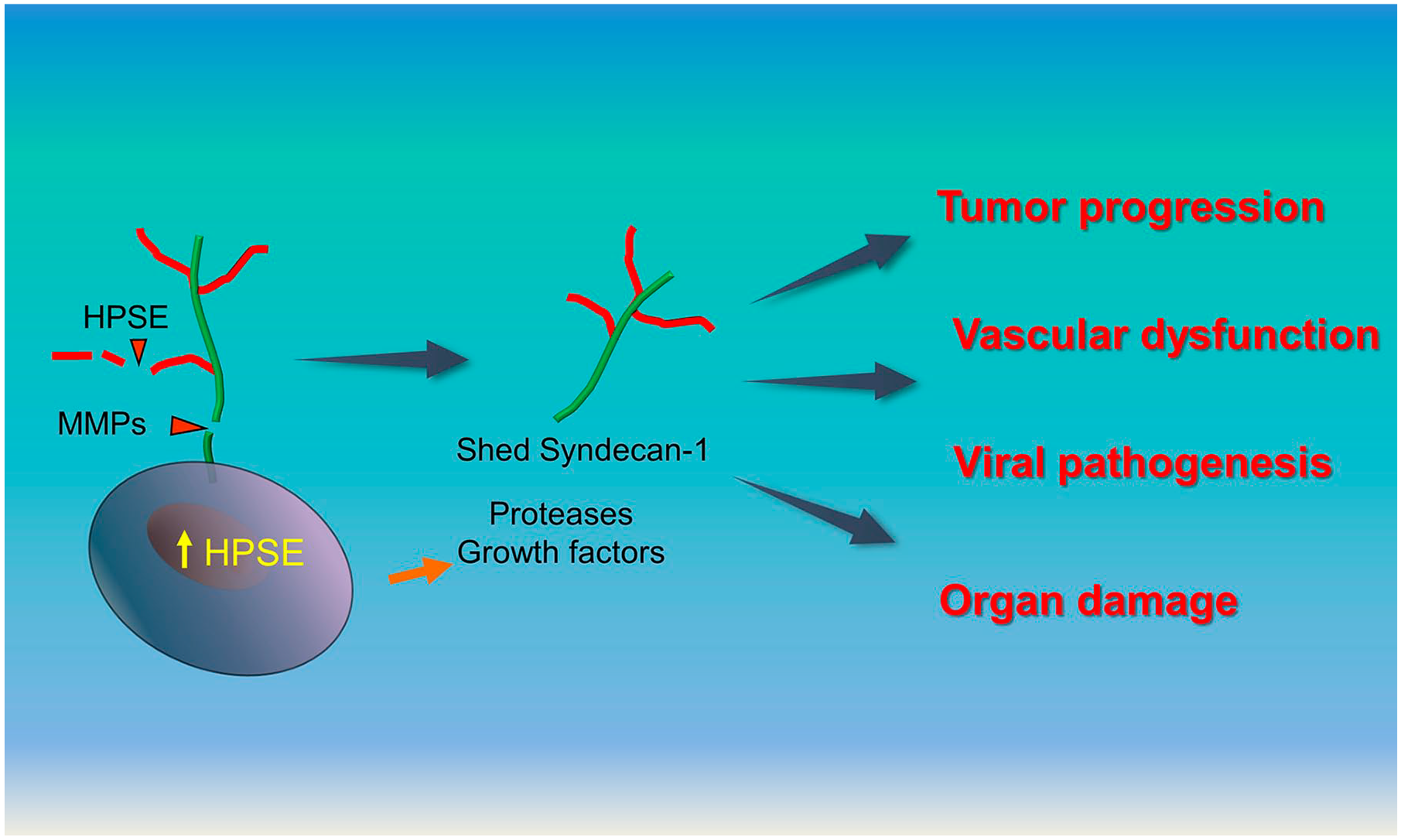
Role of the heparanase/shed syndecan-1 axis in disease pathogenesis. Heparanase (HPSE) is often upregulated by cells during the pathogenesis and progression of disease. Heparanase upregulates gene transcription thereby enhancing expression of proteases and numerous effector molecules (e.g., growth factors, chemokines). When secreted, the matrix metalloproteinases (MMPs) upregulated by HPSE, such as MMP-3, MMP-7, and MMP-9, can cleave the core protein of syndecan-1 resulting in its shedding from the cell surface. Additionally, heparanase enzyme activity shortens heparan sulfate chains of syndecan-1 rendering the syndecan core protein more assessable to cleavage by proteases. Once shed syndecan-1 is released it can act locally or distally with various effector molecules (e.g., growth factors) or nucleate complexes at the cell surface to promote signaling events that impact tumor progression, dysregulate vascular permeability, regulate viral ingress and egress and mediate organ damage.
The Heparanase/Shed Syndecan-1 Axis Regulates Cancer Progression
The complexity of cancer can be attributed to multiple factors. For example, individual cells within the tumor can vary from one another enormously, and they change continuously over time, contributing to the difficulty in treating cancer. Another complexity is attributed to the extensive cross talk between tumor cells and between tumor and host cells that fuels aggressive tumor behavior. Although initiation of cancer is largely driven by oncogenic mutations, later steps in cancer progression such as clonal expansion, angiogenesis, invasion, and metastasis are greatly influenced by the activity of growth factors and their signaling receptors. 29 Tumor cells require a continuous supply of growth factors (e.g., fibroblast growth factor [FGF], VEGF, HGF) for their growth, survival, and proliferation. Through their action, growth factors and other effector molecules activate multiple signaling pathways in tumor cells and their associated stromal cells, which collectively contribute to aggressive tumor progression.
Because of their heterogeneity and versatility, proteoglycans serve as important functional components at the cancer cell surface, within the tumor stroma and throughout tumor-derived ECM. Extensive posttranslational modifications on heparan sulfate chains of proteoglycans, particularly the addition of sulfate groups, not only impart negative charge but also increase their high degree of structural diversity and functional heterogeneity. This structural heterogeneity among heparan sulfates facilitates growth factor–binding specificity and endows proteoglycans with the ability to fine tune signaling within the tumor microenvironment. 10
Concerning cancer progression, syndecan-1 is the most extensively studied heparan sulfate proteoglycan.30–32 It is highly expressed on the surface of many types of tumor cells and in some cases within tumor stroma as well. Syndecan-1 regulates multiple functions, including tumor cell attachment, growth, proliferation, and angiogenesis. Shed syndecan-1 released from the cancer cells with intact heparan sulfate chains carrying multiple growth factors acts in paracrine fashion to promote aggressive tumor progression. In addition to heparan sulfate chains on shed syndecan-1, the core protein is also involved in the activation of multiple signaling pathways that contribute to tumor growth and metastasis.30,33 Furthermore, shed syndecan-1 binds to the ECM where it can act to trap tumor cells, contributing to the initiation of a tumor niche. It also can form growth factor concentration gradients that contribute to tumor invasion and metastasis. What follows is a discussion of the multiple roles shed syndecan-1 plays in tumor pathobiology and how heparanase contributes to this shedding, thereby fueling tumor progression. We focus predominantly on multiple myeloma where the heparanase/syndecan-1 axis has been extensively studied.
Myeloma Tumor Invasion
Fibronectin (FN) and vitronectin (VN) are ECM glycoproteins that are enriched in the bone marrow. They act as substrates for the very late antigen (VLA)-4 integrin that is expressed on the surface of myeloma cells. Work in Alan Rapraeger’s laboratory at the University of Wisconsin–Madison demonstrated that human myeloma cells engineered to express a high level of heparanase (HPSE-high) when grown on FN or VN exhibit a highly polarized morphology and display a spreading phenotype compared with myeloma cells expressing low heparanase (HPSE-low) that fail to spread. 34 In addition, HPSE-high cells have an enhanced capacity to invade through Matrigel. Myeloma cells expressing enzymatically inactive heparanase did not spread or invade, indicating that the enzymatic activity of heparanase is required for the spreading and invasive phenotype. In addition, inhibition of heparanase enzyme activity by Roneparstat, a modified heparin that binds to and blocks the enzyme’s catalytic site,35,36 diminishes the spreading and invasive phenotype of myeloma cells. Surprisingly, this effect of heparanase on cell spreading and invasion was traced to the ability of heparanase to enhance syndecan-1 shedding from the cell surface. This was demonstrated by blocking the activity of MMP-9, a syndecan-1 sheddase whose expression, as mentioned above, is elevated by heparanase. 27
How does shed syndecan-1 promote the spreading and invasive phenotype? It was discovered that when syndecan-1 is shed, it exposes a cryptic juxtamembrane site on the syndecan-1 core protein that then complexes with both VEGF receptor-2 (VEGFR2) and VLA-4 on the myeloma cell surface. 34 This leads to VEGFR2 intracellular signaling and triggering of downstream activation of Rac-GTPase, resulting in polarized migration and myeloma cell invasion. Peptides known as synstatins that mimic regions within the cryptic core protein domain of syndecan-1 can selectively block its binding to either VEGFR2 or VLA-4, thereby inhibiting the formation of the syndecan-1/VEGFR2/VLA-4 complex and preventing cell spreading and the invasive phenotype. This further confirms the crucial role of shed syndecan-1 in promoting the spread and invasive phenotype of myeloma cells seen when the heparanase level is elevated. Moreover, these results are consistent with previous studies demonstrating that high expression of either heparanase or soluble syndecan-1 dramatically increased myeloma tumor invasive behavior in vivo.37,38
Angiogenesis
Newly formed vascular networks supply oxygen and nutrients to cancer cells and act as a conduit to rid tissue of waste products. This process of angiogenesis is one of the key events that fuels cancer cell proliferation and dissemination. Endothelial cells present in the inner lining of blood vessels become activated in response to proangiogenic factors secreted by tumor cells and stromal cells, thereby initiating neovascularization. Early studies examining heparanase in myeloma patients revealed that high levels of heparanase in the myeloma bone marrow correlated closely with elevated microvessel density. 39 Additional studies in animals confirmed this association and led to investigation of the mechanism(s) whereby heparanase acts to stimulate angiogenesis. In vitro studies revealed that addition of conditioned medium from HPSE-high cells, or addition of recombinant heparanase, enhanced endothelial invasion through Matrigel. 40 Enzymatically inactive heparanase failed to promote endothelial invasion. Because of the link between heparanase and syndecan-1 shedding, the possibility of syndecan-1 playing a role in heparanase-enhanced endothelial invasion was investigated. Interestingly, it was found that shed syndecan-1 plays a dual role in promoting angiogenesis. The first mechanism is related to the ability of VEGF to bind to syndecan-1 heparan sulfate. Immunoprecipitation of syndecan-1 from medium conditioned by HPSE-high cells diminished the ability of the media to stimulate endothelial invasion. Similarly, addition of a VEGF-blocking antibody to the conditioned medium inhibited endothelial invasion. Treatment of conditioned media with heparinase III, a Flavobacterium-derived enzyme that extensively degrades heparan sulfate chains, also led to inhibition of endothelial invasion. Mechanistically, conditioned medium from HPSE-high cells was shown to enhance phospho-ERK levels in the endothelial cells and addition of ERK inhibitors abolished endothelial invasion. Together, these results indicate that shed syndecan-1 from HPSE-high cells binds to VEGF and facilitates activation of the VEGF receptor on endothelial cells. 40 In addition to promoting VEGFR signaling, it has been shown that heparanase can enhance expression of VEGF by myeloma, breast cancer, and glioma cells,40,41 further demonstrating the pivotal role played by heparanase in driving tumor angiogenesis.
The second mechanism by which heparanase-induced shedding of syndecan-1 can drive angiogenesis is via the same mechanism described above for tumor cell invasion. Just as with the myeloma cells, shed syndecan-1 induced by heparanase can complex with VLA-4 and VEGFR2 on endothelial cells to stimulate their invasive behavior, and this can be inhibited by synstatins. 34 Thus, via this single mechanism, heparanase promotes both tumor cell metastasis and angiogenesis. This was a breakthrough for the heparanase field of research because it was previously thought that heparanase promoted invasive behaviors mainly through its ability to degrade ECM to allow cells to actively migrate to new sites. Although matrix degradation may be one of the functions of heparanase, clearly the mechanism initiated by heparanase enhancement of syndecan-1 shedding and subsequent stimulation of invasive behavior of both tumor and endothelial cells reveals important new insight into how heparanase has a major impact on the progression of some tumors.
Osteolysis
Osteolysis is one of the major skeletal complications causing significant morbidity in the majority of myeloma patients and multiple other cancers that metastasize to bone. Bone is enriched with a variety of cytokines and growth factors that provide an extremely fertile environment for tumor growth. During the development of metastases within bone, cancer cells collaborate with large multinucleated bone-resorbing cells called osteoclasts. Soluble factors released by cancer cells and stromal cells enhance the osteoclast formation, thereby fueling bone destruction. 42 Breast cancer cells expressing high heparanase were shown to shed elevated levels of syndecan-1 with subsequent enhancement of osteolysis in vivo.26,43 Peripheral blood mononuclear cells (PBMC) from normal human donors when incubated with conditioned medium from HPSE-high breast cancer cells significantly enhanced osteoclast formation compared with PBMC incubated with conditioned media from HPSE-low breast cancer cells. Pretreatment of conditioned medium from HPSE-high cells with heparinase III or immunodepletion of shed syndecan-1 before adding to PBMC abolished osteoclast formation, indicating the role of shed syndecan-1 and its heparan sulfate chains in osteoclast formation. 43 Conditioned media from breast cancer cells expressing enzymatically inactive heparanase did not enhance osteoclast formation, indicating the requirement of heparanase enzymatic activity. In addition, enhanced osteoclast formation and bone resorption were observed when PBMC were grown on human bone slices in the presence of conditioned media from HPSE-high cells compared with incubation with conditioned medium from HPSE-low cells. The size and depth of resorption pits were the same, but the number of resorption pits formed in the presence of HPSE-high conditioned medium was significantly higher than that formed by HPSE-low conditioned medium. Furthermore, compared with controls, elevated bone resorption was observed in mice bearing HPSE-high-expressing tumors generated by implanting HPSE-high cells in the mammary fat pad. 44 Histomorphometric analysis of long bones isolated from HPSE-high tumor mice confirmed a significantly greater number of osteoclasts per millimeter of bone surface consistent with increased bone-resorption markers TRAP-5b and collagen telopeptides in serum when compared with HPSE-low tumor-bearing mice. Interestingly, similar to what was seen in breast cancer models, HPSE-high myeloma tumor cells also enhance osteolysis and bone homeostasis dramatically, although the role of shed syndecan-1 in the myeloma model studied has not been determined.45,46
HGF signaling is known to enhance growth, motility, and angiogenesis in many cancer types including multiple myeloma.47,48 Studies using myeloma cells have demonstrated an important role for the heparanase/shed syndecan-1 axis in regulating HGF signaling and its potential impact on osteolysis. Syndecan-1 shed by HPSE-high myeloma cells binds to HGF, and this complex activates the HGF receptor c-Met on the surface of cancer cells and stromal cells. Activation of c-Met on the osteoblast-like cell line Soas-2 stimulates interleukin (IL)-11 expression and subsequent autocrine stimulation of the IL-11 receptor, leading to upregulated expression and secretion of receptor activator of nuclear factor-κB ligand (RANKL) to promote osteoclastogenesis. 49 Addition of a small molecule inhibitor of c-Met abolished the HGF-mediated expression of IL-11 and RANKL. In addition, immunoprecipitation of shed syndecan-1 from HPSE-high conditioned media or treatment of the conditioned media with heparinase III 50 reduced the expression of IL-11 and RANKL. Interestingly, it was also discovered that heparanase expression by myeloma cells stimulates their expression and secretion of HGF. Thus, heparanase plays a dual role in fueling this osteoclastogenesis feedforward loop by both stimulating HGF expression and syndecan-1 shedding. 49
Shed Syndecan-1 Regulates Histone Acetyltransferase and Shuttles Growth Factors to the Nucleus
Syndecan-1 has been found in the nucleus of several different tumor types and can be localized there via a nuclear localization sequence found in its cytoplasmic tail.22,30,51,52 However, less is known about the ability of shed syndecan-1 to localize within the nucleus and its functional importance. To examine this, ARH77, a human lymphoblastoid cell line that lacks syndecan-1 expression, and ST2, a murine bone marrow–derived stromal cell line, were incubated with medium from HPSE-high myeloma cells that contains elevated levels of human shed syndecan-1. Shed syndecan-1 present in the conditioned medium was rapidly translocated into the nucleus of those cells. 53 The majority of the nuclear shed syndecan-1 was observed in discrete patches within the euchromatin, the area of active gene transcription. Moreover, it was found that shed syndecan-1 in the nucleus of ST2 cells binds to the histone acetyltransferase (HAT) enzyme p300 via heparan sulfate chains and diminishes HAT activity and histone H3 acetylation. Pretreatment of HPSE-high conditioned media with heparinase III decreased HAT activity and histone H3 acetylation, confirming the importance of intact heparan sulfate chains on shed syndecan-1 core protein. Further importance of sulfate groups on heparan sulfate was determined by growing the HPSE-high myeloma cells in medium containing sodium chlorate, which decreased the sulfation of heparan sulfate chains on shed syndecan-1 and abolished its nuclear localization. In addition, shed syndecan-1 via its heparan sulfate chains binds to HGF and shuttles the growth factor into the nucleus. 53 This represents a previously unappreciated mechanism, whereby the heparanase/shed syndecan-1 axis can regulate HAT activity and translocate growth factors to the nucleus.
Chemoresistance
In colorectal cancer, syndecan-1 shedding induced by MMP-7, MMP-9 or heparanase enhances the activation of epidermal growth factor receptor (EGFR) phosphorylation and the downstream Ras–Raf–MEK–ERK pathway, thereby increasing chemoresistance. 54 The authors speculated that shed syndecan-1 may be able to bind and present soluble heparin-binding EGF-like growth factor to EGFR better than surface-bound syndecan-1. A possible role for the shed proteoglycan was further implicated by the finding that shed syndecan-1 levels are higher in colorectal cancer patients than in normal controls, and that levels were diminished following surgery to remove the tumor. 54 Moreover, patients with high preoperative serum levels of syndecan-1 were less responsive to chemotherapy and had lower rates of disease-free survival than patients with low preoperative levels of syndecan-1.
In colon cancer, complex changes occur in the expression pattern of syndecan-1 and heparanase during progression from well-differentiated to undifferentiated tumors. Gene silencing and overexpression in Caco-2 cells revealed that heparanase expression and activity were upregulated in syndecan-1-depleted cells likely due to upregulation of the transcription factor Egr1. 55 Syndecan-1 depletion also increased stemness and invasiveness of Caco-2 cells in heparanase-dependent manner. Upregulated expression of heparanase resulted in increased resistance to radiotherapy and chemotherapy.
Reduced expression of syndecan-1 has also been observed in patients with ulcerative colitis, attributed to disrupted healing of colonic ulcers. Similarly, syndecan-1 knockout (KO) mice revealed a substantial increase in mortality vs wild-type mice subjected to the dextran sodium sulfate (DSS) model of inflammatory bowel disease (IBD). 56 Notably, chronic IBD increases the risk of colon cancer. Colitis-associated cancer can be experimentally modeled in mice by application of the carcinogen azoxymethane (AOM) and subsequent induction of chronic colitis with DSS. Syndecan-1 KO mice developed more severe inflammation during chronic DSS colitis, associated with increased recruitment of inflammatory cells, elevated IL-6 expression and activation of STAT3, increased crypt damage, and increased weight loss compared with wild-type mice. Notably, syndecan-1 KO mice formed larger tumors than their wild-type controls in the AOM/DSS model. It appears that the increased inflammation and tissue damage in the absence of syndecan-1 drives colon cancer progression via enhanced signaling through the IL-6/STAT pathway. 56 Although syndecan-1 is downregulated in IBD and colon cancer, heparanase is upregulated in the inflamed and tumor tissue in these diseases and in the experimental AOM/DSS mouse model. 57 Notably, when transgenic mice that overexpress heparanase were subjected to DSS colitis, high heparanase expression preserved the inflammatory conditions, along with increased expression of tumor necrosis factor-α (TNF-α) and Cyclin D1 (thus resembling syndecan-1 KO mice in this model) and a substantially increased recruitment of TNF-α-expressing macrophages. Similar to syndecan-1 KO mice, tumors were larger in heparanase transgenic mice, and tumor angiogenesis was enhanced. Overall, these data suggest that heparanase drives a vicious cycle that promotes colitis and chronic inflammation–associated tumorigenesis. 57 It is intriguing to speculate that at least part of the impact of overexpression of heparanase in these models leads to increased syndecan-1 shedding, thus contributing to inflammation. In addition, heparanase-enhanced shedding of syndecan-1 likely decreases cell surface syndecan-1, which may further contribute to tumor progression and chemoresistance.
Interestingly, it was found that when HPSE-high myeloma cells are exposed to chemotherapeutic drug and enter apoptosis, they shed high levels of syndecan-1. 58 This shedding of syndecan-1 was blocked by ADAM (a disintegrin and metalloproteinase) inhibitors or by small interfering RNA (siRNA) targeting ADAMs. In addition, it was demonstrated that drug-resistant myeloma cell lines exhibit elevated levels of both heparanase and shed syndecan-1 compared with their wild-type counterparts. 50
Another example in which the heparanase/shed syndecan-1 axis may be involved in chemoresistance is in breast cancer patients treated with the human epidermal growth factor receptor 2 (HER2) blocking antibody trastuzumab. Heparan sulfate on the surface of breast cancer cell lines aids in capturing trastuzumab and facilitates its interaction with HER2. Notably, MCF7 breast cancer cells transfected with the gene for heparanase are resistant to trastuzumab killing. 59 This is likely due to heparanase-mediated enhanced shedding of syndecans from the cell surface. Shedding of syndecan-1 not only diminishes the amount of heparan sulfate at the cell surface available for binding to trastuzumab, but also the released heparan sulfate may bind to the antibody and compete for its binding to cell surfaces. 59
Shed Syndecan-1 as a Cancer Biomarker
Expression of syndecan-1 varies among different cancer types and also between normal cells and cancer cells. These expression signatures are directly associated with tumor aggressiveness, patient survival, and clinical outcome. Heparanase expression is also known to be upregulated in cancer cells compared with normal cells. Cancer cells expressing high levels of both cell surface syndecan-1 and heparanase release elevated levels of shed syndecan-1 into the biological fluids. 33 As mentioned earlier, there are multiple mechanisms through which shedding of syndecan-1 occurs, including heparanase-mediated upregulation of MMPs. Several reports have demonstrated that shed syndecan-1 released into biological fluids can be quantified and used as a biomarker in various cancers. 32 For example, studies have shown higher levels of soluble syndecan-1 in multiple myeloma patients compared with healthy controls. In addition, it has also been shown that baseline levels of soluble syndecan-1 at the time of diagnosis in patients who responded to chemotherapy were lower than those in patients who have not responded to chemotherapy. These findings indicate that shed syndecan-1 levels can be used as a biomarker for selecting cancer patients who most likely will respond to particular type of chemotherapeutic treatment and also in predicting tumor relapse.60–62
The Heparanase/Shed Syndecan-1 Axis in Systemic Inflammatory States
During the initial response to systemic insults like trauma or infection, the human innate immune system activates circulating leukocytes, platelets, and vascular endothelium to upregulate and release a host of zymogens and enzymes, including heparanase.63,64 This paves the way for host defenses (in the case of infectious insults) and tissue repair. The vascular endothelium then serves as the interface between the immune system and the end organs to modulate tissue perfusion and function. 65 However, during overwhelming states of infection or injury, the unregulated activity of local and circulating enzymes causes damage to the endothelial glycocalyx, a protective layer lining the vascular luminal surface.66–68 This leads to compromised vascular function and propagation of multiorgan failure.
The endothelial glycocalyx layer is a gel-like matrix anchored to the endothelial cell surface that contributes to a multitude of endothelial functions, including regulation of vascular tone and permeability, coagulation, leukocyte and platelet adhesion, and propagation of inflammation. 69 The glycocalyx is comprised largely of proteoglycans bearing glycosaminoglycan chains (e.g., heparan sulfate, chondroitin sulfate) and impregnated with various regulatory proteins (e.g., antithrombin III, tissue factor pathway inhibitor, VEGF, FGF). 70 The syndecan family of heparan sulfate proteoglycans is the most ubiquitous in the endothelial glycocalyx and serves critical functions in endothelial cell signaling.
In the inflammatory milieu that accompanies trauma and sepsis, the endothelial glycocalyx is deranged by a host of circulating and locally released enzymes. 71 The role of heparanase during systemic inflammation has received increasing attention recently. As the only mammalian endoglucuronidase with heparan sulfate–degrading activity, heparanase trims heparan sulfate–releasing fragments of various lengths from endothelial surface proteoglycans (Fig. 2). This trimming of heparan sulfate chains renders the proteoglycan core protein more accessible to enzymatic cleavage by MMPs and thrombin.28,72 In the process of glycocalyx degradation, typically unexposed cell adhesion molecules and cytokine receptors become available for binding and activation, thus propagating inflammation.68,73–75 Moreover, heparan sulfate fragments released by heparanase activate toll-like receptor (TLR) 4 signaling 76 and accompany growth factors and cytokines to facilitate ligand recognition by cognate cell surface receptors. 77
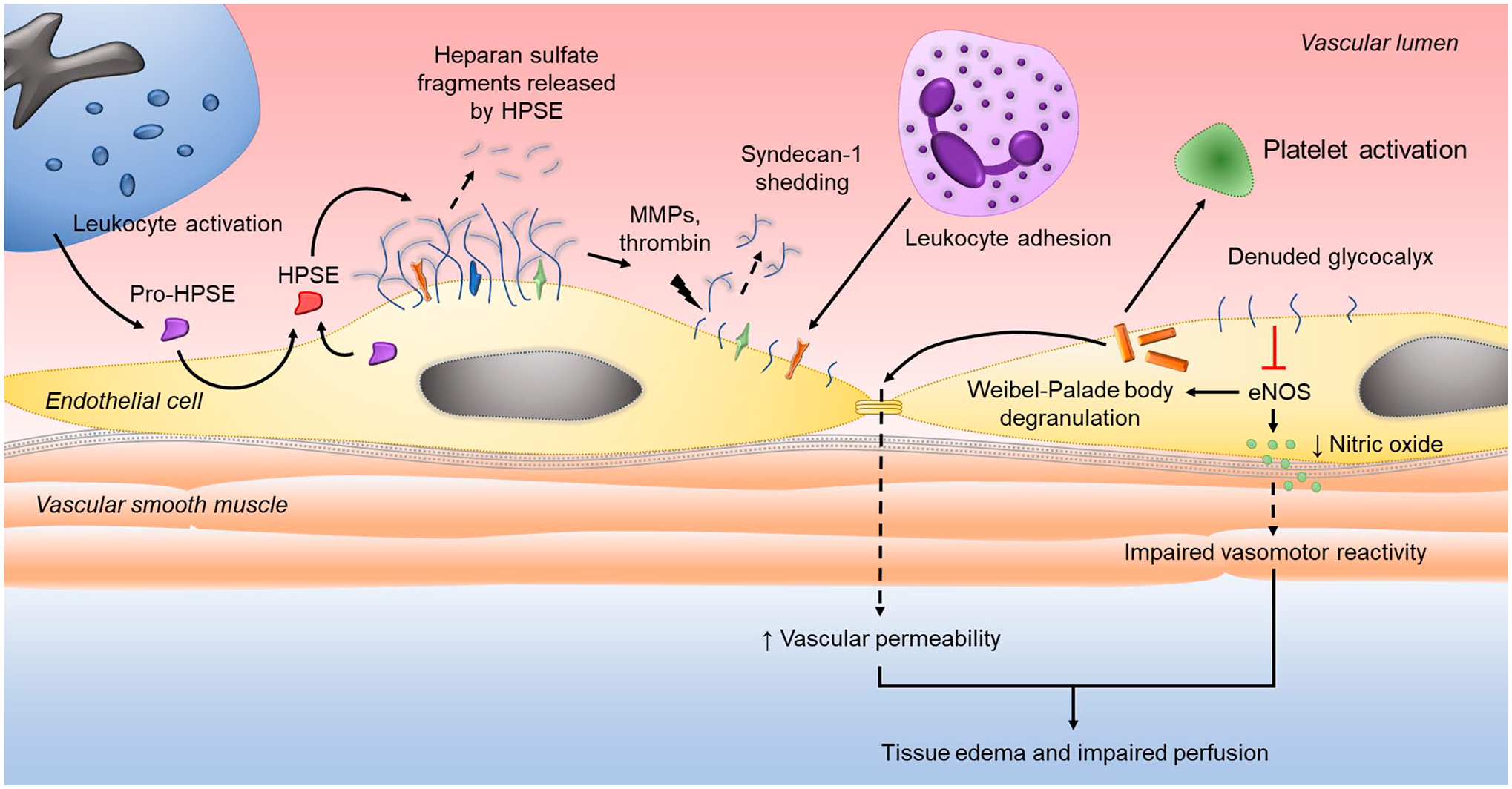
Heparanase and syndecan-1 regulation of vascular dysfunction during systemic inflammation. Latent heparanase (pro-HPSE) is released by immune and endothelial cells during the innate response to infection or injury, internalized by endothelial cells, and processed in lysosomes to the enzymatically active form of HPSE. Secreted HPSE then cleaves heparan sulfate from the endothelial glycocalyx layer. Trimming of heparan sulfate exposes proteolytic cleavage sites on syndecan-1, making the core proteoglycan susceptible to enzymatic degradation by MMPs and thrombin. Loss of syndecan-1 ectodomains from the glycocalyx layer exposes cell surface adhesion molecules and receptors to facilitate leukocyte adhesion and diapedesis. The denuded glycocalyx layer impairs intracellular production of endothelial nitric oxide synthase (eNOS) that, in turn, promotes the release of bioactive proteins from Weibel-Palade bodies that disrupt intracellular junctions and activate platelets. Reduced eNOS activity also diminishes nitric oxide production leading to impaired vascular reactivity. Thus, the downstream consequences of heparan sulfate cleavage culminate in an activated and inflamed endothelium that results in tissue edema and hypoperfusion that, if not corrected, propagates end-organ injury.
Direct injury to the pulmonary epithelium was recently reported to induce shedding of heparan sulfate into the airspace, coinciding with the induction of MMPs. 78 Inhibition of MMPs attenuated heparan sulfate shedding and improved lung function, suggesting that heparan sulfate shedding may impair lung recovery. Augmentation of airway heparanase activity significantly worsened postinjury outcomes, confirming the importance of epithelial glycocalyx integrity to lung recovery. It appears that MMP-associated heparan sulfate shedding contributed to delayed lung recovery in part by the release of large, highly sulfated fragments that sequestered lung-reparative growth factors such as HGF.
In addition to its enzymatic activity at the endothelial surface during systemic inflammation, heparanase may also exhibit intracellular endothelial activity similar to its role in cancer pathophysiology. Through ERK activation and intracellular regulation of syndecan-1 expression, 22 heparanase may contribute to MMP-9 and VEGF upregulation seen in sepsis79,80 and trauma.81,82 Heparanase may also promote the upregulated secretion of exosomes that is seen in critically ill patients83,84 by activating the syndecan–syntenin–ALIX complex that engages the endosomal-sorting complex required for transport (ESCRT).85,86 Through loss of heparan sulfate and syndecan-1 shedding from the glycocalyx, heparanase may also affect endothelial nitric oxide synthase (eNOS) activity.87,88 Decreased eNOS activity may then promote untoward alterations in vascular tone and the unpropitious exocytosis of Weibel–Palade bodies.89,90 Exocytosis of these endothelial cell–derived intraluminal vesicles causes the release of von Willebrand factor 91 and angiopietin-2 92 that go on to activate circulating platelets and promote endothelial destabilization, respectively (Fig. 2). Although these mechanisms, proven in other fields, are logical and have clinical import, at present their applicability in the innate immune response to infection or systemic injury requires further study.
Preclinical studies demonstrate an active role of heparanase in the process of trauma-induced endothelial surface injury. In a mouse model of trauma-hemorrhage and resuscitation, increased plasma heparanase activity was observed, corresponding with a concomitant rise in plasma levels of syndecan-1. 67 Though not specific to the endothelium, syndecan-1 and heparan sulfate plasma levels were shown to inversely correlate with glycocalyx thickness and were associated with measures of microvascular dysfunction, 67 thus supporting the utility of these circulating biomarkers as indicators of glycocalyx disintegrity. In similar in vivo models of trauma-hemorrhage, administration of chemically modified, non-anticoagulant heparin during resuscitation improved cardiovascular, hepatocellular, and immunological functions93,94 and decreased susceptibility to sepsis. 95 In light of the activity of heparanase at the endothelial surface following trauma, together these findings suggest that heparanase contributes to the trauma-mediated pathophysiological response that, if not ameliorated, can culminate in multiorgan dysfunction.
Recent clinical evaluations corroborate these laboratory findings after trauma. Heparanase enzymatic activity and syndecan-1 shedding, reflected by plasma heparan sulfate and syndecan-1 levels, respectively, are apparently increased in trauma patients compared with healthy individuals.96–98 Furthermore, patients with concomitant hemorrhagic shock have significantly higher levels of circulating heparan sulfate compared with those with isolated injuries alone. 99 As shown in preclinical trauma models, circulating glycocalyx biomarkers measured in trauma patients appear to reflect endothelial damage based on the correlation of plasma syndecan-1 with endothelial cell–specific surface markers like thrombomodulin.97,100 Thus, not surprisingly, trauma patients presenting with elevated plasma syndecan-1 levels, indicative of endothelial injury, also exhibit higher circulating proinflammatory cytokine levels,101,102 greater multiorgan dysfunction, 103 and higher mortality. 101 Moreover, patients with elevated syndecan-1 levels after trauma manifest worse coagulation abnormalities 104 and subsequently require more blood product transfusions. 97
Plasma heparanase activity appears to rise similarly in sepsis. Several clinical evaluations of circulating glycosaminoglycan signatures demonstrate increased levels of plasma heparan sulfate early in sepsis,105–107 which correlate with the degree of illness.105,108 These findings are corroborated in a study of human and murine sepsis describing increased heparanase activity in plasma from adults with sepsis and increased heparanase expression from harvested tissues of septic mice. 109 Furthermore, septic patients with lung injury have increased levels of circulating heparan sulfate fragments detected by mass spectroscopy.68,110 Similarly, those with sepsis-induced acute kidney injury (AKI) display increased urinary concentrations of heparan sulfate 111 that may reflect both filtered vascular endothelial glycocalyx fragments and local glomerular glycocalyx degradation by heparanase. Enzymatically active heparanase has been shown to promote lung injury by facilitating neutrophil adhesion via endothelial glycocalyx degradation, 68 although the role of heparanase in sepsis-induced AKI may involve separate processes that remain unclarified. 112 Taken together, these findings suggest that heparanase is active at the endothelial surface during sepsis and is clinically important. Indeed, when heparanase was inhibited in preclinical models of sepsis, lung injury and renal dysfunction were attenuated.68,112
Elevations in circulating levels of syndecan-1 are similarly observed in multiple sepsis populations and appear to correspond with heparan sulfate levels. In a Swedish study of critically ill adults admitted for sepsis, glycosaminoglycans and syndecan-1 were significantly elevated at the time of presentation compared with healthy controls. 108 Intriguingly, total glycosaminoglycan levels did not correlate with syndecan-1 levels. However, in a follow-up study, this group further characterized the glycosaminoglycan species, clarifying that heparan sulfate levels were significantly elevated compared with controls and correlated with cardiovascular sequential organ failure assessment scores. 107 Notably, however, associations between heparan sulfate and syndecan-1 levels were not evaluated. A secondary analysis of plasma samples from 56 patients enrolled in the ProCESS Microcirculatory Flow Ancillary Study identified a significant correlation between syndecan-1 and heparan sulfate levels measured 6 hr after study enrollment. 105 As heparan sulfate and syndecan-1 are not unique to the endothelial surface layer, Hippensteel et al. 105 also evaluated correlations between plasma heparan sulfate and soluble thrombomodulin levels as thrombomodulin corresponds more specifically to endothelial surface layer degradation. They observed a similarly significant correlation. Moreover, in 20 Finnish adults with sepsis compared with healthy controls, elevations of syndecan-1 correlated with elevated vascular adhesion protein-1, another endothelial cell–specific biomarker. 113 Together, these data implicate the heparanase/shed syndecan-1 axis in vascular pathophysiology, although further mechanistic evaluation is needed to fully understand its impact on the endothelial glycocalyx during sepsis.
Role of the Heparanase/Shed Syndecan-1 Axis in Viral Pathogenesis
Understanding viral entry into and egress from cells is critical to the development of therapeutic approaches to combat viruses. Reports have demonstrated that heparanase and/or heparan sulfate proteoglycans including syndecan-1 likely play important roles in the pathogenesis of many types of viruses, including, among others, human papillomavirus (HPV), herpes simplex virus, dengue virus, human immunodeficiency virus, hepatitis C virus, and coronaviruses.114–119 Syndecans (particularly syndecan-1) are highly expressed on epithelial keratinocytes and are important in mediating the attachment of HPV to cells. 120 It was demonstrated that shedding of syndecan-1 plays an active role in HPV16 infection. 121 Inhibition of normal processing of syndecan-1 and its heparan sulfate by MMPs or by heparanase greatly diminished cellular uptake and infection, while exogenous heparan sulfate–degrading enzymes could activate infection. 122
Heparanase-dependent syndecan-1 shedding also plays a critical role in the development of endothelial hyperpermeability, leading to vascular leak and shock caused by severe dengue virus infection. 123 The nonstructural viral protein (NS1) released by dengue-infected cells binds to heparan sulfate on endothelial cells and promotes increased heparanase expression/activity and syndecan-1 shedding. In NS1-infected endothelial cells, inhibition of lysosomal processing or enzymatic activity of heparanase attenuates syndecan-1 shedding and preserves monolayer integrity. Interestingly, addition of exogenous syndecan-1 itself was shown to increase permeability of human microvascular endothelial cells, indicating that syndecan-1 shed in response to NS1 infection is capable of modulating endothelial barrier function. 123 These in vitro observations are supported by clinical findings that plasma syndecan-1 levels strongly associate with measures of severe vascular leakage in dengue-infected patients. 119
HSV-1 and HSV-2 are a family of enveloped, double-stranded DNA viruses that infect the large majority of humans worldwide. HSV-1 infection can be widespread and attack various epithelial tissues of the human body, including mouth, gums, skin, brain, and eyes. Syndecan-1 and syndecan-2 heparan sulfate chains are critical for the initial binding of HSV-1 to target cells. 124 Expression of these proteoglycans is upregulated upon viral infection, and reducing their presence diminishes HSV-1 binding to cells and infection. Interestingly, heparanase is also upregulated in cells following their infection with HSV-1 where it facilitates viral egress by degrading heparan sulfate chains, leading to the release of newly made virus attached to the cell surface. 125 Recently, as a second mechanism of facilitating viral egress from cells, it was demonstrated that newly made HSV-1 viral particles on the cell surface are liberated by heparanase-induced shedding of syndecan-1 from infected cells.115,126 This is similar to the mechanism demonstrated in myeloma cells where heparanase promotes syndecan-1 shedding by upregulating MMP-9. However, in the case of HSV-1-infected cells, heparanase-induced shedding is mediated by upregulation of MMP-3 and MMP-7. 126 This dynamic relationship between viral attachment, viral egress, and heparanase expression and the resulting shedding of syndecan-1 provide new therapeutic targets for regulation of viral infection, including potential relevance to the current COVID-19 pandemic. 127
Heparanase/Shed Syndecan-1 in Kidney Disease
Each kidney is composed of approximately 1 million nephrons, which constitute the functional unit of this organ. A single nephron is composed of the glomerulus, proximal tubule, distal tubule, collecting duct, and the associated vasculature. Heparan sulfate is one of the main polysaccharides in the glomerular endothelial glycocalyx and contributes to the glomerular filtration barrier. Moreover, the glomerular glycocalyx serves an integral role in preventing proteinuria by maintaining adequate glomerular intercellular pore size for filtration and charge negativity to repel similarly negatively charged plasma proteins. Upregulation of endogenous heparanase by glomerular cells leads to disruption of this protective glycocalyx. 128 Albuminuria, the hallmark of glomerular diseases, occurs through a multistep process in which heparanase, immune cells, and dysregulated signaling between podocytes and endothelial cells lead to loss of glycocalyx barrier function.129–131 In diabetic kidney disease, heparanase-mediated degradation and subsequent remodeling of heparan sulfate in the ECM of the glomerulus are key mechanisms driving disease development.132–135 Patients with these diseases have increased urinary excretion of heparanase 136 and heparan sulfate. 137 One mechanism by which heparanase might contribute to development of diabetic nephropathy is through its effects on inflammation. During inflammation, macrophages can secrete cathepsin L which facilitates the processing and activation of proheparanase in the extracellular environment. Active heparanase itself can further activate macrophages by generating shedding of heparan sulfate fragments from cell surfaces that can function as ligands for TLR and/or receptor for advanced glycation end product (RAGE) signaling, thereby contributing to a vicious feedback loop. 138 Inhibition of glomerular cathepsin L and heparanase may therefore lead to restoration of the glomerular glycocalyx and barrier function. These findings are further supported by the attenuation of AKI with heparanase inhibition in states of sepsis 112 and renal ischemia/reperfusion. 139
Another important target for heparanase is the proximal tubular epithelial cells. These cells have multiple functions including fluid handling, solute handling, and acid base handling. Exposure of human proximal tubule epithelial cells (HK-2) to albumin and advanced glycation end products leads to downregulation of syndecan-1, resulting in the reduction of heparan sulfate only in the presence of heparanase. 140 This in turn results in the upregulation of FGF-2. FGF-2 further acts in an autocrine manner, inducing higher expression of heparanase and MMP-9. 141 This cascade of events triggers a phenotypic change in the proximal tubule of the kidney, which resembles epithelial–mesenchymal transition, a key initial step in the development of interstitial fibrosis. Progressive fibrosis leads to accelerated tubular dysfunction, eventually leading to end-stage kidney disease seen in diabetes mellitus. 141 Notably, the diabetic milieu is one of the strongest inducers of heparanase release, suggesting that blocking heparanase activity with inhibitors such as roneparstat or sulodexide is a potential approach to the treatment of kidney diseases. 141
Serum Syndecan-1 as a Biomarker to Predict Impending Renal Failure
Urinary glycosaminoglycans/proteoglycans have long been associated with several kidney diseases as well as diabetic nephropathies as their levels increase more readily than albuminuria. In particular, heparan sulfate, a key component of the glomerular basement membrane responsible for its charge-dependent permeability, is excreted into urine at higher concentrations during the early kidney remodeling events caused by the altered glucose metabolism in diabetes 137 and in sepsis-mediated glomerular injury. 142 Shed syndecan-1, a marker of glycocalyx damage, can thus be used as a potential biomarker to predict impending renal failure.
Heart failure is frequently complicated by AKI, worsening the patient’s prognosis. The concentration of syndecan-1 is valuable in assessing the risk of developing AKI and in-hospital mortality. 143 The postoperative urinary syndecan-1 levels were significantly higher in pediatric patients with severe AKI after cardiac surgery. 144 Similarly, in adult patients with acute decompensated heart failure, plasma syndecan-1 level was a good predictor of risk of impending renal failure and mortality. 145 High plasma syndecan-1 level also predicted renal failure in kidney transplant patients. 146 Given the strong association between heparanase expression and syndecan-1 shedding, it is likely that much of the increase in plasma syndecan-1 levels during kidney injury is the result of heparanase activity within the kidney. These and other studies underscore the potential of emerging therapeutic strategies of inhibiting heparanase, as well as the diagnostic value of detecting products of heparanase activity for prognostication and treatment of kidney diseases.
Future Perspectives
There is a significant amount of data demonstrating important roles for both heparanase and shed syndecan-1 in the pathogenesis of numerous diseases. Although it was suspected for many years that the cleaving of heparan sulfate chains by heparanase could influence heparan sulfate function, the impact of heparanase on syndecan-1 shedding and the resulting effects on cell behaviors were not anticipated. However, it has become clear that this mechanism is in play in many pathological states and stands as an important axis that holds potential for therapeutic targeting. The finding that heparanase inhibitors can diminish syndecan-1 shedding adds credence to this idea and supports further development of these drugs, many of which are now in preclinical testing or clinical trials.147,148 Another approach would be to target shed syndecan-1 with synstatins 34 or with antibodies that interfere with syndecan function. Beyond the potential of therapeutics, it will be important to further study the heparanase/shed syndecan-1 axis more extensively in non-cancer pathologies. More work needs to be done in the area of inflammation, where shed syndecan-1 can potentiate the activity of the multiple factors that drive the inflammatory process. There have been reports of shed syndecan-1 activity in, for example, inflammatory lung injury 149 and ulcerative colitis. 150 However, a role for heparanase in upregulating shedding in these examples has not been investigated. Shed syndecan-1 can also promote bacterial virulence 14 and, as discussed in this review, the life replication of viruses. A better understanding of how the heparanase/shed syndecan-1 axis regulates the viral life and replication cycle is critical, particularly given the current concern over coronavirus infections. Finally, although it is known that heparanase can regulate the expression of genes that drive MMP production and thereby promote syndecan-1 shedding, the function of nuclear heparanase remains an area ripe for investigation and may provide further clues as to how the heparanase/shed syndecan-1 axis helps drive disease pathogenesis and progression.
Footnotes
Competing Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
SR and RDS designed the review, and SR, JRR, RPR, SKB, KT, IV, and RDS wrote the review.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by a grants from the National Institutes of Health CA211752 (to R.D.S.), the United States – Israel Binational Science Foundation (jointly to R.D.S. and I.V.), and the U.S. Department of Defense W81XWH1810108 (J.R.R.).