
Abstract
Approximately 5% to 10% of all bone fractures do not heal completely, contributing to significant patient suffering and medical costs. Even in healthy individuals, fracture healing is associated with significant downtime and loss of productivity. However, no pharmacological treatments are currently available to promote efficient bone healing. A better understanding of the underlying molecular mechanisms is crucial for developing novel therapies to hasten healing. The early reparative callus that forms around the site of bone injury is a fragile tissue consisting of shifting cell populations held together by loose connective tissue. The delicate callus is challenging to section and is vulnerable to disintegration during the harsh steps of immunostaining, namely, decalcification, deparaffinization, and antigen retrieval. Here, we describe an improved methodology for processing early-stage fracture calluses and immunofluorescence labeling of the sections to visualize the temporal (timing) and spatial (location) patterns of cellular and molecular events that regulate bone healing. This method has a short turnaround time from sample collection to microscopy as it does not require lengthy decalcification. It preserves the structural integrity of the fragile callus as the method does not entail deparaffinization or harsh methods of antigen retrieval. Our method can be adapted for high-throughput screening of drugs that promote efficacious bone healing:
Keywords
Introduction
Fracture healing is a complex regenerative process that occurs in multiple stages, entailing the close coordination of several cell types. Following the initial hematoma formation and acute inflammation, a reparative callus forms at the site of injury, and healing occurs through a combination of intramembranous and endochondral ossification. Intramembranous ossification occurs along the periosteal and endosteal surfaces of the callus periphery, where mesenchymal stem cells (MSCs) and progenitors directly differentiate into osteoblasts that secrete the primary bone matrix, essentially forming a “hard callus” around the entire callus periphery. In contrast, endochondral bone formation occurs at regions juxtaposed to the fracture gap, which are less mechanically stable wherein resident and newly infiltrated MSCs differentiate first into chondrocytes that undergo hypertrophy and secrete cartilage, which undergoes calcification to produce a “soft callus.” Vascular invasion ensues, stimulating recruitment of osteoclasts that remodel the calcified cartilage. This coincides with the infiltration of the soft callus by a second wave of MSCs and osteoprogenitors that will give rise to osteoblasts, which secrete osteoid or bone matrix that calcifies or matures to form the secondary bone within the callus. “Coupled” cycles of osteoclast and osteoblast activities follow to reshape the newly formed secondary bone to the structure of the original cortical bone. These events overlap significantly in time and space, and represent an interplay of signaling mechanisms and a continuum of shifting cell populations within the fracture callus.1–4 However, the precise molecular mechanisms governing this highly coordinated healing process remain largely unknown.
Immunolabeling is a highly effective method to investigate distribution of cell types and protein expression patterns in tissue slices. Although immunohistochemistry of paraffin-embedded bone samples is an attractive method, it requires a lengthy decalcification process. Complete decalcification of murine long bones takes several weeks. Moreover, the harsh methods of deparaffinization and antigen retrieval of paraffin-embedded samples detrimentally affect the structural integrity of the early reparative fracture callus tissue. 5 In comparison, immunofluorescence (IF) labeling of cryosections prepared from freshly frozen fracture calluses is much quicker as it does not require decalcification and entails gentler processing of the callus tissue. Even frozen fracture calluses are challenging to section due to the high likelihood of tissue damage or detachment, especially those from early time points of healing. On the contrary, histological staining of the cryosections with cationic dyes requires decalcification, a lengthy process that can disturb the fracture callus. The length of decalcification is critical for preserving tissue integrity while minimizing the high background signals emanating from the intact calcified bone matrix.
Here, we report an improved method for the processing of cryo-preserved early-stage fracture calluses and IF labeling of the cryosections to study the distribution of protein markers associated with bone healing, through single antibody labeling and multiplexing. To preserve structural integrity, we generated 8-µm-thick cryosections for IF labeling and 12-µm-thick sections for safranin O/fast green histological staining. These sections were collected and directly stained on an adhesive cryofilm tape avoiding the transfer of the delicate sections to glass slides. 6 We also modified the antigen unmasking and decalcification steps to optimize IF labeling with multiple antibodies and histological staining of proteoglycan content, respectively. These improved methods helped preserve tissue integrity and enabled a detailed imaging of the entire callus following IF labeling or histological staining using a wide-field fluorescence microscope and without the need for a confocal microscope. The methods described herein provide us with an improved tool to visualize and quantify the spatial and temporal distribution of specific cell types and molecular signals within the fracture callus at different stages of healing. They will help unravel a better understanding of the molecular mechanisms governing fracture healing and could be used to screen for novel therapeutics aimed at accelerating this process.
Materials and Methods
Mice and Closed Fracture Generation
All animal procedures were approved by the Institutional Animal Care and Use Committee at Indiana University School of Medicine (IUSM). All procedures were performed in compliance with National Institutes of Health guidelines on the use and care of laboratory and experimental animals. C57BL/6J mice were purchased from Jackson Laboratories (Bar Harbor, ME) and housed in the IUSM Laboratory Animal Resource Center under a 12-hr light/12-hr dark cycle. Food and water were provided ad libitum. Unilateral closed fractures were generated on the right femurs of male C57BL/6J mice at 10 weeks of age and stabilized using an intramedullary rod as described previously.7,8
Cryopreservation of Fracture Calluses and Sectioning
Mice were euthanized on days 7 and 14 after fracture, and fractured femurs were immediately harvested and fixed in 2% paraformaldehyde for 48 hr at 4C. Samples were rinsed in phosphate-buffered saline (PBS) and transferred to 30% sucrose for an additional 48 hr at 4C. The intramedullary rod was carefully removed from the fixed and undecalcified femurs using a pair of forceps and needle-nosed pliers. Care was taken not to damage the soft callus. The femurs were then embedded in optimal cutting temperature (OCT) compound with extreme care and flash-frozen in a dry ice and n-hexane (–72C) bath. Frozen samples were stored at −80C until cryosectioning. Cryosections of 8 µm thickness were prepared for IF labeling and toluidine blue staining, whereas thicker 12-µm sections were prepared for safranin O/fast green staining using a cryomicrotome (Leica CM1950; Leica Biosystems, Nussloch, Germany), fitted with a gold standard blade (DT315G50; C.L. Sturkey, Inc., Lebanon, PA). An adhesive cryofilm tape (Section-lab Co. Ltd.; Hiroshima, Japan) was used to collect the sections. 6 The cryofilm tape was positioned at the base of the OCT block such that longitudinal bone sections were directly collected on the adhesive side of the tape (Fig. 1A). Sections on cryofilm tapes were stored in covered plastic trays at −20C for up to a year before histological staining or IF labeling. Cryofilm tapes containing callus sections were placed on individual slides for support such that the tissue sections were facing up during histological staining and IF labeling. These steps helped keep the fractured bone intact.
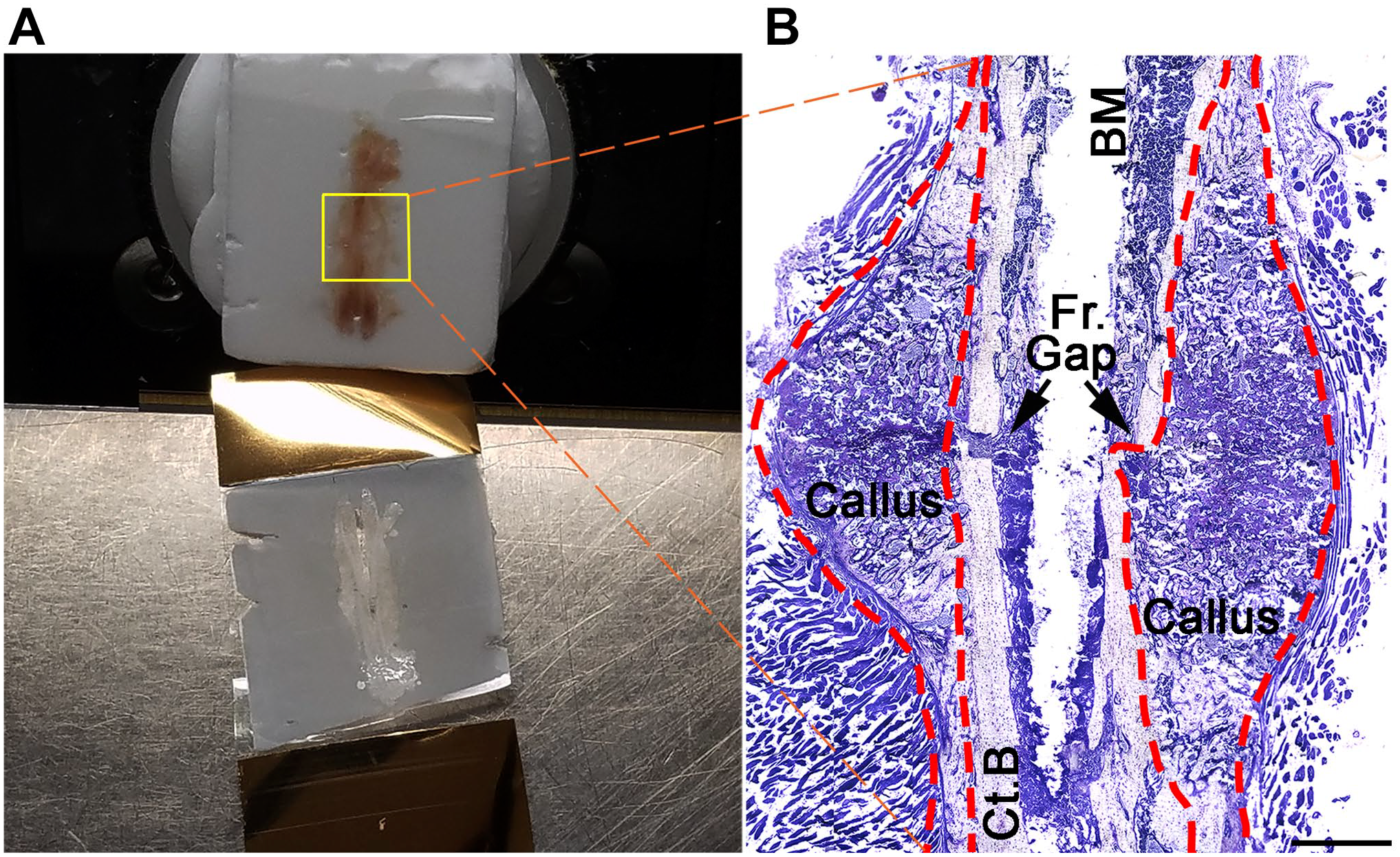
Cryosectioning and verifying the structural integrity of fracture callus sections. (A) The OCT block containing the fracture callus is fixed to a circular metal disk attached to the stage of the cryotome. The section is directly collected on an adhesive cryofilm tape, as shown in the bottom of the image. (B) A composite image of a day 7 murine fracture callus (outlined with red dotted lines) from an 8-µm cryosection stained with toluidine blue. Black arrows indicate the site of injury resulting in the Fr. gap. Scale bar shown in (B) is 1 mm. Abbreviations: OCT, optimal cutting temperature; Fr., fracture; BM, bone marrow; Ct.B, old cortical bone.
Histological Staining
To perform toluidine blue staining, artificial wells were drawn around the cryosections with a histopen. The cryosections on the tape were rehydrated in distilled water for 5 min, incubated in 0.01% toluidine blue solution for 10 s, and excess stain washed off by rinsing with distilled water. The stained tissue was mounted onto coverslips with Super Cryo-Mounting Medium (SCMM; Section lab), before capturing images using light microscopy (Fig. 1B).
Safranin O/fast green staining required a brief decalcification step which impaired the structural integrity of callus tissue in 8-µm sections (Supplementary Fig.). For this reason, thicker 12-µm sections were prepared for safranin O/fast green staining. After rehydrating with distilled water as above, the cryosections were decalcified in 10% EDTA for 5 min at room temperature (RT) and washed once with water for 10 min. The samples were then incubated in 0.1% safranin O for 20 min, followed by one quick rinse in 1% acetic acid. The samples were counterstained with 0.1% fast green for 5 min and washed once with 1% acetic acid for 1 min. The tissue was mounted onto coverslips with SCMM, and images were captured using light microscopy.
IF Labeling of Cryosections
Artificial wells were drawn around the samples with a histopen. Samples were rehydrated with PBS for 10 min and permeabilized with 0.2% Tween 20 in PBS for 5 min. The sections were washed twice with PBS for 10 min each, prior to antigen retrieval, where applicable. When needed, antigen retrieval was achieved by incubating the samples in 1% hyaluronidase for 30 min at RT or in citrate buffer (0.01 M citrate, pH 6.0) for 30 min at 50C (Table 1). Samples were then washed twice in PBS at RT for 10 min each. Autofluorescence was quenched by incubating the samples with Tris-Glycine solution (1 M Tris base, pH adjusted to 7.2–7.4 with 0.1 M glycine) for 30 min at RT, followed by three 15-min washes at RT in PBS. Samples were then incubated in blocking buffer (3% BSA/5% normal donkey serum) for 1 hr at RT, followed by two 10-min washes in PBS at RT. Normal donkey serum was used for blocking as the secondary antibodies used in this study were raised in donkeys. Additional blocking with anti-mouse IgG was performed when primary antibodies raised in mouse were used to minimize background staining. In this case, the samples were incubated at RT with unconjugated AffiniPure Fab Fragment Goat Anti-mouse IgG (H+L) (1:10 dilution; Jackson ImmunoResearch, West Grove, PA) for 1 hr, followed by two 10-min washes in PBS. Primary antibodies were diluted in blocking buffer (see Table 1 for information on antibodies used in this study), and 100 µl of the respective primary antibody dilution was added on the callus sections, followed by overnight incubation at 4C in a humid chamber. Sections were washed with washing buffer (PBST: 1× PBS, 0.2% Tween 20) three times for 15 min at RT. The samples were then incubated for 2 hr with the appropriate fluorescence-conjugated secondary antibodies diluted in blocking buffer (Table 1) at RT in dark, followed by four 15-min washes using PBST. Antibody multiplexing was performed to investigate colocalization of target proteins through simultaneous incubation of unlabeled primary antibodies that originated from different species. Briefly, primary antibodies were diluted in 100 µl blocking buffer, and allowed to incubate on callus sections at 4C overnight, in a humid chamber. This was followed by three 15-min washes at RT in PBST. Corresponding secondary antibodies conjugated with different colored fluorophores were diluted in 100 µl blocking buffer as indicated in Table 1 and placed on callus sections for a 2-hr incubation at RT, followed by four 15-min washes with PBST. For nuclear staining, the sections were incubated with DAPI (4′, 6-diamidino-2-phenylindole, 5 mg/ml stock solution diluted 1:10,000 in distilled water) (D9542; Millipore Sigma, St. Louis, MO) for 10 min in the dark. The sections were quickly rinsed once in 1× PBS and mounted with VECTASHIELD antifade mounting medium (H-1000; Vector Laboratories, Burlingame, CA) on coverslips facing the silver side of the cryofilm tape such that the tissue was sandwiched in between the tape and the coverslip. Coverslips were used to avoid air bubbles during mounting and form a tight interface. This minimized background and yielded cleaner images. IF-labeled images were acquired using a Leica DMi8 fluorescence microscope and processed using Leica LAS-X software (Leica DMi8, Leica Microsystems CMS GmbH, Wetzlar, Germany).
List of Primary and Corresponding Secondary Antibodies, Dilutions, and Information on Antigen Retrieval Method Used in This Study.
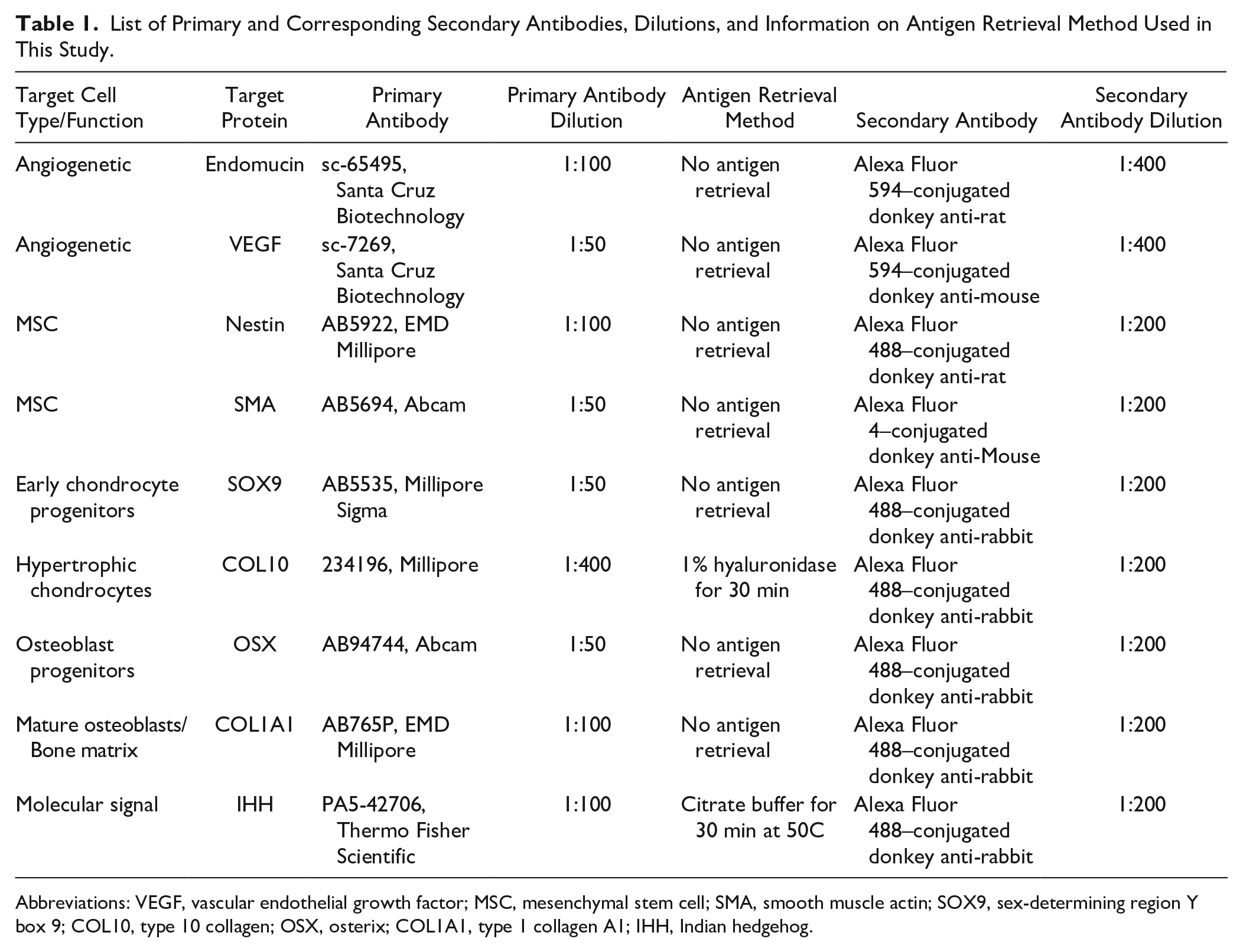
Abbreviations: VEGF, vascular endothelial growth factor; MSC, mesenchymal stem cell; SMA, smooth muscle actin; SOX9, sex-determining region Y box 9; COL10, type 10 collagen; OSX, osterix; COL1A1, type 1 collagen A1; IHH, Indian hedgehog.
Results
Cryosectioning of Early-stage Fracture Calluses for IF
To preserve tissue integrity, we generated OCT-embedded blocks of frozen callus tissue and captured the sections on adhesive cryofilm tapes (Fig. 1A). Thickness of cryosections generated from early-stage fracture calluses was very critical for the successful outcome of IF labeling and histological staining. For instance, the traditional 5-µm-thick cryosections resulted in tissue disintegration during sectioning or following processing, whereas thicker sections impeded a clear visualization of cell morphology within the callus. Through trial and error, we concluded that sections of 8 µm thickness were thick enough to retain tissue integrity during antigen retrieval processes and withstand the repeated washes associated with IF labeling, but thin enough to enable the visualization of cellular details during microscopic interrogation of immunolabeled sections. Preservation of the overall structural integrity and cell morphology within the callus sections was determined through toluidine blue staining of serial sections (Fig. 1B).
IF Labeling of Cell Distribution and Signaling Markers in Early-stage Fracture Calluses
IF labeling was performed directly on cryotapes containing the 8-µm callus sections. Antigen retrieval was necessary to unmask the epitopes for some primary antibodies used in the study (Table 1). For example, antigen retrieval was necessary to unmask the epitope for an antibody against Indian hedgehog (IHH), a signaling molecule required for chondrocyte maturation, cartilage vascularization, and osteoblast differentiation during embryonic skeletal development and bone healing 9–14 (Fig. 2). However, antigen retrieval steps were not necessary for the antibody against osterix (OSX), a marker for osteoprogenitors (Table 1). We immunostained cryosections prepared from fracture calluses freshly isolated on days 7 and 14 after fracture with primary antibodies against markers for angiogenesis (endomucin 1 [EMCN]), chondrocyte progenitors (sex-determining region Y box 9 [SOX9]), osteoprogenitors (OSX), and bone matrix (type 1 collagen A1 [COL1A1]) (Fig. 3). Tissue integrity was very well preserved after the IF procedure in these 8-µm cryosections, and cellular details were readily visualized using a simple wide-field fluorescence microscope (Fig. 3). Using our modified cryosectioning method and multiplexing, we investigated the colocalization of OSX-positive osteoprogenitors invading the callus with vascular endothelial growth factor (VEGF)-positive endothelial cells (Fig. 4A). The majority of VEGF-positive cells in the day 7 postfracture callus were also OSX-positive, suggesting that the bulk of VEGF expression during the early stages of healing came from OSX-expressing early osteoblasts (Fig. 4A). In contrast, there was no overlap among type 10 collagen (COL10)-positive hypertrophic chondrocytes and COL1A1 expression in the early reparative callus (Fig. 4B), suggesting that the hypertrophic chondrocytes in the day 7 fracture callus do not express type 1 collagen.
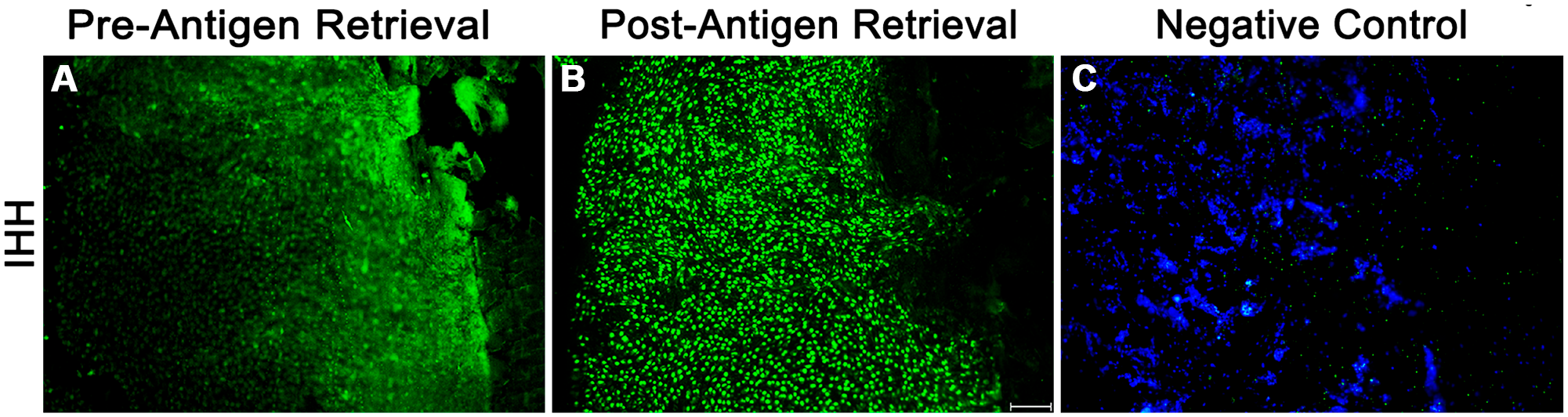
Effect of antigen retrieval on IHH immunofluorescence on callus cryosections. Eight-µm cryosections prepared from day 7 fracture calluses were stained directly on the adhesive cryotape with a primary antibody against IHH, followed by a secondary antibody conjugated to Alexa Fluor 488. (A) and (B) IHH immunolabeling in serial sections without or with antigen retrieval, respectively. (C) Secondary antibody–only stained serial section (negative control). Nuclei were stained with DAPI. Representative scale bar is 100 µm in length. Abbreviations: IHH, Indian hedgehog; DAPI, 4′, 6-diamidino-2-phenylindole.
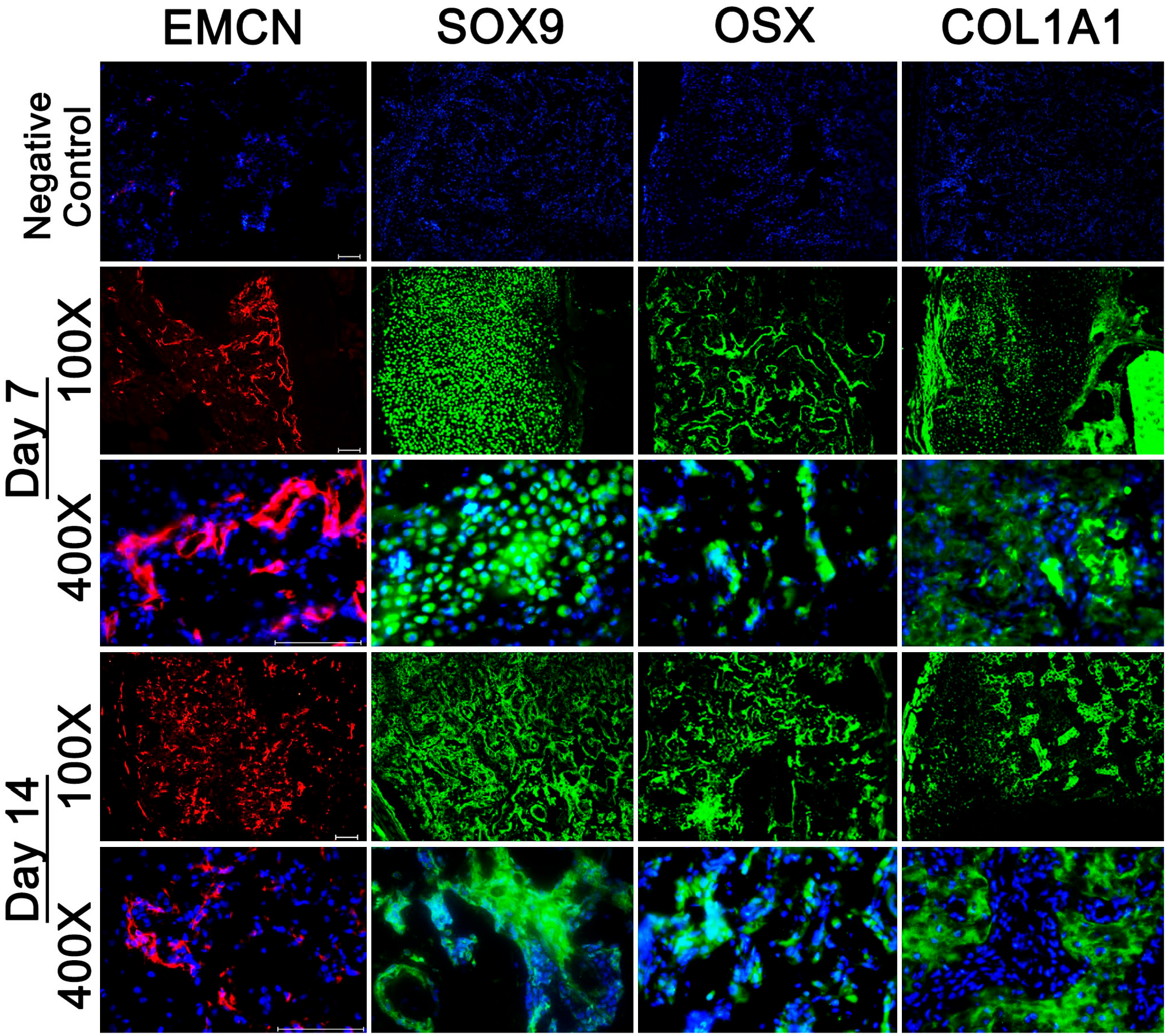
Spatial and temporal patterns of marker protein expression within the fracture callus. Eight-µm cryosections of fracture calluses were incubated with primary antibodies against EMCN, SOX9, OSX, and COL1A1 and subsequently with Alexa Fluor 594–conjugated (EMCN) or Alexa Fluor 488–conjugated (all others) secondary antibodies. Nuclei were stained with DAPI. Low (100×) and high (400×) magnification images from day 7 and day 14 fracture callus sections are shown to provide an overall view of the pattern of expression of these marker proteins within the callus as healing proceeds. Negative controls are day 7 cryosections stained with only the secondary antibody and DAPI. Scale bars are 100 µm in length. Abbreviations: EMCN, endomucin 1; SOX9, sex-determining region Y box 9; OSX, osterix; COL1A1, type 1 collagen A1; DAPI, 4′, 6-diamidino-2-phenylindole.
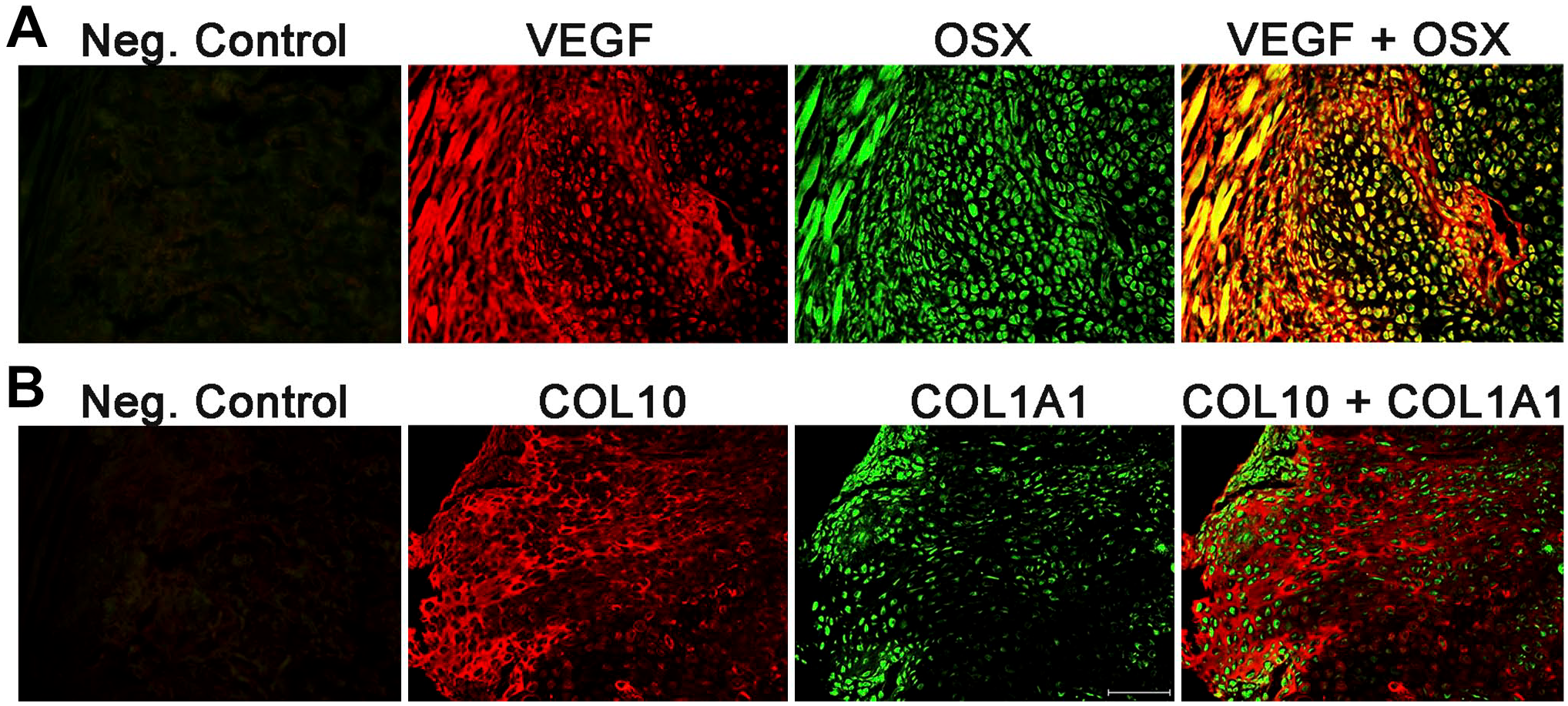
Multiplexing of day 7 postfracture callus sections. Simultaneous staining with primary antibodies against VEGF and OSX (A) or against COL10 and COL1A1 (B) was performed on 8-µm cryosections generated from a day 7 fracture callus. Secondary antibodies conjugated to Alexa Fluor 594 (VEGF and COL10) and Alexa Fluor 488 (OSX and COL1A1) were used. The presence of yellow color in the overlay (far right panel) images indicates colocalization of signals. Negative controls are serial sections stained only with the two secondary antibodies. Images were taken at 200× magnification. Representative scale bar is 100 µm in length. Abbreviations: VEGF, vascular endothelial growth factor; OSX, osterix; COL10, type 10 collagen; COL1A1, type 1 collagen A1.
During the early stages of bone healing (days 4–14 after fracture), MSCs infiltrate the callus tissue. However, the exact timing of their recruitment into injury site and their mobilization patterns within the callus during bone fracture healing are not fully understood. Studying the recruitment and mobilization of MSCs during fracture healing is difficult due to the lack of reliable markers that accurately identify murine MSCs, which are a heterogeneous population. We performed IF labeling of the cryosections for the presence of two proteins that are highly expressed in stem/progenitor cells that give rise to osteoblasts, namely, smooth muscle actin (SMA) and Nestin.15–18 We observed that SMA-positive cells are predominantly located at the periosteal regions of the day 7 fracture callus, but migrate to the central callus region by day 14 (Fig. 5). In contrast, Nestin-positive progenitor cells are more widely distributed throughout the callus at days 7 and 14 (Fig. 5). These results suggest that SMA and Nestin are markers for differing progenitor populations within the callus.
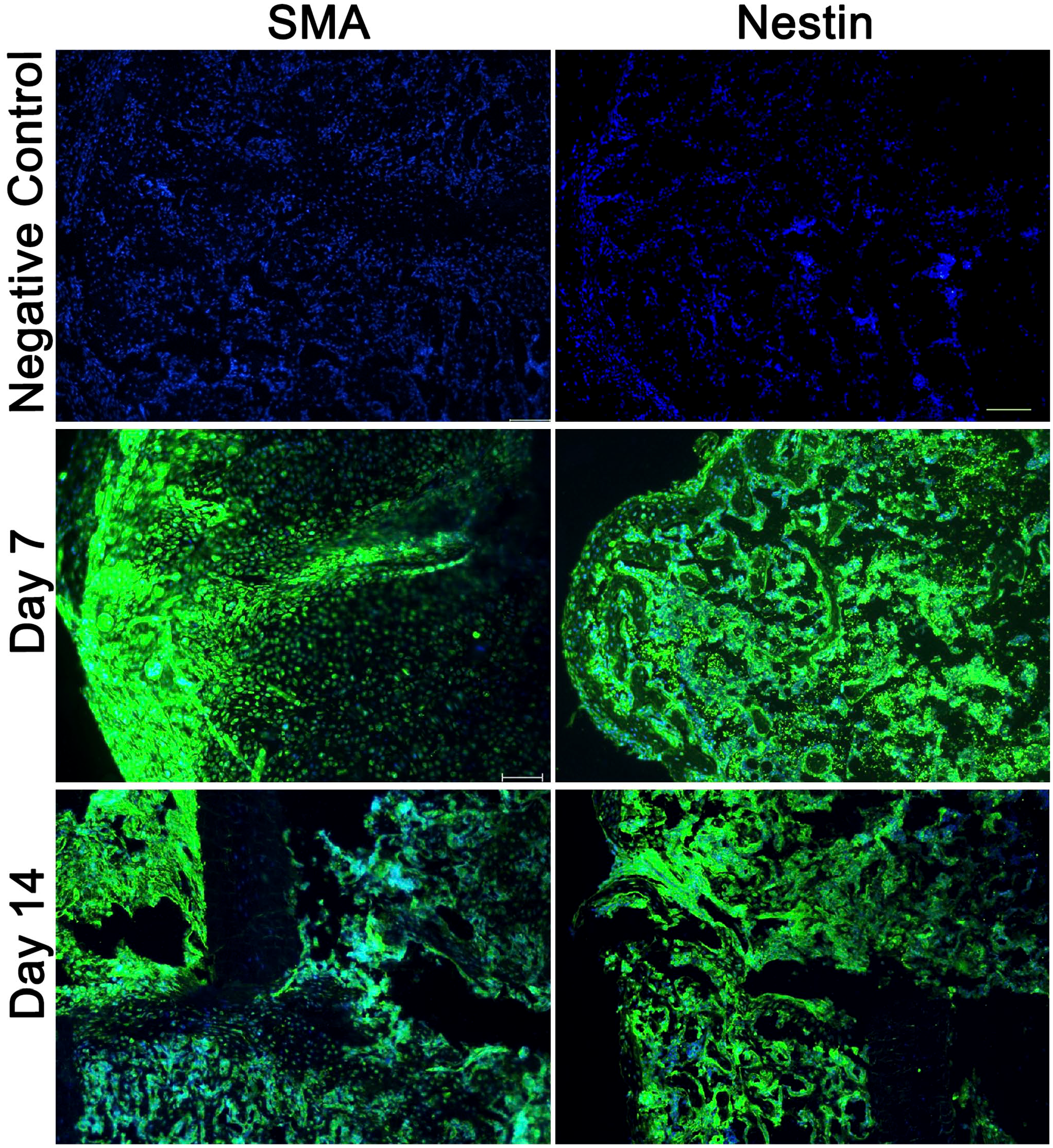
Immunofluorescence staining shows patterns of two different MSC populations within the reparative fracture callus. Cryosections (8 µm) from day 7 and day 14 fracture calluses were stained with primary antibodies against SMA or Nestin, followed by a secondary antibody conjugated to Alexa Fluor 488. Nuclei were stained with DAPI. Negative controls are day 7 cryosections (8 µm) stained with only the secondary antibody and DAPI. Images were captured at 100× magnification. Representative scale bar is 100 µm in length. Abbreviations: SMA, smooth muscle actin; MSC, mesenchymal stem cell; DAPI, 4′, 6-diamidino-2-phenylindole.
Cryosectioning of Early-stage Fracture Calluses for Histology
Safranin O stains proteoglycans orange, and fast green enables a contrasting view of the surrounding stroma within the reparative callus. Decalcification enables better binding of fast green to bone. However, the integrity of the tissue within the 8-µm cryosections from day 7 calluses was compromised following decalcification (Supplementary Fig.). We empirically determined that thicker 12-µm cryosections can withstand decalcification using 10% EDTA for 5 min prior to safranin O/fast green staining. Moreover, staining for proteoglycan deposition with 12-µm cryosections was sufficient to provide high-quality and detailed imaging of cartilage tissue and hypertrophic chondrocytes within the day 7 fracture callus (Fig. 6).
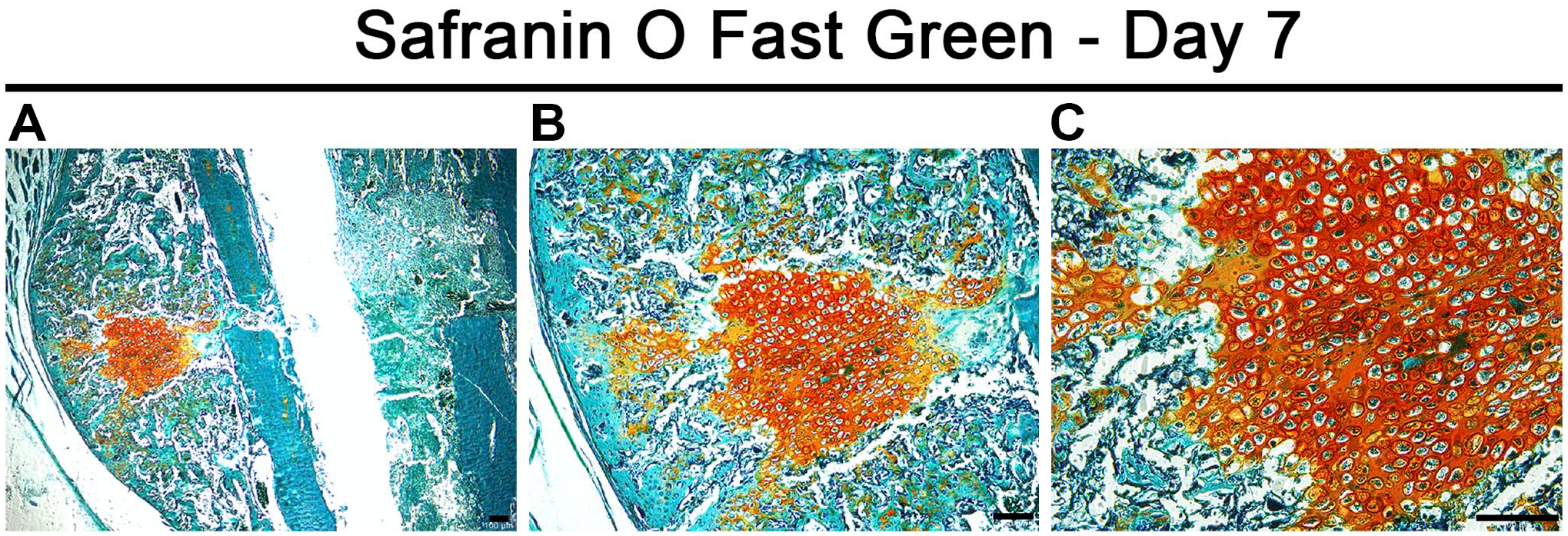
Endochondral ossification within a day 7 fracture callus. Representative images of a day 7 fracture callus cryosection of 12 µm thickness stained histologically with safranin O/fast green are shown. Safranin O stains cartilage orange, and fast green stains other tissues including bone green. Images were taken at (A) 40×, (B) 100×, and (C) 200× magnification. The lower magnification images provide an overview of the location and extent of cartilage within the day 7 callus, whereas the higher magnification image provides an appreciation of the morphology of hypertrophic chondrocytes within the central callus. Scale bars are 100 µm in length.
Discussion
Bone healing is orchestrated by a cascade of signaling processes that modulate the shifting of cell populations within the fracture callus. One way to investigate the identity of the molecular signals that regulate healing is to perform a comprehensive analysis of gene and protein expression using RNA and protein isolated from whole calluses collected at varying time points after fracture. Despite being a powerful investigative tool, this method is not useful to identify the cell types within the callus that express a given signaling protein at a given time point of healing or to investigate whether a particular cell type is localized at the callus periphery or near the fracture gap. IF, on the contrary, enables the determination of the spatial and temporal patterns of cell type fluctuations or molecular signals expressed by the cells localized in the various regions of the callus at any given time point during the healing process. However, the early reparative callus is also very fragile and not easily amenable to sectioning and/or harsh tissue processing procedures. We improved the methodology to cryosection early-stage fracture calluses and perform IF labeling and histological staining to enable the evaluation of cell and molecular mechanisms that govern the healing process. Cryopreservation allowed the sectioning of calluses isolated on days 7 and 14 after bone fracture without the need for decalcification, a step required for paraffin embedding. Complete decalcification of adult murine long bones takes 3 weeks. Thus, our technique significantly reduces the wait time required for freshly isolated fracture callus specimens to be ready for sectioning. This method of cryosectioning can also be applied to process hardened calluses from later stages of bone healing. Furthermore, it eliminates the need for deparaffinization and harsh methods of antigen retrieval, which could impair the structural integrity of the early-stage fracture calluses.
Eight-µm cryosections of the calluses, prepared as we describe here, enabled IF imaging of various cell types as well as signaling proteins (IHH) that govern bone repair. We were able to perform multiplexing of antibodies to understand the colocalization of different cell types and molecular signals within the callus during early time points following a fracture. Through this technique, we also identified the localization patterns of two different types of stem/progenitor cells within the fracture callus during early time points of healing. SMA-positive cells originated from the callus periphery at day 7 after fracture and moved into the central callus as healing proceeded. On the contrary, Nestin-positive progenitors had a more even distribution throughout the fracture callus regardless of the time point examined. Finally, we successfully performed safranin O/fast green staining of the fracture callus cryosections by preparing slightly thicker 12-µm slices and a short decalcification step. Altogether, these improvements allowed us to drastically reduce the time it takes to generate sections and study the early cellular and molecular events associated with bone healing.
In conclusion, we describe an improved method to perform IF and histological staining of cryosections prepared from undecalcified fracture calluses, which enables the detection and quantification of marker proteins regulating various cell and molecular processes associated with bone healing. This method is easy to follow and can be used to unravel novel signaling mechanisms that orchestrate the healing process, which could then be targeted to accelerate fracture healing in patients.
Supplemental Material
2019-00145R1_Production_Supplemental_Figure_1_online_supp – Supplemental material for An Improved Methodology to Evaluate Cell and Molecular Signals in the Reparative Callus During Fracture Healing
Supplemental material, 2019-00145R1_Production_Supplemental_Figure_1_online_supp for An Improved Methodology to Evaluate Cell and Molecular Signals in the Reparative Callus During Fracture Healing by Anuradha Valiya Kambrath, Justin N. Williams and Uma Sankar in Journal of Histochemistry & Cytochemistry
Footnotes
Acknowledgements
The authors thank Dr Keith W. Condon for help with histology and guidance on immunofluorescence.
Competing Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
AVK developed the methodology, performed experiments, took images, analyzed the data, and wrote the manuscript; JNW analyzed the data and made figures; US conceived and supervised the study, designed the experiments, and edited the manuscript.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Research reported in this publication was supported by the Department of Defense (DoD) United States Army Medical Research and Materiel Command and Congressionally Directed Medical Research Program under Award Number PR121604, and the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health (NIH) under Award Number R01AR068332 (both to U.S.). J.N.W. is supported through a Comprehensive Musculoskeletal T32 Training Program from NIAMS/NIH (AR065971). The content is solely the responsibility of the authors and does not necessarily represent the official views of the DoD or the NIH.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.