
Abstract
Gastric adenocarcinoma develops in metaplastic mucosa associated with Helicobacter pylori infection in the stomach. We have sought to evaluate the precise lineage changes in the stomachs of insulin-gastrin (INS-GAS) mice infected with H. pylori and/or intestinal flora (Altered Schaedler’s Flora; ASF). Stomachs from groups infected with H. pylori contained progressive spasmolytic polypeptide-expressing metaplasia (SPEM) compared with germ-free and mice infected with ASF alone. The overall phenotype of the H. pylori-infected mice was dominated by Ulex europaeus lectin (UEAI)-positive foveolar hyperplasia that was distinct from GSII/CD44v9-positive SPEM. However, in the mice with H. pylori co-infected with ASF, we identified a subpopulation of UEAI-positive foveolar cells that co-expressed intestinal mucin 4 (MUC4). These regions of foveolar cells were variably positive for CD44v9 as well as TFF3. Interestingly, an intravascular lesion identified in a dual H. pylori/ASF-infected mouse expressed both UEAI and Muc4. Finally, we identified an increase in the number of tuft cells within the mucosa of H. pylori-infected groups. Our findings suggest that H. pylori infection promotes foveolar hyperplasia as well as metaplasia, while co-infection may promote progressive foveolar and metaplastic lesions as well as dysplasia. Grading of gastric lesions in mice as preneoplastic requires multiple immunostaining markers to assign lineage derivation and behavior.
Introduction
Gastric adenocarcinoma is one of the leading causes of cancer-related deaths in the world. 1 Epidemiological studies in humans, as well as experiments in rodents, have associated Helicobacter pylori with peptic ulcers, gastric atrophy, and intestinal-type gastric adenocarcinoma.2–5 However, only a small percentage (1–3%) of individuals that are infected with H. pylori ever develop neoplasia related to its presence, suggesting that other factors are involved in the process. In addition, a subset of patients that presents with gastric cancer has no evidence of H. pylori infection. 6 The exact mechanism, however, requires further investigation. 7
Chronic H. pylori infection leads to atrophic gastritis and hypochlorhydria.8,9 This low-acid state in the stomach can promote colonization of the gastric mucosa with intestinal flora. Previous studies have demonstrated that non-H. pylori bacteria, including those considered as pathogenic or commensal intestinal flora, can colonize the stomach and represent an additional gastric cancer risk, particularly in H. pylori-infected, susceptible individuals.10,11 Using the transgenic, hypergastrinemic insulin-gastrin (INS-GAS) mouse model of H. pylori-accelerated gastric carcinogenesis, Lertpiriyapong et al. evaluated if a restricted microbiota limited to 3 species of Altered Schaedler’s Flora (restricted ASF or rASF), including ASF356 Clostridium species, ASF361 Lactobacillus murinus, and ASF519 Bacteroides species, was sufficient to contribute to the gastric intestinal neoplasia incidence after development of gastric atrophy secondary to H. pylori infection. 12 They concluded that colonization with H. pylori and a restricted microbiota consisting of only 3 species of commensal bacteria promoted neoplastic changes in gnotobiotic male INS-GAS mice to a similar extent as mice colonized with complex microbiota. Previous studies had shown that INS-GAS mice develop spasmolytic polypeptide-expressing metaplasia (SPEM) and invasive gland lesions that penetrate into the submucosa. 13 These lesions were potentiated with co-infection with H. felis.
The changes observed in dual H. pylori/ASF-infected mice were analyzed primarily by H&E staining and were dominated by cystic intramucosal lesions that were considered dysplastic. 12 To differentiate between inflammation, dysplasia, hyperplasia, and neoplasia, a gastric histologic activity index (GHAI) was utilized. Based on H&E staining, rASF mice and mice colonized with normal intestinal flora developed mildly increased severity of all pathomorphological features included in the GHAI scoring system. INS-GAS mice infected with only H. pylori (mHp), at 7 months post-infection, developed moderate inflammation, diffuse glandular hyperplasia, severe oxyntic atrophy, SPEM, and moderate to severe dysplasia, including cytological atypia and architectural abnormalities. Mice co-colonized with rASFHp and IFHp had more severe pathology, including high-grade glandular architectural and cytological abnormalities that were classified as gastrointestinal intraepithelial neoplasia with invasion into the lamina propria, classified as intramucosal carcinoma. 12
Because the previous studies evaluated mucosal lesions based on histological criteria alone, we sought to evaluate in greater detail the exact cell lineages contributing to putative metaplastic and neoplastic lesions in the stomachs of INS-GAS mice with H. pylori colonization without or with ASF co-colonization. We found that the gastric mucosae of INS-GAS mice infected with H. pylori without or with ASF co-infection were dominated by Ulex europaeus lectin (UEAI)-positive foveolar hyperplasia. Stomachs from the groups infected with H. pylori contained more extensive SPEM compared with germ-free mice and mice infected with ASF alone. While scattered TFF3 and intestinal mucin 2 (MUC2)-expressing cells were observed in H. pylori-infected mice, we did not observe any evidence for intestinal metaplasia, as assessed by expression of Cdx1 or Cdx2 or production of morphological goblet cells. While the mucosal phenotype in the mice co-infected with H. pylori and ASF was dominated by foveolar hyperplasia, we identified a subpopulation of foveolar cells that co-expressed MUC4 and UEAI. Some cells in these regions also showed expression of TFF3. In addition, we identified an increase in the number of tuft cells within the mucosa of H. pylori-infected mice. Our findings suggest that H. pylori infection is the major promoter of SPEM and foveolar hyperplasia in the stomachs of infected mice, but further dual infection with intestinal flora leads progression of metaplasia and foveolar hyperplasia with the identification of dysplastic lesions by histological staining.
Materials and Methods
Animals
Archival tissue samples were examined from the previous investigation. 12 Animal use was approved by the Massachusetts Institute of Technology Committee on Animal Care. Four experimental groups of INS-GAS mice on an FVB/N background [Tg(Ins1-GAS)1Sbr] included germ-free control (n=4), rASF (n=4), Hp (n=4), and rASFHp (n=4). Transgenic INS-GAS mice were propagated by in-house breeding and maintained in a facility accredited by the Association for Assessment and Accreditation of Laboratory Animal Care, International under the environmental conditions of a 10:14 light/dark cycle, 68 ± 2F and relative humidity range of 30–70%. GF and rASF INS-GAS mice were maintained in sterile isolators and housed in solid-bottomed polycarbonate cages on autoclaved hardwood bedding. Autoclaved diet (Prolab RMH 2000; PMI Nutrition International; St. Louis, MO) and autoclaved water were provided ad libitum. Isolators were evaluated bi-monthly and confirmed negative for microbial contaminants by aerobic, anaerobic, and fungal culture. IF INS-GAS mice were group-housed in microisolator polycarbonate caging on hardwood bedding with ad libitum access to reverse osmosis water, the same autoclaved diet, and were barrier-maintained. Mice were fasted overnight, and euthanized by carbon dioxide. All infected mice were followed for 210 days after inoculation. Because previous investigations have suggested that gastritis and development of GIN in the INS-GAS model is male-predominant, 14 we focused on data from male mice.
Immunostaining
For all immunostaining studies, 5 µm sections of paraffin-embedded material were used. Sections were deparaffinized, rehydrated, and submitted to antigen retrieval using Target Retrieval solution (Dako North America, Inc.; Carpinteria, CA) in a pressure cooker. Blocking was performed using Protein Block Serum-Free (Dako North America, Inc.) for 90 min at room temperature. The primary antibody incubation was performed in Antibody Diluent with Background Reducing Components (Dako North America, Inc.) overnight at 4C. The primary antibodies used were as follows: rabbit anti-MUC2 (1:500, cat. sc-15334; Santa Cruz Biotechnology; Dallas, TX), mouse anti-MUC4 (1:100, cat. sc-33654; Santa Cruz Biotechnology), goat anti-clusterin α (1:2000, cat. sc-6420; Santa Cruz Biotechnology), rabbit anti-Ki67 (1:1000, cat. 12202; Cell Signaling; Danvers, MA), mouse-specific rat anti-CD44v9 (1:10,000; a gift from Prof. Hideyuki Saya, Keio University; Tokyo, Japan), rabbit anti- doublecortin-like kinase 1 (DCLK1; 1:2000, cat. Ab109029; Abcam; Cambridge, MA), and rat anti-MMP-7 (1:500; Vanderbilt Antibody and Protein Resource; Nashville, TN). For detection with immunofluorescence, secondary antibodies (1:500) conjugated with Cy2, Cy3, and Cy5 (Jackson ImmunoResearch Laboratories; West Grove, PA) or FITC-UEAI lectin (1:2000; Sigma; St. Louis, MO) or Alexa 647-conjugated Griffonia simplicifolia lectin II (GSII lectin; 1:2000; Molecular Probes; Eugene, OR) were incubated for 1 hr at room temperature. After incubation with 4′,6-Diamidino-2-phenylindole (DAPI) for 5 min, slides were mounted with ProLong Gold Antifade Reagent (Invitrogen; Carlsbad, CA). Fluorescence imaging was analyzed using an Axio Imager 2 microscope (Carl Zeiss AG; Oberkochen, Germany) or a Leica/Aperio Versa 200 automated fluorescence slide scanner (Leica Biosystems; Buffalo Grove, IL) in the Vanderbilt Digital Histology Shared Resource.
Quantitative Analysis
Fluorescently immunostained tissue slides were imaged on a Leica/Aperio Versa 200 automated slide scanner (Leica Biosystems–Vanderbilt Digital Histology Shared Resource). Tissue sections were imaged at 20× magnification to a resolution of 0.323 µm/pixel. Images were analyzed using the Cell Profiler cell image analysis program. To quantify the number of CD44v9- and Ki67-positive cells, tissue sections were divided into 1 mm sections, and the number of positive cells per section was identified. An average of >5 mm of stomach corpus tissue was analyzed per mouse. To quantify DCLK1-positive cells, we measured total positive pixels on the entire slide and divided by the total area of tissue scanned. Experimental groups contained 4 mice. All graphs and statistics were completed in GraphPad Prism, using one-way ANOVA with Bonferroni’s post hoc multiple comparison tests to determine significance.
Results
SPEM Was Present in All Groups and Increases With H. pylori Infection
SPEM was evaluated using immunostaining with antibodies against CD44v9, which is strongly expressed on the basolateral membrane in SPEM.15,16 In addition, we stained with GSII lectin, a marker of mucous neck cells and SPEM. As previously reported, 13 INS-GAS mice develop SPEM after 6 months of age, and scattered CD44v9 and GSII dual-positive cells were observed at the bases of some corpus glands in uninfected INS-GAS mice (Fig. 1A). We quantified the number of cells expressing CD44v9 and found that SPEM significantly increased with H. pylori infection (Fig. 1B). Additional infection with ASF did not alter the number of CD44v9-positive cells.
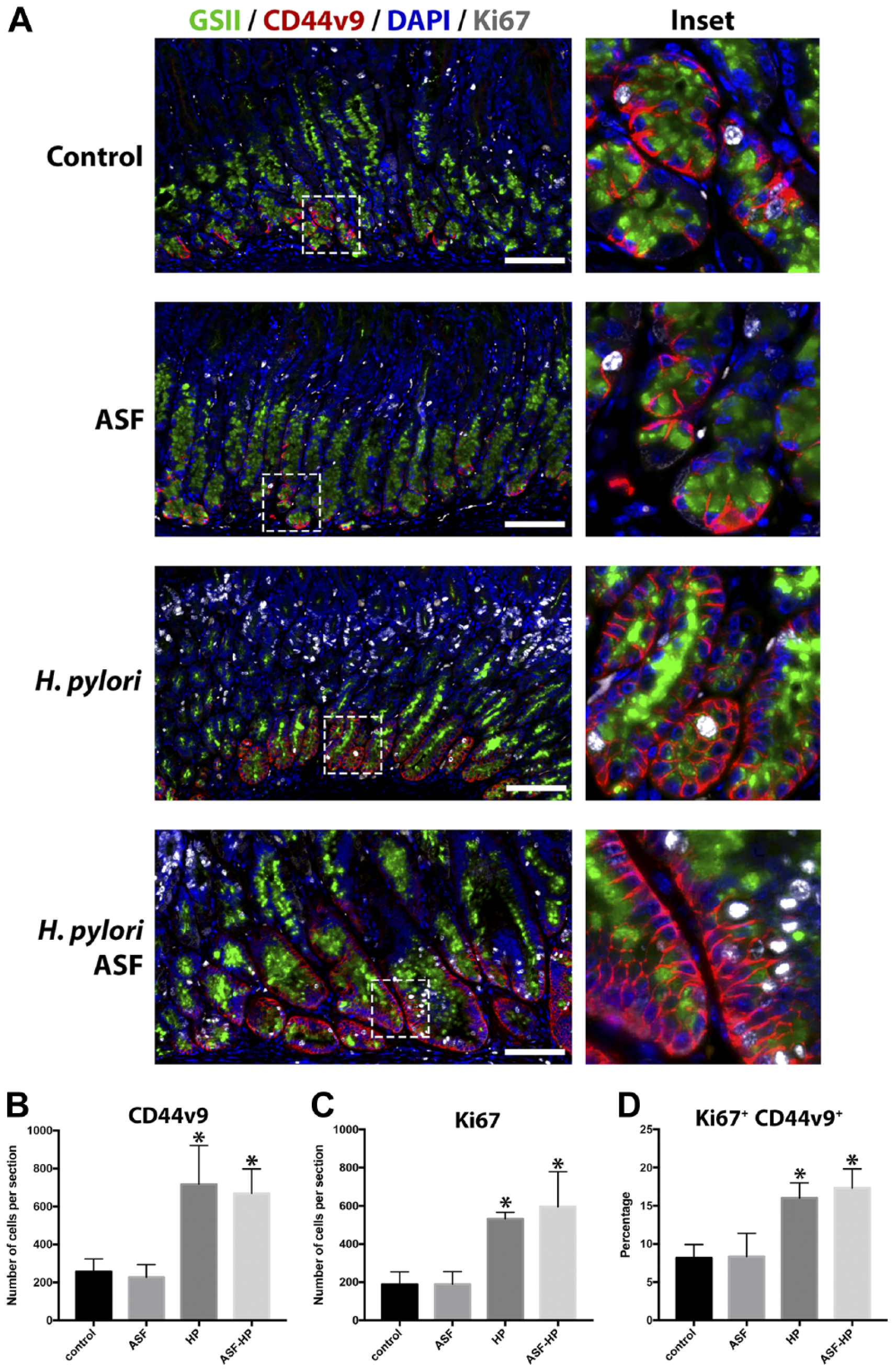
Increased SPEM in INS-GAS transgenic mice infected with H. pylori. (A) Representative sections of stomach stained with CD44v9 (red), GSII lectin (green), DAPI (blue), and Ki67 (white) to determine proliferative SPEM lesions at the base of glands. (B) Quantification of CD44v9-positive cells as determined by the number of CD44v9-positive cells per 1 mm section of stomach (*HP p=0.01, ASF-HP p=0.02 compared with uninfected control). (C) Quantification of proliferative cells as determined by the number of Ki67-positive cells per 1 mm section of stomach (*HP p=0.02, ASF-HP p=0.008 compared with uninfected control). (D) Percent of CD44v9-positive cells that are co-positive for Ki67 (*HP p=0.02, ASF-HP p=0.009 compared with uninfected control). Significantly higher levels of proliferation and CD44v9-positive cells were observed in the H. pylori-infected groups. Scale bars = 100 µm. Abbreviations: SPEM, spasmolytic polypeptide-expressing metaplasia; INS-GAS, insulin-gastrin; GSII lectin, conjugated Griffonia simplicifolia lectin II; DAPI, 4′,6-Diamidino-2-phenylindole; HP, H. pylori; ASF, Altered Schaedler’s Flora.
To evaluate proliferation in the epithelium, we used Ki67 staining to visualize dividing cells. We quantified the number of cells expressing Ki67 and found that proliferation significantly increased with H. pylori infection (Fig. 1C). Ki67-positive cells were observed throughout the foveolar regions of the stomach glands. To determine whether the SPEM cells are dividing, indicating proliferation in the metaplastic areas, we evaluated the percentage of CD44v9 cells dual-positive for Ki67. We found that H. pylori infection significantly increased the number CD44v9-positive SPEM cells that are dividing (Fig. 1D). We observed no additional increase in proliferation in mice with dual H. pylori /ASF infection.
Foveolar Hyperplasia Is Significantly More Extensive in the H. pylori Infected Groups
Because most of the Ki67-positive cells were found in the foveolar area, we evaluated foveolar hyperplasia in the 4 conditions. Foveolar hyperplasia, a histopathological manifestation of reactive gastritis and oxyntic atrophy with elevations in gastrin, was evaluated using fluorescence staining to examine binding of UEAI lectin (Fig. 2A). We quantified UEAI staining foveolar cells as a percent of mucosal height in each condition. Figure 2B demonstrates that mice monocolonized with H. pylori or co-colonized with H. pylori and ASF both showed similar increases in foveolar cells, suggesting that H. pylori infection promotes foveolar hyperplasia in INS-GAS mice. Nevertheless, we did observe expanded intramucosal cystic lesions in the mice co-colonized with H. pylori and ASF.
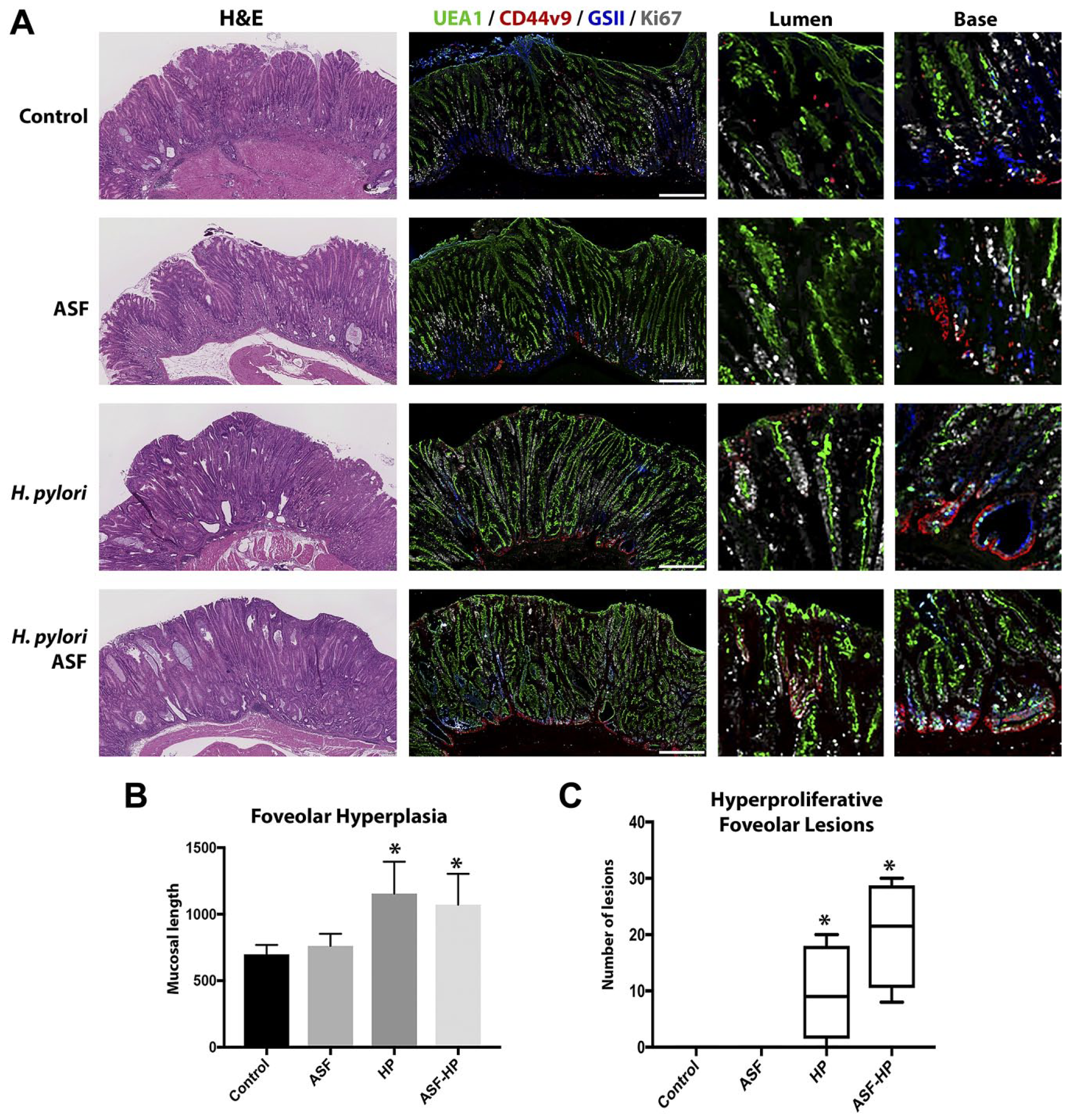
Increased foveolar hyperplasia in INS-GAS transgenic mice infected with H. pylori . (A) Representative sections of stomach stained with H&E (at left) and immunostained for CD44v9 (red), UEA1 lectin (green), GSII lectin (blue), Ki67 (white) to determine foveolar hyperplasia. Magnified insets from the lumen and base to the right. (B) Length of foveolar hyperplasia was calculated by measuring the thickness of the total tissue (μm) and subtracting the thickness of the CD44v9-positive area (p<0.01 compared with uninfected control). (C) Quantification of hyperproliferative foveolar lesions found in the luminal area of the tissue. Lesions were identified by CD44v9- and Ki67-positive staining (*p<0.01 compared with uninfected control). HP-infected groups exhibited larger foveolar hyperplastic areas and more frequent hyperproliferative foveolar lesions. Scale bars = 250 μm. Abbreviations: INS-GAS, insulin-gastrin; GSII lectin, conjugated Griffonia simplicifolia lectin II; HP, H. pylori ; ASF, Altered Schaedler’s Flora.
Further examination of the foveolar hyperplasia identified in the H. pylori -infected groups demonstrated that hyperplastic glands contained regions luminal to the intramucosal cystic areas containing cells that were CD44v9-positive in the basolateral membranes and also Ki67-positive (Fig. 2A). No such hyperproliferative foveolar lesions were observed in the control or ASF groups. Both the H. pylori -infected group and the H. pylori /ASF dual-infected mouse stomachs contained a variable number of hyperproliferative foveolar lesions in their mucosa (Fig. 2C).
H. pylori Co-infected Groups Express Markers of Progressive Metaplasia
We have previously demonstrated that SPEM progresses in the context of chronic inflammation to expression of increasing levels of intestinal markers and increasing proliferation. 17 We, therefore, evaluated markers of intestinalization and progressive SPEM in lineages from all 4 groups using antibodies against TFF3, MUC2, MUC4, MMP7, and clusterin. Strong clusterin staining was observed in all 4 groups, especially in the basal regions of the mucosa, consistent with the presence of progressive SPEM (Fig. 3A). No TFF3 or MUC2 staining was observed in control or ASF-infected mice. However, TFF3 and MUC2 were both present in scattered cells within the mucosa of both H. pylori-infected and dual H. pylori/ASF-infected mice (Fig. 3A and B). No MMP7 expression in epithelial cells was observed in any of the groups (Fig. 3B). MUC4 expression was not observed in control and ASF-infected stomachs, but regions of MUC4-expressing cells were found in the foveolar regions of H. pylori-infected mice. More extensive regions of MUC4-expressing foveolar cells were observed in dual H. pylori/ASF-infected mice (Fig. 3B). MUC4 staining was not observed in regions of SPEM.
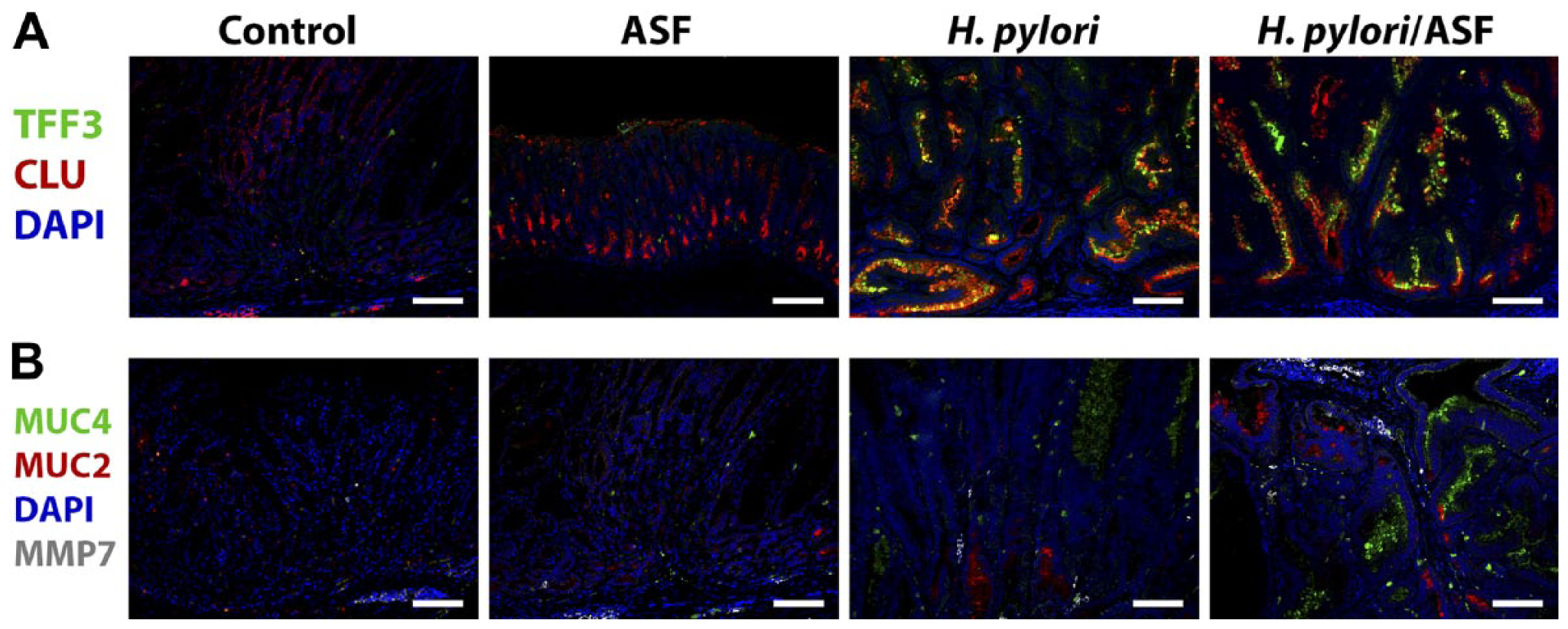
H. pylori co-infection with ASF promotes increased expression of MUC4 and TFF3 in foveolar cells. Representative sections of stomach stained with (A) TFF3 (green), clusterin (red), and DAPI (blue). (B) MUC4 (green), MUC2 (red), DAPI (blue), and MMP7 (white). Note that puncta with grayscale MMP7 staining were not in epithelial cells and mostly represented non-specific staining of red blood cells. Scale bars = 100 µm. Abbreviations: ASF, Altered Schaedler’s Flora; DAPI, 4′,6-Diamidino-2-phenylindole; MUC4, intestinal mucin 4; MUC2, intestinal mucin 2; CLU, clusterin.
As we found a significant increase in foveolar hyperplasia with H. pylori-infected mice, we also investigated whether the MUC4/TFF3-positive cells were arising in the same areas. Most of the MUC4/TFF3-positive cells were found in the UEAI-positive areas (Fig. 4). These were a separate and distinct UEAI-positive population consistent with a different surface cell population (Fig. 4). These areas were not highly proliferative as the majority of these areas were not populated with Ki67-positive cells.
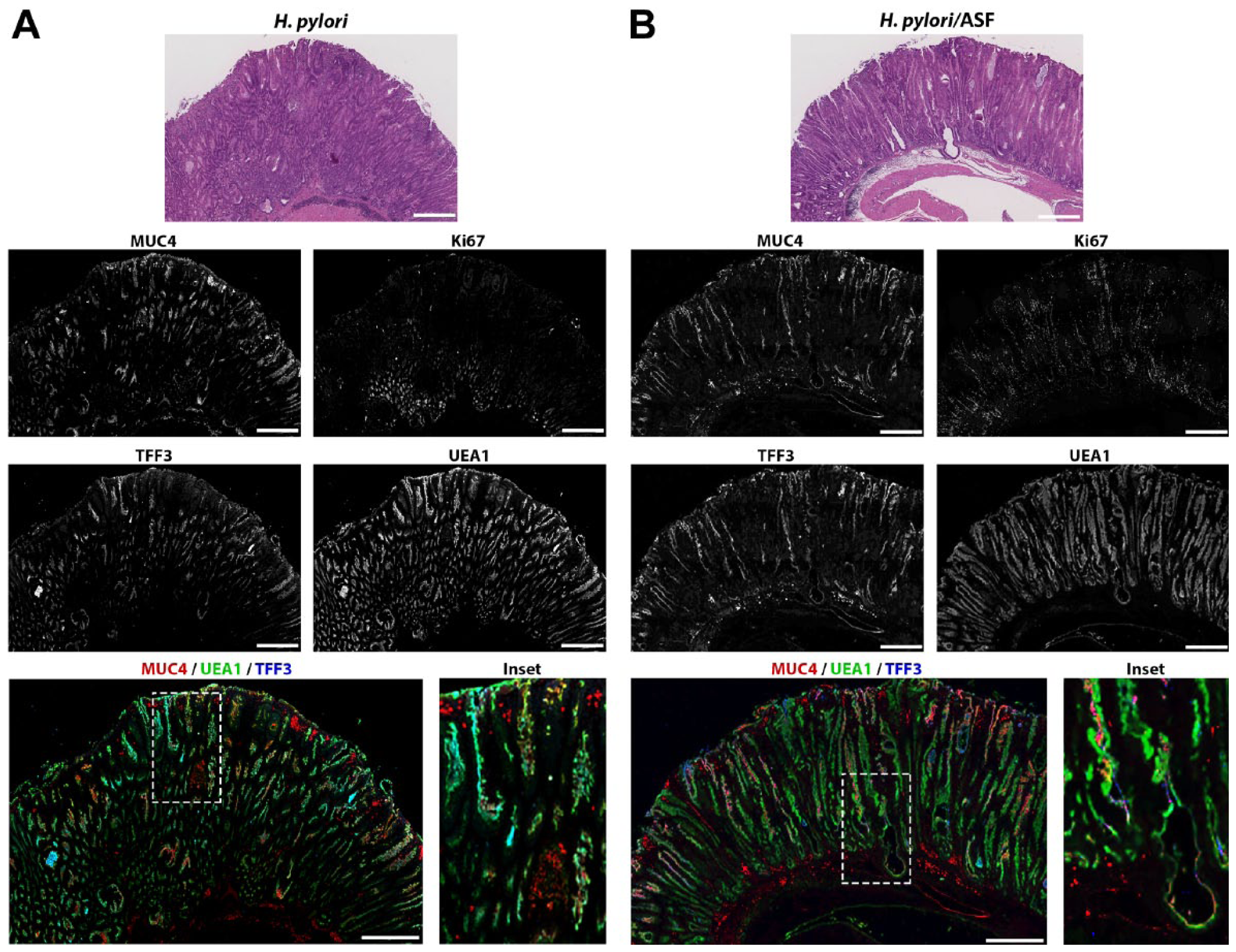
Expression of MUC4, Ki67, TFF3, and UEAI in H. pylori versus H. pylori and ASF co-infection. Serial sections of stomach were stained for H&E, MUC4/UEAI/TFF3, and Ki67. Representative sections from (A) H. pylori-infected or (B) H. pylori/ASF-infected mice. Merged image containing MUC4 (red), UEA1 (green), TFF3 (blue), with high magnification inset. Scale bars = 500 μm. Abbreviations: ASF, Altered Schaedler’s Flora; MUC4, intestinal mucin 4; UEAI, Ulex europaeus lectin.
As previously noted, especially in H. pylori/ASF-infected mice, one can identify foci of metaplastic/dysplastic epithelial cells within vessels in the submucosa.12,13 These lesions suggested aberrant extension of more aggressive dysplastic cells out of the mucosa. In one of the H. pylori/ASF-infected mouse stomach samples, an intravascular lesion identified on H&E staining was noted. We, therefore, evaluated the immunostaining characteristics of the ectopic vascular invasion (Fig. 5). Immunostaining on serial sections demonstrated that the vascular invasive epithelial cells were stained with UEAI lectin as well as with antibodies against clusterin and Muc4. Staining for TFF3 and GSII lectin was not detected, and CD44v9 was only weekly positive. Ki67-positive cells were present within the intravascular lesion.
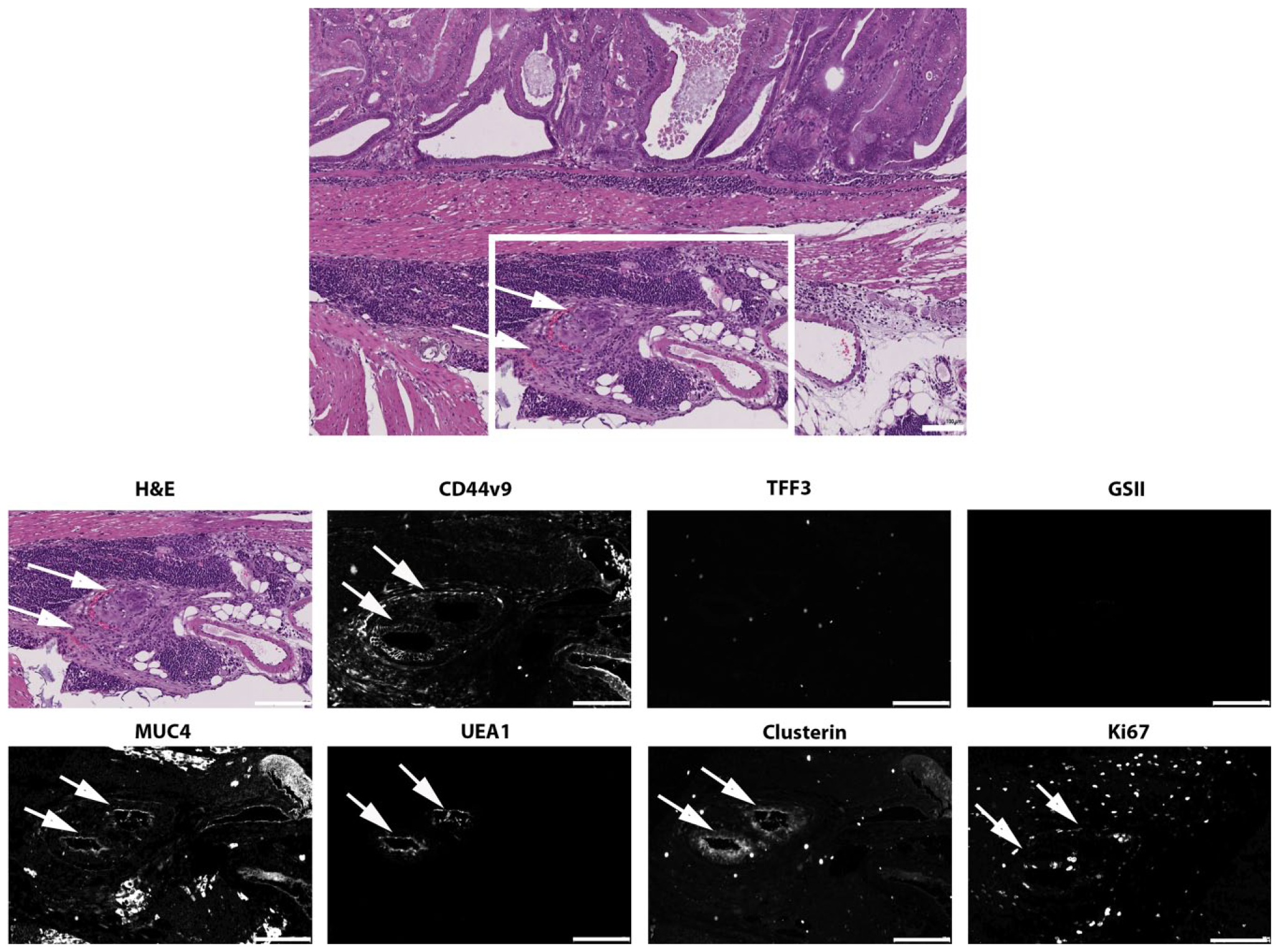
Marker expression in lesion with vascular invasion. An intravascular invasive lesion was identified by H&E staining (panel at left) in a stomach from a dual H. pylori/ASF-infected mouse. Higher magnification area showing the lesion is shown at right with staining in serial sections for CD44v9, TFF3, GSII lectin, Muc4, clusterin, UEAI lectin, and Ki67. Arrows indicate the positions of the two intravascular lesions. Scale bars = 500 µm. Abbreviations: ASF, Altered Schaedler’s Flora; GSII lectin, conjugated Griffonia simplicifolia lectin II; UEAI, Ulex europaeus lectin; MUC4, intestinal mucin 4.
Using immunocytochemistry, we also evaluated the presence of Cdx1 and Cdx2, markers of intestinal metaplasia in the stomach. However, all groups were negative for nuclear Cdx1 and Cdx2 (data not shown), suggesting the mucosal changes seen in these groups do not represent frank intestinal metaplasia.
The Number of DCLK1-positive Tuft Cells Was Significantly Increased in All Conditions
We and others have previously reported increases in the number of mucosal tuft cells in association with oxyntic atrophy, particularly in the presence of Helico-bacter infection.18,19 We, therefore, evaluated the number of DCLK1-positive tuft cells in all 4 conditions. As we have previously reported, in the normal stomach, relatively few DCLK1-positive tuft cells are visualized, usually in the isthmus zone. 19 In the control and ASF-infected INS-GAS mice, DCLK1-positive cells were dispersed throughout the upper mucosa (Fig. 5). However, in the H. pylori-infected groups, they were found in clusters, usually adjacent and luminal to CD44v9-positive SPEM regions at the bases of the mucosal glands (Fig. 5A). We found a significant increase in the number of tuft cells in all conditions, with the H. pylori groups having twice as many DCLK1-positive cells as the other groups. Co-colonization with ASF did not further increase the number of tuft cells present (Fig. 6).
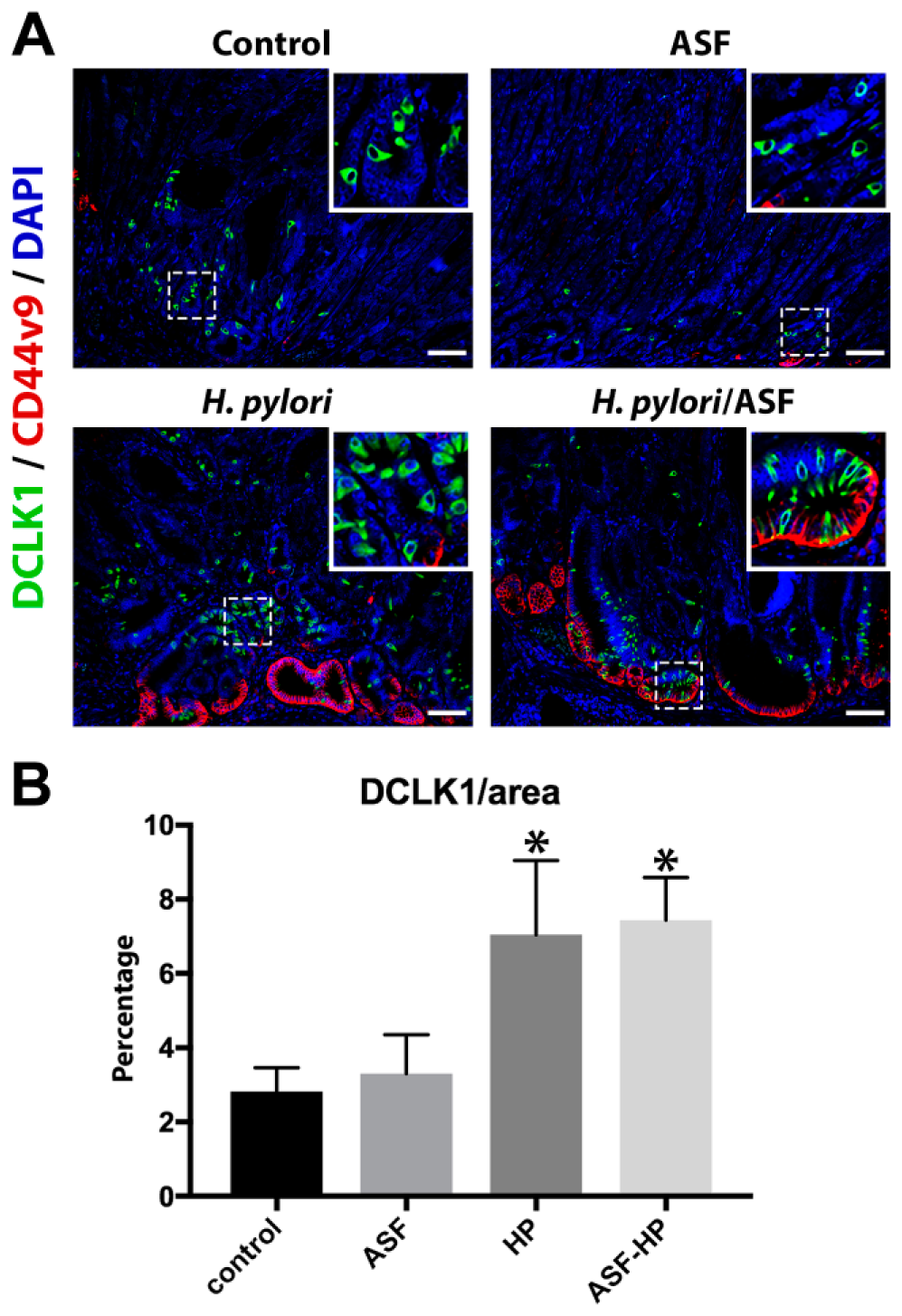
The number of tuft cells stained with DCLK1 increased with H. pylori infection. (A) Representative sections of stomach stained with DCLK1 (green), CD44v9 (red), and DAPI (blue) to visualize tuft cells. (B) Quantification of tuft cells as determined by the number of DCLK1-positive pixels divided by total area of tissue scanned (*p<0.002 compared with uninfected control). Scale bars = 100 µm. Abbreviations: DCLK1, doublecortin-like kinase 1; DAPI, 4′,6-Diamidino-2-phenylindole; HP, H. pylori; ASF, Altered Schaedler’s Flora.
Discussion
We have reevaluated with detailed lineage markers the stomachs of male INS-GAS mice infected with either H. pylori alone, ASF alone, or H. pylori combined with ASF. We found that H. pylori infection elicited most of the changes in the stomach including increased proliferation, foveolar hyperplasia, proliferative SPEM, and increased tuft cell numbers. Moreover, ASF co-infection did not change the metaplastic phenotype in the basal area of the stomach as the CD44v9-positive areas in the base of the stomach did not contain higher levels of proliferative cells. However, ASF co-infection did increase the expression of MUC4 in areas of foveolar cells.
Previous discussion about the levels of dysplasia or advanced metaplasia in INS-GAS mice have usually focused on the severity of architectural abnormalities and cytological atypia along with identification of lamina propria invasiveness or penetrating glands derived from SPEM in the basal area of the stomach corpus mucosa. 13 The predominance of changes in foveolar cells demonstrated in the INS-GAS mice infected with H. pylori without or with ASF suggests that analysis of mucosal lesions with multiple lineage-based and proliferative markers is required for interpretation of mucosal lesions. INS-GAS mice without infection spontaneously develop SPEM and penetrating submucosal lesions but induction of metaplasia is markedly accelerated in the presence of Helicobacter infection. 13 Because all of these models must involve cycles of damage and regeneration, elements of the mucosal phenotype likely reflect regeneration with mucous cell lineages of various heritage. The presence of prominent foveolar hyperplasia likely is promoted by the persistent elevations in serum gastrin in the INS-GAS mice.13,20 Regions with prominent foveolar hyperplasia often demonstrated prominent intramucosal cysts. However, these cystic areas showed low levels of proliferation. Thus, the cystic lesions themselves do not appear to indicate more advanced pathology or dysplasia. Nevertheless, we have found that, especially in H. pylori and ASF dual-infected mice, cells were present in foveolar regions closer to the surface that expressed markers of foveolar hyperplasia along with high levels of proliferation and expression of MUC4. MUC4 has previously been observed in human intestinal metaplasia, 21 and is expressed in progressive SPEM in H. pylori-infected gerbils. 22 MUC4 is also implicated as a marker of vascular invasion and lymph node metastasis in human gastric cancer. 23 Thus, it is of interest that we observed expression of MUC4 in an intravascular lesion in a dual H. pylori/ASF-infected animal. Intravascular lesions have been noted previously in the INS-GAS mouse, 13 but because the intravascular lesion identified here showed limited proliferation, it is not possible at this time to determine its exact implication for metastasis. Nevertheless, all of these results suggest that dual H. pylori/ASF infection may be affecting the lineage phenotypes not only in the context of metaplasia, but also in foveolar hyperplastic lineages.
In previous studies of dual H. pylori/ASF-infected mice, the presence of the intramucosal cellular atypia on H&E stains was considered as evidence for dysplasia. 12 The evaluation of mucosa with determinations of metaplasia, advanced metaplasia, invasive metaplasia, or dysplasia should be aided by correlative evidence of lineage marker expression and an assessment of proliferative index. In this work, we have demonstrated that the phenotype of infected INS-GAS mice was dominated by hyperplasia of foveolar cells, based on UEAI staining. At present, no immunostaining markers exist to identify dysplastic lineages in the mouse stomach, so evaluation of dysplasia relies on identification of aberrant cellular morphology based on H&E staining. Because, unlike in human disease, a direct connection of dysplasia to distinct tumor masses or distant meta-stases is not possible in present H. pylori-infection models in mice, further studies are needed to identify definitive markers that could discriminate between dysplasia and hyperplastic reactive lineages in the mouse gastric mucosa.
The recognition of the processes of damage and regeneration is further underlined by the increases in tuft cells in the mucosa with oxyntic atrophy.18,19 The role of DCLK1-positive tuft cells is not well understood in the stomach, although recent investigations suggest that they may have a role in sensing changes in different conditions such as loss of parietal cells or altered luminal pH.18,19 The striking increase in the number of DCLK1-positive cells with H. pylori infection without or with ASF infection may also indicate a possible function of these cells in sensing bacterial infection and attendant parietal cell loss and mucous cell responses of foveolar hyperplasia and metaplasia. It remains to be determined if the presence of increased tuft cell numbers has implications for progression of metaplasia toward neoplasia.
In summary, a detailed reevaluation of mucosal pathology using immunostaining markers and lectins in INS-GAS mice has revealed that, while infection with H. pylori or dual infection with H. pylori and ASF induce expanded foveolar hyperplasia, they do not cause significant expansion of SPEM. Nevertheless, we did observe evidence, especially in the dual-infected animals, upregulation of MUC4 expression in foveolar hyperplastic cells, without expression of intestinal transcription factors such as Cdx1 or Cdx2. All of these studies indicate that categorization of mucosal lesions in the body of the stomach and its translational significance would be better served by augmenting routine H&E-based histological analysis with a broad range of lineage-based and proliferative markers.
Footnotes
Competing Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
All authors have contributed to this article as follows: devised and performed experiments (CP-Z, ARM), devised experiments (EC, SM, TCW, JGF, JRG), performed experiments (RW), wrote manuscript (CP-Z, ARM), edited manuscript (RW, EC, SM, TCW, JGF), and all authors have read and approved the manuscript as submitted.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: These studies were supported by grants from a Department of Veterans Affairs (VA) Merit Review Award (I01BX000930), DOD CA160479, and National Institutes of Health (NIH) RO1 DK071590 to J.R.G. E.C. was supported by DOD CA160399 and an AACR-Debbie’s Dream Foundation Grant (PC521707). T.C.W. was supported by NIH grants R35 CA210088 and R37 DK052778, and T.C.W. and J.G.F. were supported by NIH grant U54 CA163004. J.G.F was supported by NIH grants R01 CA093405, P30 ES02109, P01 CA028842, and T32 OD010978. R.W. was supported by T35 DK007383. C.P-G. was supported by T32 CA106183. A.R.M. is supported by NIH T32 GM008554. This work was supported by core resources of the Vanderbilt Digestive Disease Center (P30 DK058404), the Vanderbilt-Ingram Cancer Center (P30 CA68485), and imaging in the Vanderbilt Digital Histology Shared supported by a VA Shared Instrumentation grant (1IS1BX003097).