
Abstract
Proteoglycans (PGs) are complex, multifaceted molecules that participate in diverse interactions vital for physiological and pathological processes. As structural components, they provide a scaffold for cells and structural organization that helps define tissue architecture. Through interactions with water, PGs enable molecular and cellular movement through tissues. Through selective ionic interactions with growth factors, chemokines, cytokines, and proteases, PGs facilitate the ability of these soluble ligands to regulate intracellular signaling events and to influence the inflammatory response. In addition, recent findings now demonstrate that PGs can activate danger-associated molecular patterns (DAMPs) and other signaling pathways to influence production of many of these soluble ligands, indicating a more direct role for PGs in influencing the immune response and tissue inflammation. This review will focus on PGs that are selectively expressed during lung inflammation and will examine the novel emerging concept of PGs as immunomodulatory regulators of the innate immune responses in lungs.
Overview
Proteoglycans (PGs) are dynamic molecules with complex macromolecular structures. The conventional understanding of PG functions relates to the ionic charge interactions of their glycosaminoglycan (GAG) chains. These GAG chains have negatively charged clusters due to sulfate-enriched domains allowing them to bind to water as well as to positively charged soluble ligands such as chemokines, cytokines, matrix metalloproteinases (MMPs), growth factors, and cell surface receptors. As a result, PGs function as structural components of the extracellular environment by acting as space filling molecules, facilitating expansion and thus regulating tissue architecture. PGs also act as regulators of signaling pathways by interacting with chemokines, cytokines, and growth factors, facilitating cell growth and migration.
There is abundant evidence that PGs are involved in more diverse interactions than just those described above. In addition to ionic charge interactions between GAG chains and immunomodulatory molecules such as chemokines, cytokines, and growth factors, it is now apparent that PGs can influence production of these molecules as well. Therefore, we propose a new paradigm for thinking about PG function—that of PGs as immunomodulatory molecules themselves. This review will present evidence to support this perspective and will highlight the pulmonary setting as there is ample evidence in the literature to support the roles of PGs in the innate immune response in the lungs as discussed below. As such, we will first describe the PG changes that occur in the transition from healthy to inflamed lungs. We will then discuss mechanisms by which lung PGs not only provide fine control of, but also directly regulate, the acute inflammatory response. In this review, we will mainly discuss the PGs most abundantly found in the lungs, namely, syndecan, perlecan, biglycan, decorin, lumican, and especially versican because this PG is selectively upregulated during acute inflammatory responses in the lungs and provides an excellent example of immunomodulatory roles regulated in structure- and context-dependent manners. Moreover, we want to note that many of the topics such as the role of heparan sulfate proteoglycans (HSPGs) in lung development and hyaluronan (HA) as a key immune modulator have been elegantly discussed in a number of excellent reviews1–7 or are the subject of other articles in this issue. Thus, we will focus on novel emerging concepts related to the role of PGs in the immune response as understood from pulmonary studies.
PGs in Developing and Healthy Lungs
The importance of HSPGs in lung development is demonstrated in genetically engineered mice lacking enzymes involved in building or modifying HSPGs such as GlcAt1, Ext1, Ext2, and Ndst1, which are embryonically or perinatally lethal due to severe defects in lung morphogenesis.1–4 The mechanisms by which HSPGs influence lung development are attributed to heparan sulfate (HS) interactions with growth factors and their receptors such as fibroblast growth factor (FGF), FGF receptors, bone morphogenic protein (BMP), and BMP receptors, among others. The influence of HSPGs in lung development has been extensively reviewed.8–10 In healthy adult human lungs, the total amounts of chondroitin sulfate/dermatan sulfate (CS/DS), HA, and HS were found to be equal. 11
Not as much is known about the importance of CS, DS, or keratan sulfate (KS) PGs in lung morphogenesis. There is evidence that the amount of CS progressively diminishes during development especially in the lung parenchyma.12,13 Versican is a chondroitin sulfate proteoglycan (CSPG) that has been shown to be important in the development of the heart where it has a critical role in the mesenchyme expansion during the formation of the ventricular septum and the endocardial cushion.14–17 This suggests that versican might be important in developing mouse lungs where it is at its most abundant on embryonic day 13.5 (Fig. 1A), a critical time for differentiation and expansion of mesenchymal progenitor cells, as well as pulmonary vascularization, right lung lobulation, and airway epithelialization. Versican expression subsequently decreases with increasing embryonic age, and very little versican is observed in lungs of healthy adult mice (Fig. 1B).18–20 The observation that versican accumulation peaks early in embryogenesis and decreases as gestation approaches has also been reported in sheep. 21 Among the major CS/DS PGs in healthy adult lungs, biglycan and decorin, but not versican, are predominant. Lumican is the major keratan sulfate proteoglycan (KSPG) in the extracellular matrix (ECM) of adult human lungs where it is localized mainly in vessel walls. 22
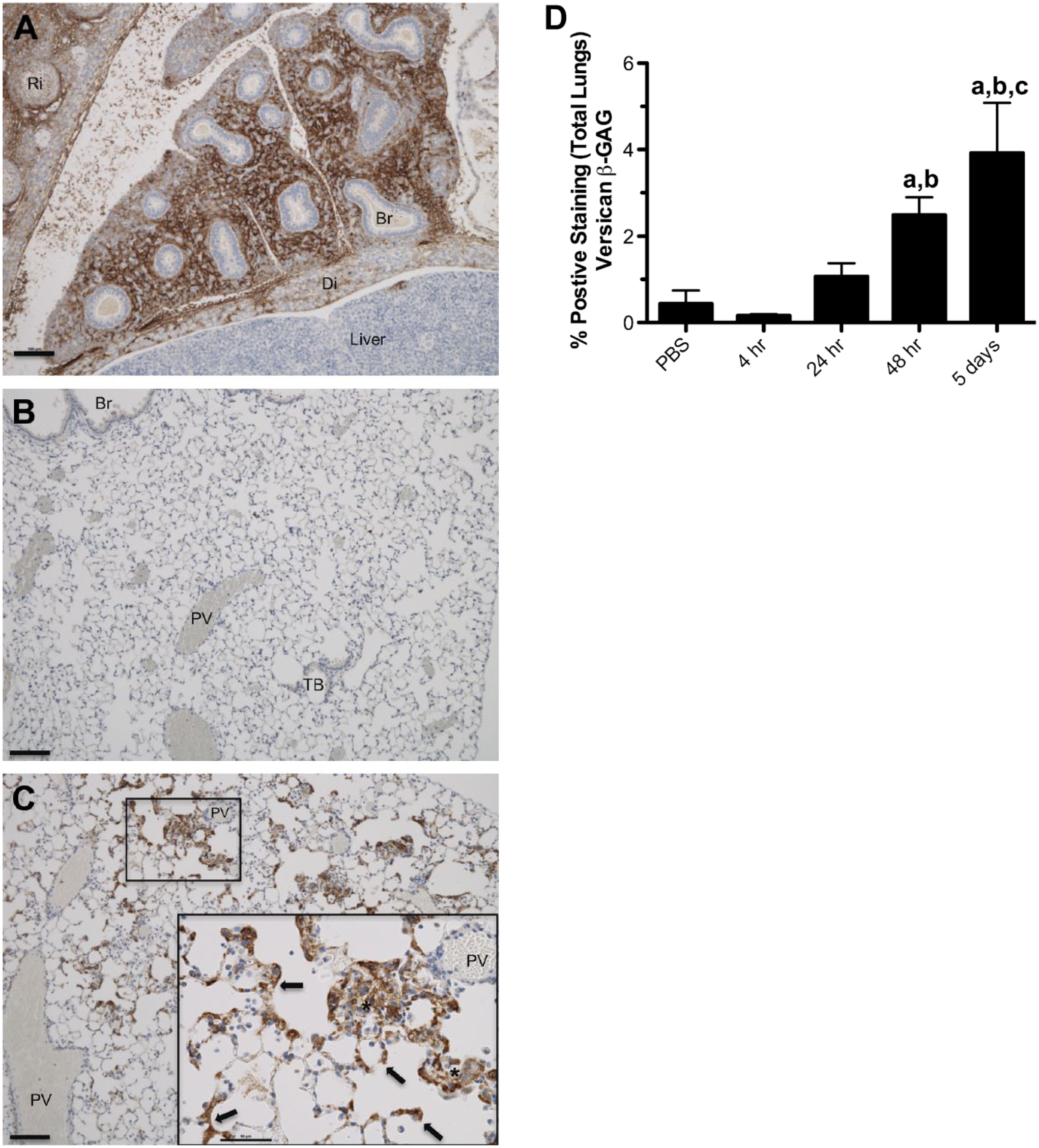
Versican accumulation during embryonic development in healthy adult lungs and in lungs of a mouse with Pseudomonas aeruginosa lung infection. (A) Versican accumulation in fetal lung tissue at E14.5 days. (B) Versican accumulation in the lungs of an 8- to 12-week-old mouse treated with PBS as vehicle control. (C) Versican accumulation from an 8- to 12-week-old mouse infected with live P. aeruginosa for 5 days. Brown = positive staining for versican β-GAG, blue = hematoxylin counterstain. The scale bars for A, B, and C = 100 μm and for C inset = 50 μm. Br = bronchiole, Di = diaphragm, Ri = rib, PV = postcapillary vein, TB = terminal bronchiole, arrows = versican staining in alveolar septa, * = area where positive staining of the alveolar septa and cells in alveolar space makes it difficult to distinguish these two anatomical compartments. (D) The amount of versican-stained lung tissue as a percentage of total lung tissue in control mice (PBS) and those exposed to live P. aeruginosa for up to 5 days. Values are the mean ± SEM (n=3–6) with a = significantly different from PBS, b = significantly different from 4 hr, c = significantly different from 24 hr and p<.00001 using one-way ANOVA with Tukey’s multiple comparison test.
PGs in Inflamed Lungs
ECM remodeling is part of the normal repair process in acute responses to injury but can contribute to chronic inflammatory situations if not properly regulated. PGs can contribute to both anti-inflammatory and proinflammatory processes depending on the specifics of the particular lung injury. In the acute phase of injury or inflammation, a well-hydrated provisional ECM is formed which acts as a temporary scaffold to allow cell migration, necessary in the initial response to the offending injury or infection and in the subsequent wound healing and resolution phases of pulmonary inflammation. 7 Whereas there is a progressive shift of 35S incorporation from HS to CS, especially CS-4 and DS following exposure of guinea pigs to lipopolysaccharide (LPS), the compositional changes appear to be selective (Fig. 2A and B). 23 For example, in studies of mice exposed to oropharyngeal LPS, a component of the cell wall of gram-negative bacteria, changes in mRNA expression of HS and CS PGs were examined. Of the four major HSPGs studied (syndecan-1, -2, -4, and perlecan), only syndecan-4 was significantly increased, with peak expression at 2–4 hr after treatment with a return to baseline at 24 hr (Fig. 2C). 24 Of the three major CS/DS PGs studied (biglycan, decorin, and versican), only versican mRNA was significantly increased in lungs, with peak expression at 6 hr after treatment (Fig. 2D). 25 The selective increases in versican, both in lung development and in acute lung injury, suggest an important role for versican in these overlapping cellular processes.12,19,26–28 Versican is also upregulated in lungs of mice exposed to the gram-negative bacteria, Pseudomonas aeruginosa (Fig. 1C and D); the toll-like receptor-3 (TLR3) agonist; polyinosinic-polycytidylic acid [poly(I:C)]; and cockroach allergen (Fig. 3).19,28–30 Whether the increases in versican expression in response to these and other inflammatory agonists are selective as in the case of LPS-induced inflammation is yet to be determined.
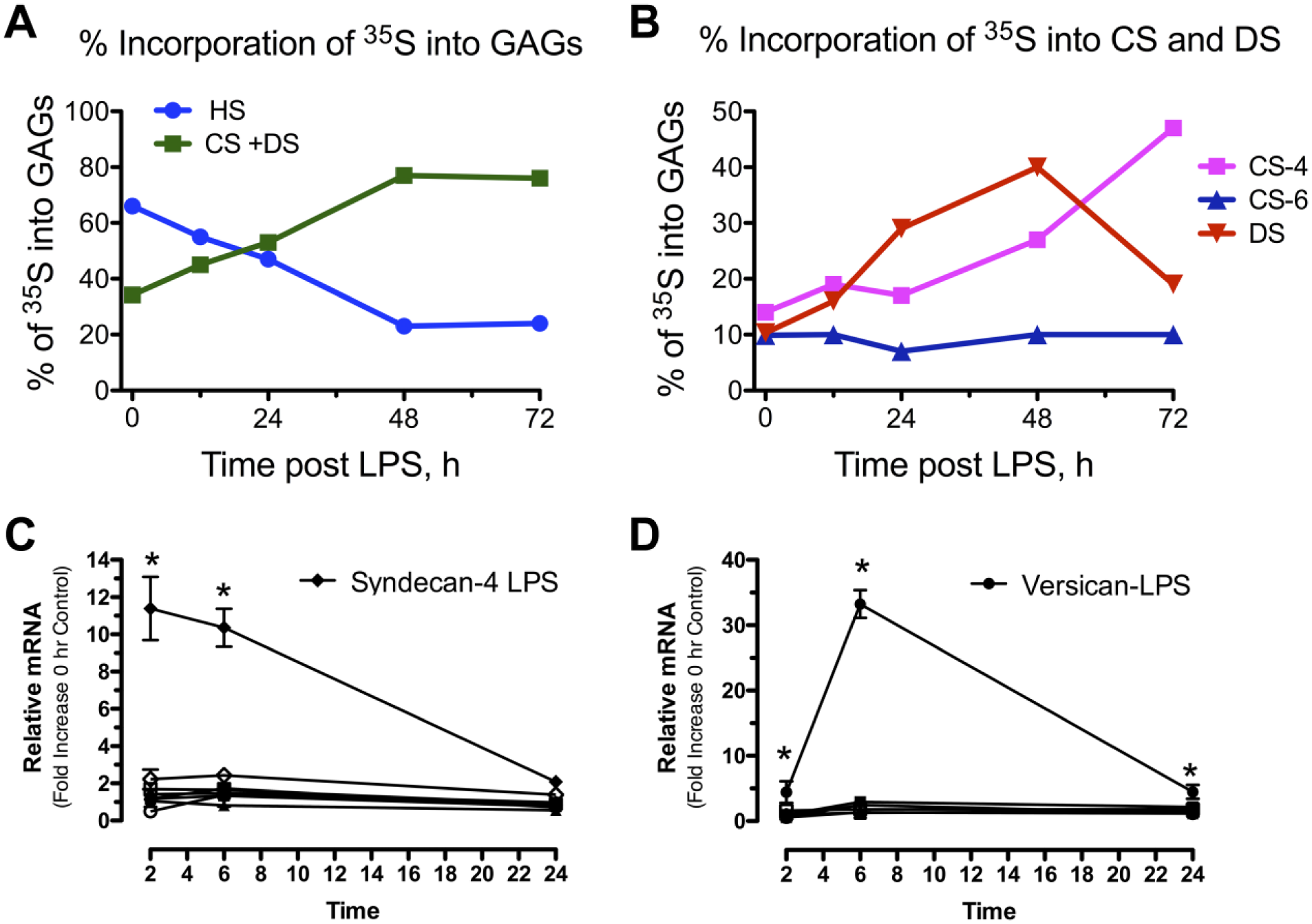
Development of a provisional matrix with selective changes in expression of HSPGs and CSPGs in lungs of mice exposed to LPS. (A) Adaptation of data extrapolated from the work of Blackwood et al., 23 showing a shift from an ECM in lungs that is composed predominately of HS to that of lungs composed predominately of CS and DS 24 h after exposure to LPS. (B) Adaptation of data extrapolated from the work of Blackwood et al., showing changes to CS-4, CS-6, and DS after exposure to LPS. (C) Changes in the relative amounts of mRNA for the HSPGs, syndecan-1, -2, -4, and perlecan, were determined using mRNA collected from whole lung homogenates and quantitative real-time PCR. (D) Changes in the relative amounts of mRNA for the CSPGs, biglycan, decorin, and versican, were determined using mRNA collected from whole lung homogenates and quantitative real-time PCR. (C, D) Comparisons of mRNA recovered from lungs of mice treated with PBS (open symbols) and 1 μg/g LPS at (closed symbols) were made at 2, 6, and 24 hr. An asterisk (*) shows groups that are significantly different (p≤0.05) when mice treated with PBS and LPS were compared. Abbreviations: HSPGs, heparan sulfate proteoglycans; CSPGs, chondroitin sulfate proteoglycans; LPS, lipopolysaccharide; CS, chondroitin sulfate; DS, dermatan sulfate; HS, heparan sulfate; PCR, polymerase chain reaction.
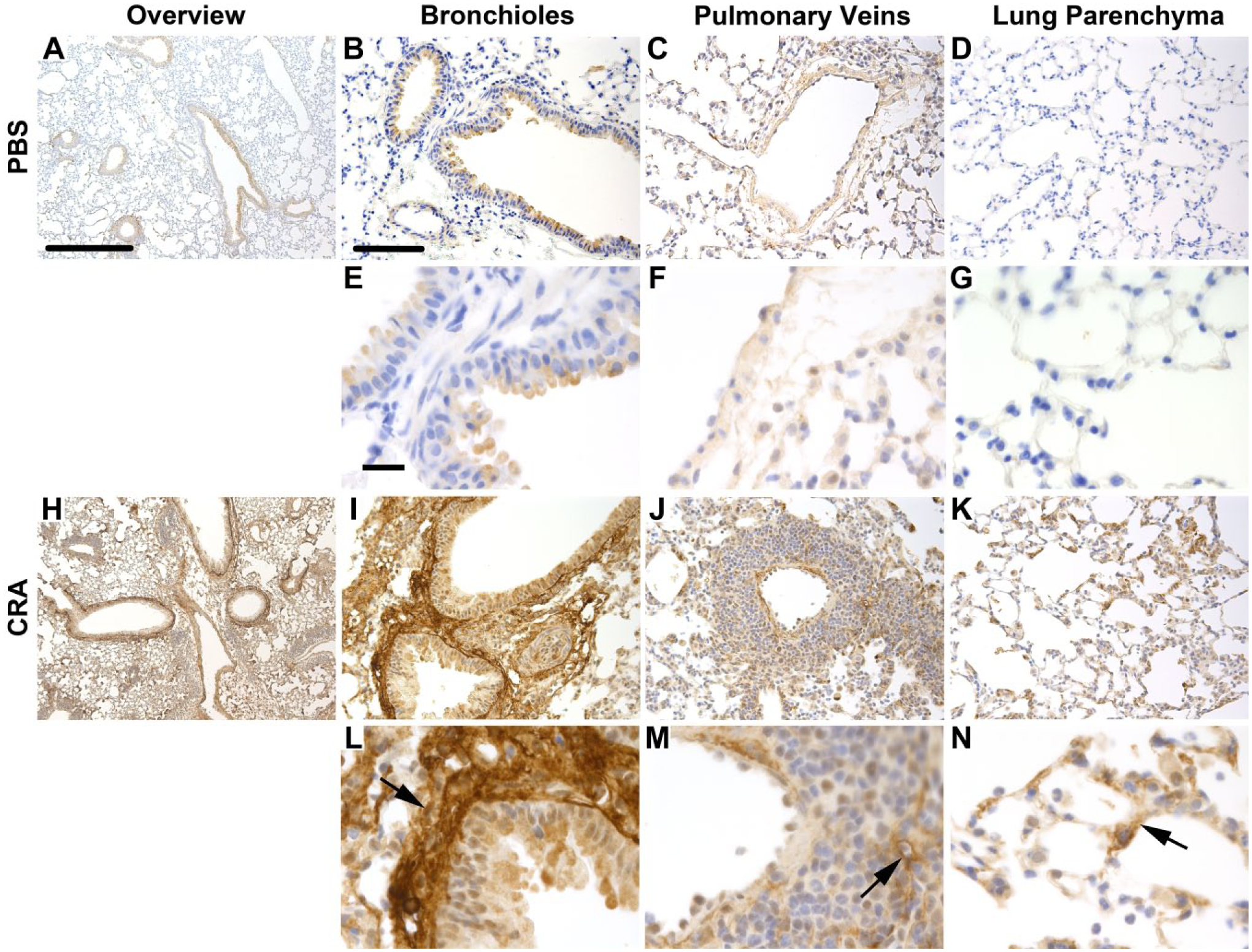
Versican staining in lung tissue sections following treatment with cockroach antigen (CRA). Depicted are representative images of control (A–G) and CRA-treated mice (H–N) stained for versican. Sections obtained from control and CRA-treated mice are shown at low magnification (Panels A and H, respectively; 50×, scale bar = 500 μm). Higher magnification images (200× B–D and I–K, scale bar = 100 μm; 630× E–F and L–N, scale bar = 20 μm) are shown for comparison, including bronchioles and pulmonary arteries (B, E, I, and L), pulmonary veins (C, F, J, and M), and lung parenchyma (D, G, K, and N). Black arrows denote increased versican staining in the subepithelial space, perivascular space, and parenchyma.
The observation that the peak expression of syndecan-4 precedes that of versican suggests differences in the mechanisms regulating the expression of these two PGs. Syndecan-4 is regulated through MyD88-dependent signaling pathways in lungs of mice treated with LPS. 24 In contrast, the increased expression of versican in lungs in response to LPS is regulated through Trif-dependent signaling. 25 The regulation of versican through a TLR4/Trif-dependent signaling pathway would explain the delayed increase in versican mRNA as compared with syndecan-4 because the Trif pathway has a slower onset of signaling.31,32 This difference in timing suggests that there are distinct roles for syndecan-4 and versican at different phases of the inflammatory response.
PG Interactions With Immunoregulatory Molecules and Immune Cells in Lungs
PGs provide fine control of innate immunity by binding to a number of recognized immunoregulatory molecules—chemokines, cytokines, growth factors, and MMPs—mostly mediated by ionic charge interactions, as extensively discussed elsewhere33–36 and summarized in Table 1. In this review, we will briefly summarize a few new findings regarding PG interactions with immunoregulatory molecules that are relevant to pulmonary inflammation.
Partial List of Cytokines, Chemokines, and Growth Factors That Bind Glycosaminoglycans.
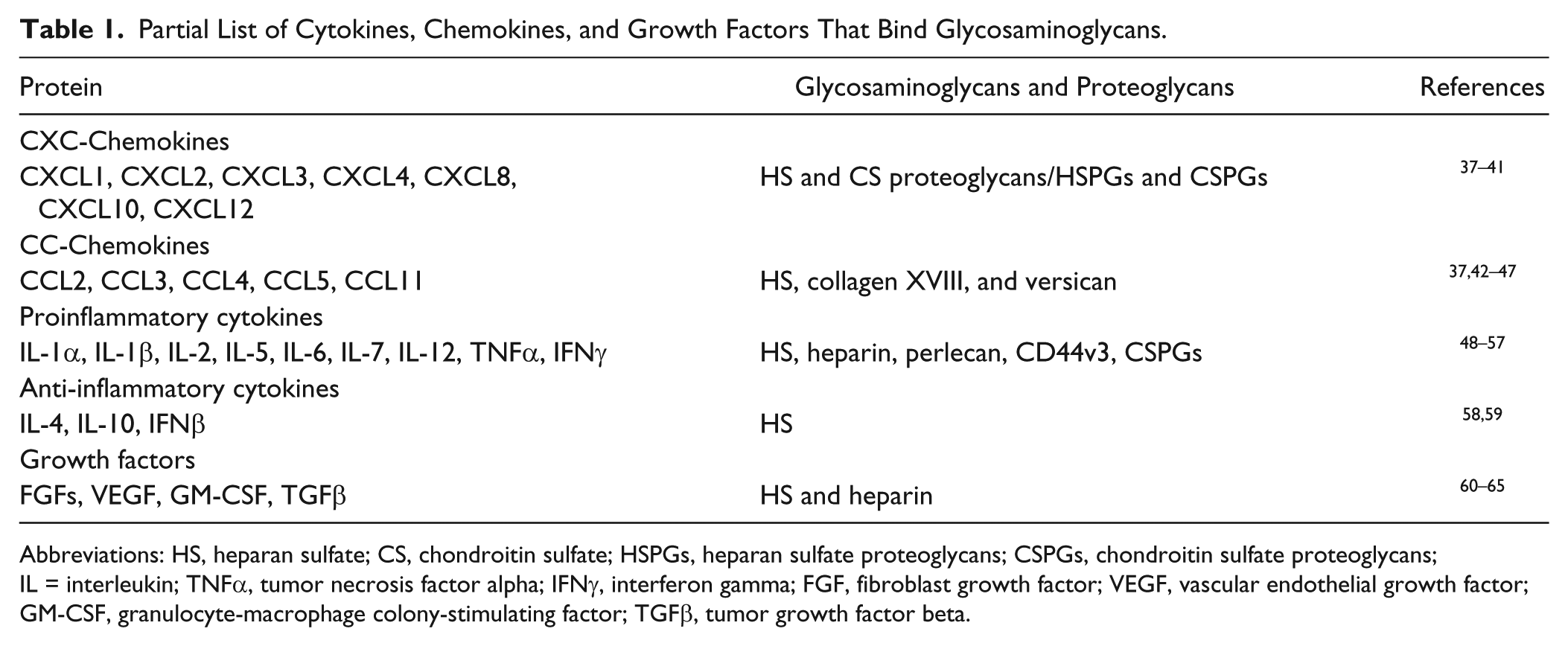
Abbreviations: HS, heparan sulfate; CS, chondroitin sulfate; HSPGs, heparan sulfate proteoglycans; CSPGs, chondroitin sulfate proteoglycans; IL = interleukin; TNFα, tumor necrosis factor alpha; IFNγ, interferon gamma; FGF, fibroblast growth factor; VEGF, vascular endothelial growth factor; GM-CSF, granulocyte-macrophage colony-stimulating factor; TGFβ, tumor growth factor beta.
Chemokines Binding to PGs
Chemokines provide important cues for the migration of leukocytes from the systemic circulation into sites of infection and injury in lungs. Recent work from several labs shows that the GAG-binding domains of chemokines provide fine control of neutrophil migration into lungs.66–68 This included work showing that recombinant human CXCL8 (IL-8) and its murine analogs, CXCL1 and CXCL2 (also known as KC and MIP-2, respectively), have differential binding to heparin, such that murine CXCL1 interacts with immobilized heparin with higher affinity and more rapid kinetics when compared with human CXCL8 and murine CXCL2 (Fig. 4). In vitro and in vivo studies showed that the rate and characteristics of gradient formation were, in part, determined by the kinetics of GAG–chemokine interactions, the affinity of the GAG–chemokine interactions, and the structure of lung tissue. These observations were used to develop a model in which chemokines that bind to GAGs with high affinity and fast kinetics form long-range, short-lived gradients, whereas chemokines that bind to GAGs with similar affinities, but slow kinetics, form short-range, long-lived gradients. 66
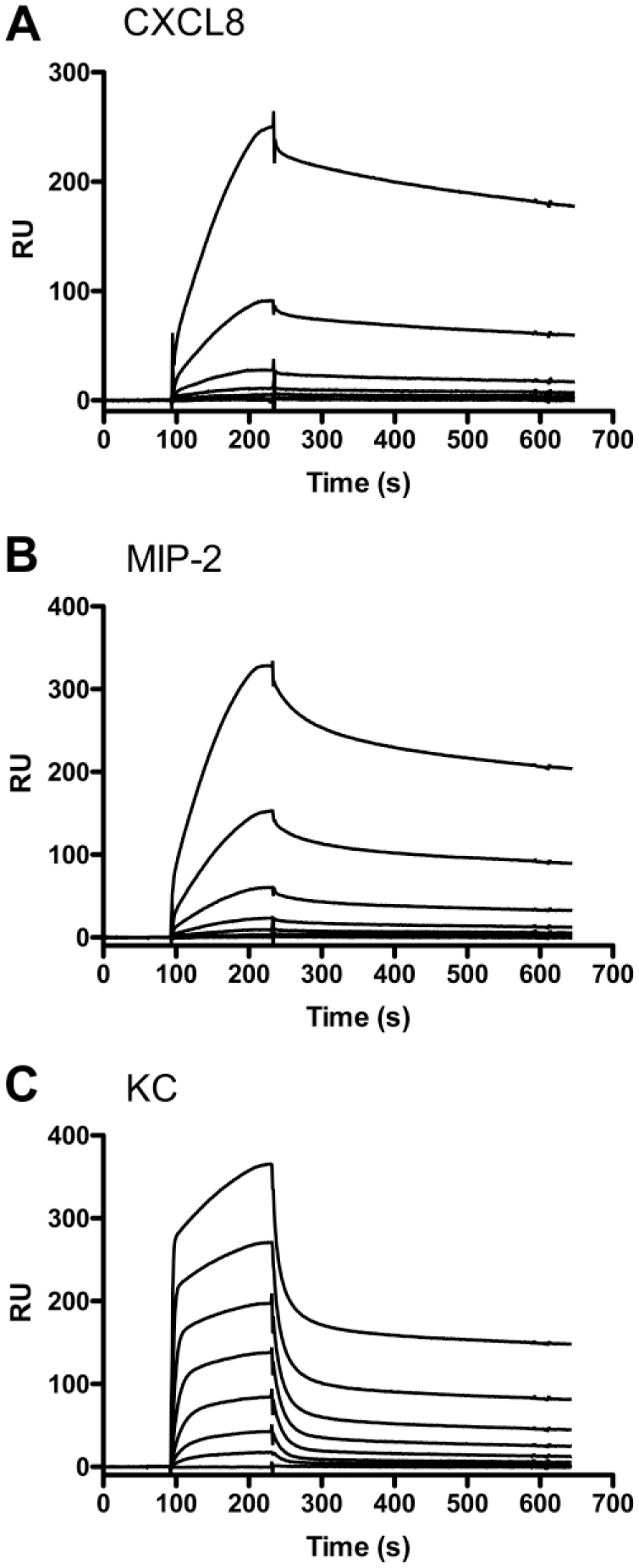
Sensograms obtained using surface plasmon resonance (SPR) reveal the chemokines bind heparin with different kinetics. The binding of various concentrations of rhCXCL8 (A), rmCXCL1/MIP-2 (B), and rmCXCL2/KC (C) to heparinized CM-4 chips is shown. (A) The concentration of CXCL8 was (upper to lower curves) 1,000, 500, 250, 125, 62.5, and 0 nM. (B) The concentrations of CXCL2/MIP-2 were (upper to lower curves) 1,000, 333, 111, 37, 12.4, and 4.12 nM. (C) The concentrations of CXCL1/KC were (upper to lower curves) 1,000, 333, 111, 37, 12.4, 4.12, 1.37, 0.46, and 0.15 nM.
Recent findings also show that GAG–chemokine interactions influence more than just chemokine gradients. For example, CXCL12 (SDF1) and CCL5 (RANTES) have been shown to directly modulate receptor ligation and leukocyte activation.69,70 In the absence of HS, the CXCL12γ isoform interacts with sulfotyrosines on its receptor, CXCR4, with higher affinity but with reduced signaling and chemotactic activities, than the CXCL12α isoform. In the presence of HS, CXCL12γ is prevented from interacting with sulfotyrosines on CXCR4, resulting in functional presentation of the chemokine to its receptor and enhanced biological activity of CXCL12γ similar to that of CXCL12α. In short, HS controls CXCL12 interactions with its receptor in an isoform-specific manner. 69 Similarly, HS alters interactions of CCL5 with its receptor, CCR1, and different HS structures exhibit different biological activities. These findings suggest another way in which GAGs provide fine control of the immune response. 70
Finally, posttranslational modifications to the NH2-terminal domain of chemokines by proteolytic degradation modifies the biological activity of chemokines by altering interactions with their high-affinity receptors. 71 Binding and sequestration of the NH2-terminal domain of CCL11 (eotaxin) and CXCL12 by GAGs protects these two chemokines from proteolytic degradation, potentiating their biological activity both in vitro and in vivo.42,72 This mechanism could serve to protect chemokines from proteolytic degradation in protease-rich inflamed tissue environments.
Cytokines Binding to PGs
HSPGs produced by macrophages bind to type I interferons (IFNs) and regulate bioavailability of IFNβ for its receptors IFNAR1 and 2. Reduction of macrophage-generated HSPGs increases bioavailability of IFNβ, resulting in activation of macrophages and worsening of chronic inflammation in diseases such as atherosclerosis. 73 In contrast, HSPGs, such as syndecan-1, sequester TNFα and IFNγ.74,75 The loss of HSPGs in a mouse model of protein-losing enteropathy liberates these cytokines, which exacerbates loss of HSPGs and instigates further protein leakage, indicating important roles of this interaction for maintaining barrier function. 76 These observations have been extended to lungs where PGs and GAGs in the endothelial cell glycocalyx form a hydrated endothelial surface layer that contributes to the vascular barrier function that excludes proteins and fluid.8,77,78 The ability of GAGs to provide a barrier to fluids and proteins such as albumin is attributed to their negative charge. Therefore, it is not surprising that loss of HA and HS from the endothelial glycocalyx results in pulmonary edema.77,78
Growth Factors Binding to PGs
As with chemokines and cytokines, interactions of growth factors with GAGs can influence their bioavailability. For example, the binding of FGFs to HSPGs serves to sequester this ligand near the site of action, providing a stable reservoir for paracrine signaling. 79 On the other hand, endocrine FGFs have reduced affinity for HSPGs, resulting in a ligand that diffuses more readily, resulting in a longer, more diffuse gradient of the ligand. Likewise, HSPGs regulate BMP signaling such that binding of HS to BMP causes internalization and inhibition of BMP signaling, whereas receptor-BMP binding activates downstream signaling, thus regulating BMP-mediated morphogenesis during lung development. 80
HSPGs have also been identified as co-receptors for vascular endothelial growth factor (VEGF).81,82 Studies show that cell-associated HSPGs function as a common modulator of VEGF binding to its receptors/co-receptors, and that sulfated HS is required for efficient signaling. 83 Moreover, recent studies suggest an important role for perlecan in angiogenesis. Mast cells generate a shortened form of perlecan containing domain V and functional endorepellin, which stimulates endothelial cell adhesion, proliferation, and migration. 84 Studies have found differences in GAG structure, cellular adhesion, and signaling capabilities of FGF1 and FGF2 depending on whether perlecan was generated by vascular smooth muscle cells or by endothelial cells. 85 These findings suggest that the HSPGs provide complex regulation of angiogenesis as well as morphogenesis, in structure- and cell source-dependent manners.
MMPs Binding to PGs
PGs can interact with both MMPs and their substrates in multiple ways that influence proteolytic processing, as exemplified by interactions with MMP7 (also known as matrilysin). 86 PGs can serve as anchors for MMP7 at the cell surface, within secretion granules, or in the pericellular environment, to compartmentalize MMP7 activity toward its substrates within defined locations. PGs also act as modulators of MMP activation, and specific highly sulfated GAG structures markedly enhance processing of pro-MMP7 to its active proteolytic form. 86 As previously mentioned, PGs are also able to interact with the substrates of MMPs, including chemokines and growth factors, to protect them from proteolytic cleavage.34,87 Such interactions protect heparin binding growth factors, cytokines, and chemokines from proteolytic degradation.42,72,88–90 However, inhibition of proteolytic degradation can have contrasting implications depending on the specific molecules involved. As an example, both HS and heparin bind to IFNγ to protect this cytokine from proteolytic cleavage, thereby enhancing the activity of IFNγ.48,91 In contrast, the binding of HSPGs to IFNβ results in sequestration of the cytokine and inhibition of its activities. 73 MMPs, on the other hand, can release sequestered chemokines by cleaving the PG, as reported in studies showing that syndecan-1/CXCL1 complexes, which are shed by MMP7 and are able to promote neutrophil migration into the airspaces of the lungs.92,93 Thus, PGs can affect MMP activity by influencing both MMP and substrate availability.
Leukocytes Binding to PGs
PGs and GAGs play critical roles in both promoting and interfering with leukocyte infiltration in inflamed tissues.94,95 For example, heparin’s anti-inflammatory effects are shown to be mediated by blocking selectin-initiated leucocyte adhesion to the endothelium by inhibiting CD11b/CD18- and selectin-mediated leukocyte binding.96,97 In contrast, upregulation of endothelial CD44v3, a unique HSPG variant of this cell surface receptor, and its interactions with CD11b/CD18 on neutrophils facilitates neutrophil adhesion and migration across epithelial cells during inflammatory episodes.98,99 Similarly, interactions of endothelial-derived lumican with CD11b and CD18 on neutrophils facilitates neutrophil migration during inflammation. 100 In addition, lumican increases macrophage phagocytosis of the gram-negative bacteria, P. aeruginosa, suggesting a role for bacterial clearance from lungs. 101 Lumican interaction with pathogens and pattern recognition receptors (PRRs) will be further discussed below.
Previous work has shown that HA accumulation in the ECM at sites of inflammation provides a substrate for leukocyte adhesion by binding to CD44 on leukocytes.102–108 This is mediated by HA-binding molecules such as versican, inter-α-trypsin inhibitor (IαI), and TNFα-stimulated gene 6 (TSG-6) which crosslink HA into cable-like structures.102–108 Work performed in vitro has demonstrated that HA-dependent monocyte binding to poly(I:C)-stimulated lung fibroblasts can be abolished by blocking antibodies against the HA-binding region of versican.29,105 These studies were extended in an in vivo model of lung inflammation by showing that versican deficiency resulted in reduced Has2 expression and HA accumulation in lung tissue coupled with reduced total cell counts in bronchoalveolar lavage fluid (BALF) and an overall dampened inflammatory cytokine expression profile. These findings confirm that versican is critical to generating a specialized HA-enriched ECM that binds leukocytes (Fig. 5). Intriguingly, versican inhibits T-cell binding to HA and further affects T-cell adhesion and migration, 109 suggesting that hyaladherins such as versican might also provide fine-tune regulation of the immune response by modulating HA interactions with its receptors.
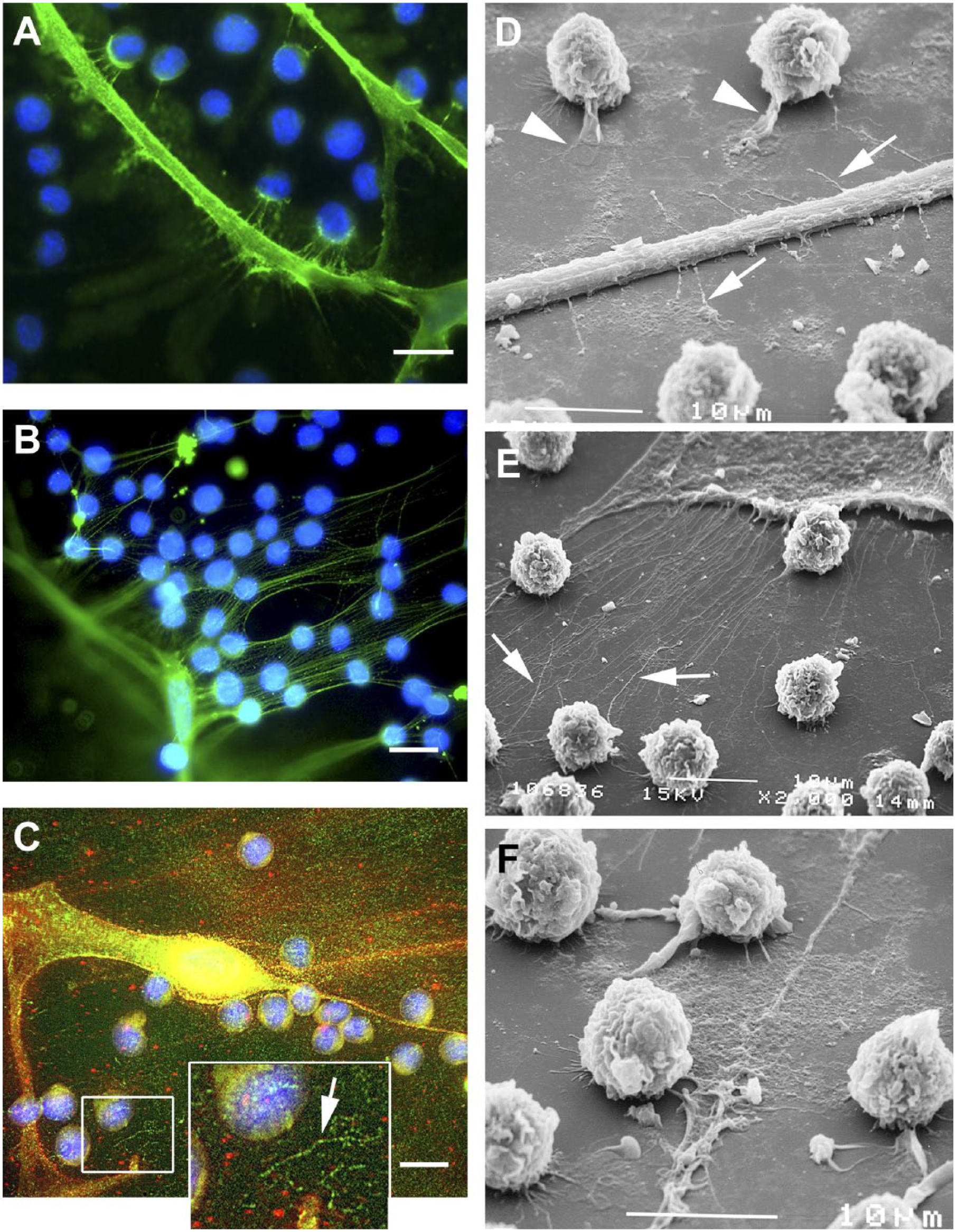
Monocyte binding to pericellular matrices of control (A, D) and poly I:C-treated (B, E, C, F) fibroblasts. (A, B) Hyaluronan is stained green and nuclei are stained blue (4′,6-diamidino-2-phenylindole). These specimens were fixed with acetic acid–ethanol–formalin. (C) This specimen was fixed with formalin and stained for hyaluronan (red) and versican (green). Monocytes are bound in the matrix and along the uropod/tail of a partially rounded fibroblast. The inset in C shows a more-condensed aggregate of closely spaced punctate versican staining within a looser network (arrow). The size (~100 nm) and spacing (~100 nm) is consistent with each green signal representing antibody bound to one versican monomer in a compact aggregate. (D–F) Scanning electron micrographs reveal monocytes that have adhered to matrix associated with fibroblast protrusions or uropods (arrows). The monocytes have extended their own filopodia (arrowheads) into the matrix and are partially guided by the fibroblast protrusions.
PGs as Immunomodulatory Molecules in Lungs
Elsewhere in this issue, Frevert et al. present an overview of ECM-derived danger-associated molecular pattern (DAMP) interactions with PRRs and the ensuing cascade of events that either aggravate inflammation and tissue injury or promote the regeneration of injured tissue. Whether these interactions promote healing or prolong inflammation depends on multiple factors including the nature of the ECM-derived DAMPs, the cells with which they interact, the signaling pathway(s) invoked, and the specific injury or disease process.
The best-studied ECM-derived DAMPs include HA5,6,110–113 and the small leucine-rich PGs (SLRPs), biglycan and decorin.114–117 Each of these is discussed in detail in a tissue-specific context in this issue: HA in the gut (de la Motte) and SLRPs in kidneys (Nastase). For discussions of the importance of low- and high-molecular-weight HA interactions with TLRs in the context of lung injury (asthma, lung transplantation, non-infectious lung injury, or pulmonary fibrosis), see the elegant work by Paul Noble’s group.5,118 Similarly, for discussions of biglycan and decorin interactions with PRRs in the pulmonary context, see the work of Liliana Schaefer and colleagues.114–117
Lumican is another SLRP that promotes the innate immune response. Although not a DAMP, per se, lumican interacts with CD14 to facilitate LPS sensing by TLR4. 119 In P. aeruginosa lung infections, lumican-deficient (Lum−/−) mice showed increased bacterial persistence in lungs with a dramatic increase in mortality. 101
Versican as a DAMP
Versican as a DAMP has been demonstrated to be a critical regulator of immune responses in a number of recent studies discussed below. Studies by Kim et al. and Tang et al. are important in this regard.120,121 Kim et al. showed that versican secreted by Lewis lung carcinoma (LLC) cells interacts with TLR2 and its co-receptors, TLR6 and CD14, to activate bone marrow–derived macrophages (BMDMs) in vitro, resulting in secretion of TNFα and IL-6. 120 Of these cytokines, which are typically considered proinflammatory, TNFα was shown to be important for LLC metastasis. Furthermore, mice injected with versican-silenced LLC cells that were transduced with short hairpin RNA (shRNA) specific for the V1 isoform of versican had significantly fewer and smaller lung tumor nodules and, most importantly, improved survival compared with mice injected with control shRNA. 120 Thus, this study demonstrated that versican–TLR2 interactions have proinflammatory and premetastatic consequences in LLC. Similarly, Tang et al. examined the impact of LLC cell secretory products to engage TLR2 on preconventional dendritic cells (pre-cDCs). 121 These studies showed stimulated secretion of IL-6 and IL-10, increased expression of cell surface receptors for these cytokines, and increased sensitivity of tumor DCs to these cytokines, indicating immunosuppression of tumor DCs. They found that negating versican, either by addition of versican antibodies to media from LLC cells or by transduction of LLC with versican shRNA, resulted in decreased secretion of IL-6 and IL-10, with subsequent increased secretion of IL-12 and improved DC function. These findings suggest that versican–TLR2 interactions in LLC promote tumor DC dysfunction. Both of these studies establish versican as a ligand for TLR2 and support pathological consequences of their interactions in lung cancer.120,121 The role of versican–TLR2 interactions in the innate immune response in lungs is not known.
It is interesting to consider biglycan–TLR interactions in comparison with the consequences of versican–TLR interactions. As previously discussed, biglycan can promote a proinflammatory response through activation of TLRs and inflammasomes. Biglycan also activates Trif-dependent signaling pathways after engaging TLR4, which results in decreased production of CCL5 and decreased recruitment of T cells. These data suggest that biglycan has a potential role in the resolution of inflammation and the transition from innate to acquired immunity. 122 When the pathways elicited by versican and biglycan are compared, the major differences are the TLR adaptor molecules that are activated. Versican binds to TLR2 and activates MyD88-dependent signaling,120,121 whereas biglycan binds to TLR4 and activates Trif-dependent signaling. 122 This raises interesting questions in that versican and biglycan activate two separate signaling pathways, suggesting that these CSPGs have different functions during the resolution of tissue inflammation and injury.
While intact versican interacts and activates TLR2, resulting in protumor properties, Hope et al. demonstrated that versikine, a degradation product of versican, acts as a DAMP with antitumor properties. 123 Versikine results from the action of ADAMTS-type proteases at the Glu441–Ala442 bond within the V1 isoform of versican. 124 The resulting 70-kDa N-terminal fragment represents the G1 domain of V1 and lacks the G2 and G3 domains which include GAGβ-, EGF-, lectin- and complement regulatory protein-like functionality. Just as intact versican, versikine was found to engage TLR2 on macrophages.123,125,126 Versikine also stimulated production of a number of molecules that can promote innate myeloma sensing and T-cell inflammation through induction of type I IFN-stimulated genes (e.g., MX1 and STAT1), macrophage activation (IL-6 and IL-1β), T-cell chemotactic mediators (CCL2), and myeloid-derived suppressor cell homeostasis (IRF8). Taken together, these studies establish that both intact versican and a proteolytic degradation product of versican have immunomodulatory properties and suggest that the antitumor properties of versikine might antagonize the protumor properties of intact versican. The juxtaposition of these findings indicates that the consequences of ECM-derived DAMP interactions with PRRs can have sharply differing outcomes depending on the contextual specifics. While Hope et al. examined the immunoregulatory properties of versikine in myeloma, ADAMTS-type proteases are highly expressed in cancers of many organs, including those in lungs. Additional work is warranted to define the relevance of versican and versikine-derived DAMP responses specifically in the context of the immune response in lungs.
Versican as a Regulator of Pulmonary Innate Immune Responses
In addition to the abilities of versican to modulate the inflammatory response via interaction with TLR PRRs, a growing number of studies demonstrate the ability of versican to modulate the innate immune responses. We will discuss several studies with a focus on the ability of versican to modulate cytokine expression. In particular, we will address the outcomes from our studies of acute pulmonary inflammation in two unique versican-deficient mouse models.29,127
The cellular sources of the ECM protein, versican, have been considered to be stromal cells, primarily smooth muscle cells and fibroblasts, as nicely summarized by Gunilla Westergren-Thorsson’s group. 28 Therefore, it was of interest when positive immunostaining was observed in a subset of alveolar macrophages in lungs of mice treated with LPS (Fig. 6). Experiments performed in vitro using classically activated murine BMDMs treated with LPS showed that macrophages of the M1 phenotype had increased expression of versican mRNA and accumulation of versican protein. 25 In contrast, alternative activation of BMDMs to a less proinflammatory M2 state, with IL-4 plus IL-13, or with IL-10, did not increase the expression of versican mRNA. Similarly, the versican gene is differentially expressed in M1 macrophages, as opposed to M2 macrophages, as they differentiate from monocytes. 128 In addition, versican gene upregulation in monocytes/macrophages was found in a number of proinflammatory states, such as myocardial infarction, 129 coronary stenosis, 130 autoimmunity,131–133 and hypoxia.134,135 Recent studies identified versican as a part of signature gene set common for classical monocytes (CD14++ CD16−) and classical CD1C+ dendritic cells. 136
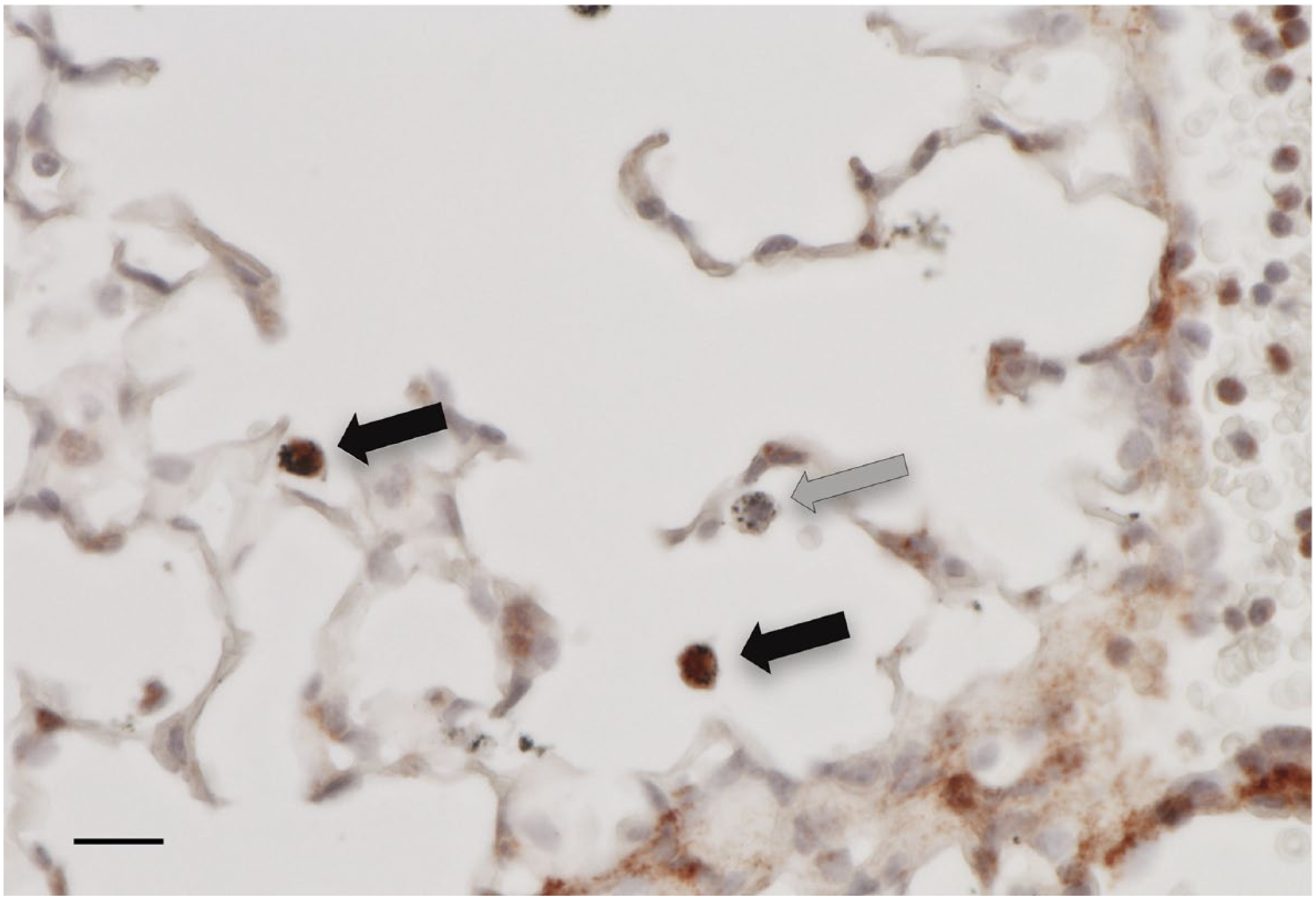
Increased expression of the ECM protein, versican, by alveolar macrophages in lungs of mice exposed to lipopolysaccharide. Immunohistochemistry for versican (red) showing alveolar macrophages that have positive staining for versican (black arrows). In contrast, several alveolar macrophages, identified by colloidal carbon uptake, do not show positive staining for versican (gray arrows). For these studies, the IT instillation of Escherichia coli 0111:B4 LPS (1 μg/g) was performed in mice anesthetized with 3–4% isoflurane. The mice were euthanized 6 hr after exposure to LPS, and the lungs were fixed in neutral buffered formalin and processed into paraffin. Immunohistochemistry was performed with an antibody specific for the β-GAG domain of versican and the tissues were counterstained with hematoxylin. The scale bar is 10 µm. Abbreviations: ECM, extracellular matrix; IT, intratracheal; LPS, lipopolysaccharide.
The TLR agonists, LPS and poly(I:C), stimulate versican expression in both murine BMDMs and alveolar macrophages in vitro 25 and in murine alveolar macrophages, as well as in stromal cells, in vivo.29,127 We explored the mechanism of regulation of versican expression in macrophages through in vitro studies. Engagement of TLR4 or TLR3, by LPS or poly(I:C), respectively, triggered a signaling cascade involving the Trif adapter molecule, type I IFNs and their receptor, IFNAR1, leading to increased versican production. In short, versican was shown to be a type I IFN-stimulated gene (ISG) in macrophages (Fig. 7), suggesting an anti-inflammatory role for macrophage-derived versican. This pathway is likely distinct from that described by Rahmani et al. and Baarsma et al., in which canonical Wnt/β-catenin/T-cell factor signaling was responsible for the increased expression of versican in airway smooth muscle cells treated with tumor growth factor beta-1 (TGFβ1).137,138 Considering these two signaling pathways that mediate versican expression, several intriguing questions are suggested regarding the specificity of the regulation mechanisms. Are these mechanisms agonist-, cell-, or signaling pathway-specific? Is versican structurally different (in terms of isoform or GAG structure) depending on the regulation mechanism? Most importantly, is versican functionally different depending on the regulation mechanism? All of these possibilities merit further study and could shed light on the complex and seemingly conflicting roles of versican in different inflammatory circumstances.
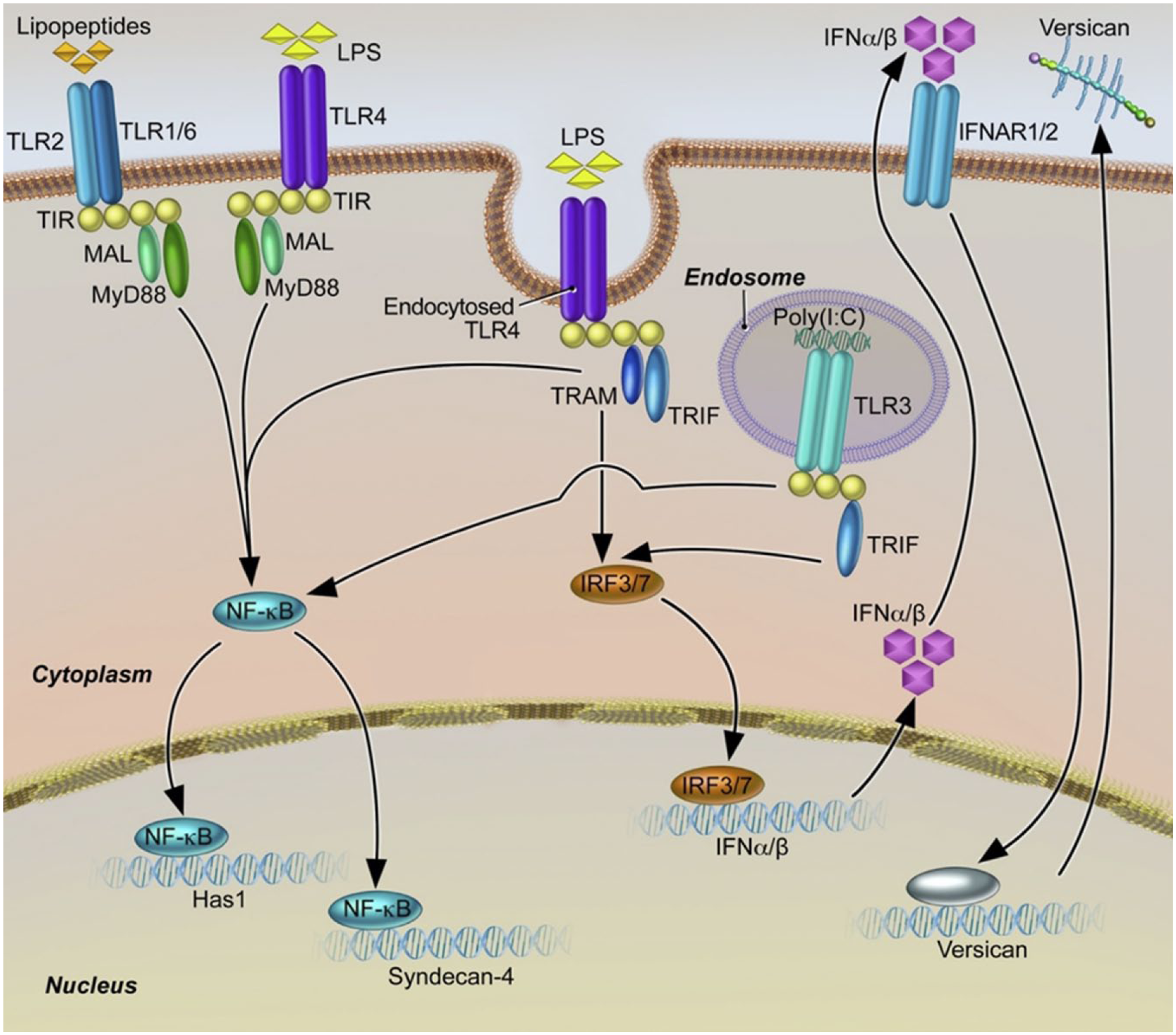
Schematic depicting pathways by which LPS and poly(I:C) regulate the expression of versican, HAS1, and syndecan-4. Engagement of macrophage toll-like receptors TLR4 and TLR3 by LPS and poly(I:C), respectively, results in enhanced versican expression. Subsequent to activation of TLR4 and TLR3, engagement of the TRIF adaptor molecule is known to activate transcription factors IRF3/7 that lead to production of type I interferons (IFNα/β) and recognition by type I interferon receptors (IFNAR1/2). Signaling events downstream of IFNAR activation lead to production of versican; the transcription factor(s) mediating expression of versican in this response are still to be determined. In contrast, expression of both HAS1 and syndecan-4 result from TLR4- and TLR2-mediated engagement of the MyD88 adaptor molecule. In addition, syndecan-4 expression results from TLR4- and TLR3-mediated engagement of TRIF and downstream signaling events which are independent of type I IFNs. Abbreviations: LPS, lipopolysaccharide; TLR, toll-like receptor; IFN, interferon.
To evaluate the role of versican in the acute pulmonary inflammatory response and to circumvent the obstacle of embryonic lethality in versican knockout mice, 14 we developed two mouse models of conditional versican deficiency. LysM Cre+/Vcan−/− mice are versican-deficient in their myeloid cells, which includes macrophages, 127 and R26Rert2Cre+/Vcan−/− mice have global deficiency of versican when treated with tamoxifen. 29 The inflammatory response was evaluated in both of these models in vitro in macrophages and in vivo in whole lungs following exposure to poly(I:C) to specifically activate the Trif/type I IFN signaling pathway. Three major findings were the result of this work: (1) macrophage-derived versican limits inflammatory cell recruitment; but (2) stromal cell–derived versican promotes inflammatory cell recruitment into lungs; and (3) macrophage-derived versican promotes expression of type I IFNs and IL-10 in vivo in whole lungs and in vitro in macrophages in both mouse models. The findings of both anti- and proinflammatory roles for versican in these studies suggest that either the cellular source or signaling pathway invoked determines which role versican plays in the inflammatory response.
Our studies also revealed a feedback regulatory process in macrophages in which type I IFNs influence the production of versican, and versican, in turn, influences the production of type I IFNs. The finding that versican deficiency contributes to decreased IL-10 expression in both of these mouse models indicates an important role for versican in regulating the production of this anti-inflammatory cytokine in the host response to lung infection. This is consistent with the recent report by Tang et al. in which versican, produced by LLC, reprograms dendritic cells to an immunosuppressive phenotype through autocrine TLR2-dependent regulation of IL-10 production. 121 Interestingly, Arimori et al. showed decreased recovery of IL-10 and increased lung pathology and mortality in mice lacking type I IFN receptors (i.e., Ifnar1−/− mice) exposed to influenza virus. 139 Our recent findings of increased inflammation and decreased IL-10 production in LysM Cre+/Vcan−/− mice 127 are similar and suggest a potential role for versican in modulating the anti-inflammatory effects of type I IFNs in mice through increased production of IL-10.
Intriguingly, overexpression of the V3 spliced variant of versican lacking GAG-binding regions has been demonstrated to retard inflammatory cytokine production (including CXCL1, CCL20, CCL2) in smooth muscle cells via blockade of epidermal growth factor receptor and NFκB signaling cascades.140,141 Overexpression of V3 in melanoma cells reduces the rate of tumor growth by decreasing the proliferative index and increasing the apoptotic rate of primary tumor cells.142–144 In addition, expression of a mutant G3ΔEGF construct (the versican G3 domain with deletion of the EGF-like repeats) also results in slower growth, increased apoptotic rate, and lower tumorigenic potential of astrocytoma cells. 145 Despite the structural differences between V3, versikine, and G3ΔEGF, it is likely that their antitumor properties are due to loss of EGF signaling. However, V3 overexpression also favored the spontaneous metastasis and development of secondary tumors in lungs, likely due to increased HA production by stromal or inflammatory cells, which was conducive to the migration of melanoma cells outside the primary tumor. 143
These findings demonstrate that versican, as a component of the provisional ECM, plays immunomodulatory roles and is differentially regulated in cell source–dependent manners, perhaps due to differential isoform expression, proteolysis, or GAG structure composition between stromal-derived versus myeloid-derived versican. Studies are underway to define these differences.
In conclusion, the extracellular space can be regarded as the final frontier in our understanding of immunomodulation as there are numerous components that work either in concert or in competition with each other to regulate normal developmental and homeostatic processes, as well as acute and chronic responses to inflammation and injury. In this review, we highlighted recent findings regarding several PGs that are selectively expressed during pulmonary inflammation, with a special emphasis on versican. This selective PG expression during the innate immune response to TLR agonists is critical in the development of a provisional matrix,7,24,25,146 providing open and hydrated spaces that facilitate cell migration, proliferation, and leukocyte infiltration. It is now evident that PGs influence the inflammatory response in a number of ways. Numerous studies show that PGs provide fine control of the innate immune response in lungs via their interactions with immunomodulatory molecules and cells by acting as DAMPs that directly activate key PRRs and signaling cascades. As such, PGs should be considered as immunomodulatory molecules in their own right. Notably, recent findings establish that the impact of a given PG in the inflammatory response is context-, cell-, and structure-dependent. In this regard, we focused on versican as an example of a PG with distinct contextual immunomodulatory properties and consequences in the acute pulmonary inflammatory responses. Thus, a more thorough understanding of the mechanisms by which PGs regulate immune cell phenotype and function is essential to designing therapeutic approaches to control pulmonary inflammation.
Footnotes
Acknowledgements
The authors thank Michael G. Kinsella, PhD, and Ingrid A. Harten, PhD, of the Benaroya Research Institute for their insight and helpful discussions. They also thank Virginia M. Green, PhD, of the Benaroya Research Institute for her assistance with the manuscript.
Competing Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
IK, MYC, TNW, and CWF contributed to the conception and preparation of the manuscript, and wrote the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was funded, in part, by the National Institutes of Health grants U19 AI125378 (T.N.W.) and 1P30 DK089507 (C.W.F.), and by USDA Cooperative Agreement, 59-2090-5-004 (C.W.F.).