
Abstract
Circulating neutrophils, rapidly recruited in response to microbial infection, form the first line in host defense. Humans express ~50 chemokines, of which a subset of seven chemokines, characterized by the conserved “Glu-Leu-Arg” motif, mediate neutrophil recruitment. Neutrophil-activating chemokines (NACs) share similar structures, exist as monomers and dimers, activate the CXCR2 receptor on neutrophils, and interact with tissue glycosaminoglycans (GAGs). Considering cellular assays have shown that NACs have similar CXCR2 activity, the question has been and remains, why do humans express so many NACs? In this review, we make the case that NACs are not redundant and that distinct GAG interactions determine chemokine-specific in vivo functions. Structural studies have shown that the GAG-binding interactions of NACs are distinctly different, and that conserved and specific residues in the context of structure determine geometries that could not have been predicted from sequences alone. Animal studies indicate recruitment profiles of monomers and dimers are distinctly different, monomer–dimer equilibrium regulates recruitment, and that recruitment profiles vary between chemokines and between tissues, providing evidence that GAG interactions orchestrate neutrophil recruitment. We propose in vivo GAG interactions impact several chemokine properties including gradients and lifetime, and that these interactions fine-tune and define the functional response of each chemokine that can vary between different cell and tissue types for successful resolution of inflammation.
Keywords
Chemokines play crucial roles in defining the innate and adaptive arms of immunity by recruiting leukocytes including neutrophils in health and disease.1,2 Circulating neutrophils are rapidly recruited in response to microbial infections and form the first line in host defense.2–5 A dysregulation in recruitment and/or impaired activation could result in runaway infection, whereas excess recruitment and/or sustained activation could result in collateral tissue damage. Therefore, all aspects of chemokine from expression levels and trafficking to receptor activity must also be highly regulated.
In humans, seven chemokines (CXCL1, CXCL2, CXCL3, CXCL5, CXCL6, CXCL7, and CXCL8), characterized by the conserved N-terminal “Glu-Leu-Arg” motif, orchestrate neutrophil recruitment (Fig. 1). These chemokines, released at the site of injury, cross the epithelium, extracellular matrix (ECM), and the endothelium to the vasculature, and direct neutrophils from the vasculature to the target site (Fig. 2). Neutrophil-activating chemokines (NACs) exert their function by signaling via CXCR2 on neutrophils and binding glycosaminoglycan (GAG) heparan sulfate (HS) on the endothelium, epithelium, and ECM.6,7 Animal models and cellular studies have established that GAG interactions dictate chemokine gradients, and that these gradients orchestrate leukocyte trafficking from circulation to the target site.8–10

(A) Sequences of human and mouse neutrophil-activating chemokines. Conserved basic residues implicated in heparin interactions are highlighted in red and shaded in gray. Chemokine-specific residues that have been shown to be involved in heparin binding are in bold blue and underlined. Quasi-conserved residues are shown in red. (B) Table showing conserved residues that are involved in heparin binding from NMR studies. Abbreviation: NMR, nuclear magnetic resonance.
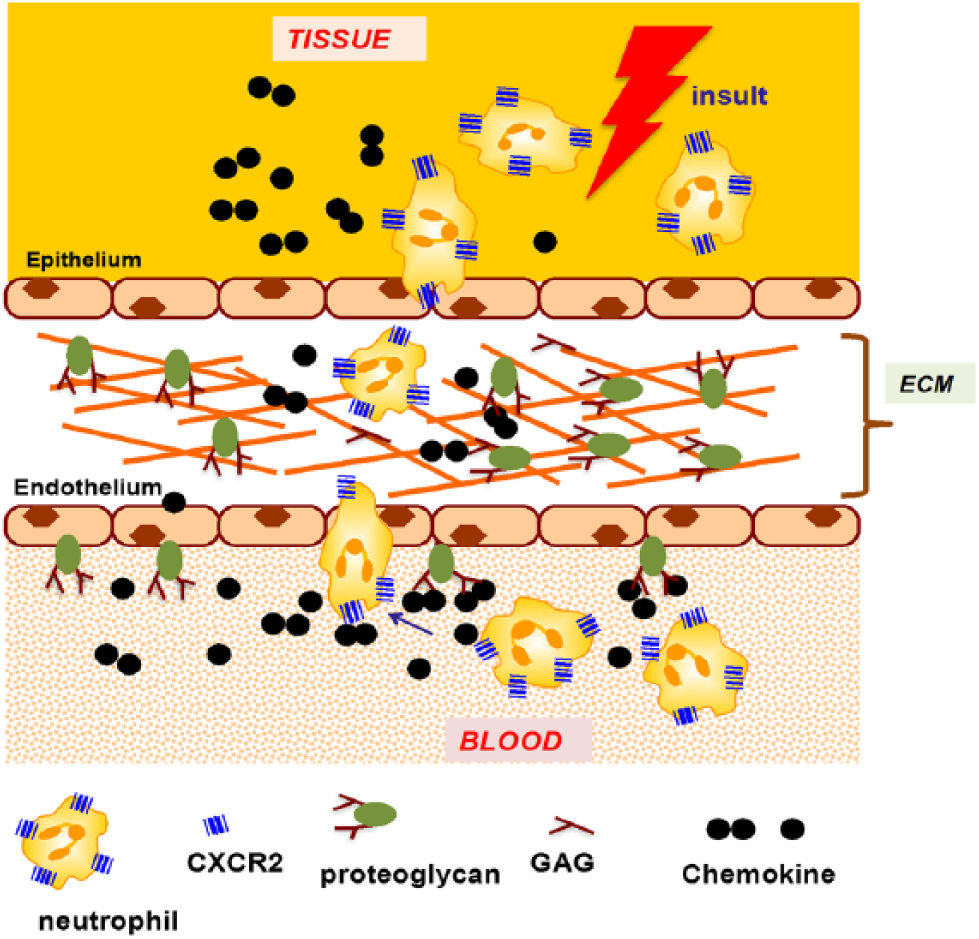
A schematic showing chemokine-mediated neutrophil recruitment. Abbreviations: ECM, extracellular matrix; GAG, glycosaminoglycan.
GAGs, such as HS, are a family of linear sulfated polysaccharides covalently attached to core proteins and are the glycan part of a family of proteins called proteoglycans (PGs).11–13 They are present on both the luminal and abluminal sides of the endothelium and epithelium and as non-covalent complexes with proteins such as collagen and laminin in the ECM. GAGs also exist freely in the glycocalyx that dominates the luminal side of the endothelium,14,15 and are also transiently generated due to cleavage by bacterial and endogenous proteases.16,17 GAGs are acidic, and chemokines are basic or contain clusters of basic residues, indicating a prominent role for electrostatic and H-bonding interactions in mediating the binding process.
HS has a modular structure with sulfated sequences separated by nonsulfated regions and is also more diverse due to differential N-sulfation and O-sulfation. Most structural and biophysical studies use heparin as a surrogate for HS for the reasons that it is more uniformly sulfated and has been shown to capture endogenous interactions.18–20 Heparin is abundantly expressed in mast cells, and so sufficient amount of size-defined oligosaccharides can be obtained from commercial vendors at a reasonable price. The basic building block of HS and heparin consists of repeating disaccharide units of
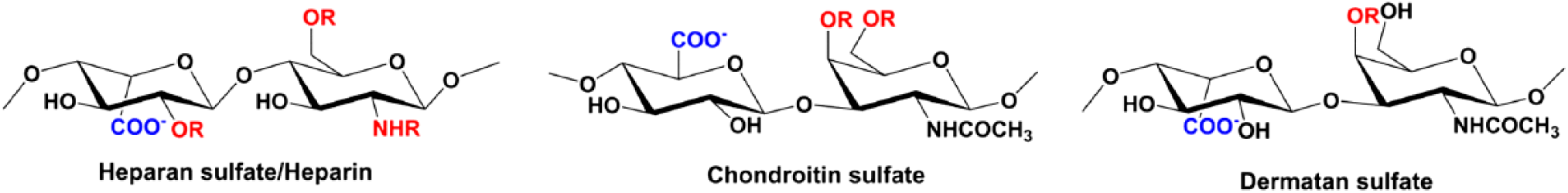
A schematic of heparin/HS, CS, and DS structures. R stands for sites where sulfation can occur. Abbreviations: HS, heparan sulfate; CS, chondroitin sulfate; DS, dermatan sulfate.
During active neutrophil recruitment, chemokine levels can vary by many orders of magnitude as a function of location such as in the vasculature and at the injury site, and as a function of time. 21 Inflammatory chemokine expression levels will be high during the early phase and lower or nonexistent during the resolution phase. Chemokines reversibly exist as monomers and dimers (Fig. 4). Therefore, in addition to the amount of chemokine, in vivo monomer and dimer levels are also dependent on the local tissue/vasculature architecture and the monomer–dimer equilibrium constant (KD-M). The KD-M of CXCL1, CXCL5, CXCL7, and CXCL8 are known, and vary between ~0.1 and 100 µM.22–24 Under normal healthy conditions, chemokine concentration is negligible (less than picomolar [pM]), and any chemokine that is present will exist as a monomer. During active recruitment, the chemokine concentration is much higher but not homogeneous and will vary at different locations. In the systemic circulation, compared with basal conditions, concentration will be higher (nanomolar [nM]), but will still predominantly exist as a monomer. However, concentration could be much higher (µM to mM) in the proximity of the membrane surface due to GAG interactions, and at sites where neutrophils extravasate into the tissue due to the small dimensions of high endothelial venules and capillaries. Therefore, monomeric and/or dimeric forms of the chemokine could exist at different locations and at different times, and continuous changes in the concentration will result in continuous changes in the monomer/dimer ratio.
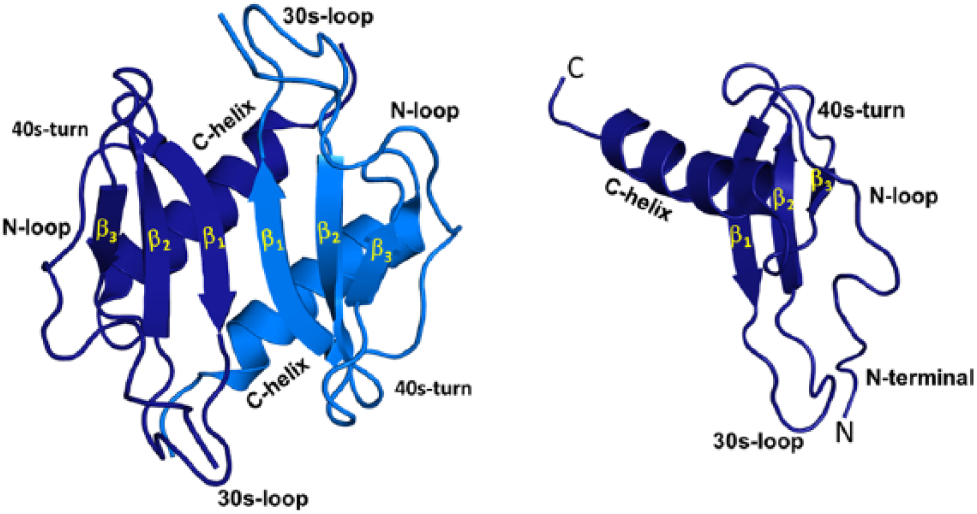
Structures of chemokine CXCL1 monomer and dimer. All NACs have the same structural fold. The individual monomers in the dimer are shown in dark and light blue for clarity. Different structural and functional regions are labeled both in the monomer and dimer. Abbreviation: NACs, neutrophil-activating chemokines.
Using designed trapped monomers that cannot dimerize and designed trapped dimers that cannot dissociate, we have shown that the neutrophil recruitment activity of chemokine monomers and dimers can be distinct and that the monomer–dimer equilibrium regulates recruitment.20,25–27 We also observe that dimer-mediated neutrophil recruitment could be quite robust, suggesting dimerization acts as an on-switch, but persistent high dimer levels are not desirable, as they could elicit massive recruitment and a runaway inflammatory response. Therefore, the ability of chemokines to continuously redistribute between monomeric and dimeric forms could be essential for regulated neutrophil recruitment in a healthy proinflammatory response. In a true disease situation, dysregulation in chemokine expression and/or disruption in monomer/dimer ratio could lead to either low or uncontrolled neutrophil trafficking resulting in unresolved inflammation and significant collateral tissue damage and disease.
A complete description of chemokine function requires knowledge of the molecular mechanisms by which chemokine monomers and dimers interact with GAGs and CXCR2. Activity measurements for CXCL1, CXCL7, and CXCL8 have shown that both monomers and dimers are potent agonists for the CXCR2 receptor.22,28–30 The molecular basis of CXCL1, CXCL5, CXCL7, and CXCL8 binding to heparin oligosaccharides has been characterized using solution nuclear magnetic resonance (NMR) spectroscopy.31–34 In particular, binding-induced NMR chemical shift changes have been shown to be quite useful for describing the molecular basis of the binding process. These studies have shown that the dimer binds heparin with higher affinity, and cellular studies have also shown that chemokine dimerization and GAG binding are coupled.35–38 Furthermore, mutants of basic residues identified as essential from in vitro heparin binding show reduced in vivo GAG binding and altered neutrophil recruitment that also vary between tissues.20,25,36–39 These studies collectively indicate GAG interactions regulate the levels of free monomer and dimer, and makeup and duration of gradients, and that GAG interactions and neutrophil recruitment are intimately coupled.
Structures of CXCL8 (also known as interleukin-8 [IL-8]), CXCL1 (also known as MGSA and Groα), CXCL5 (also known as ENA-78), and CXCL7 (also known as NAP-2) are known.23,40–43 All share a similar structural fold indicating differences in GAG interactions arise due to differences in the amino acid sequence (Fig. 1). NMR studies indicate basic residues (lysines, arginines, and histidines) are involved in binding, which is not surprising considering GAGs are highly acidic. Furthermore, these studies show not only basic residues that are conserved but also those that are unique to a given chemokine mediate binding.30–34 Most interestingly, these chemokines show a diversity of GAG-binding surfaces, indicating chemokine-specific residues play an important role in dictating the binding interactions, and that location and distribution of both conserved and chemokine-specific residues in the context of three-dimensional structure determine binding geometry. Residues implicated in binding are highlighted, and those that are conserved (in at least five out of seven sequences) are in red and shaded in gray and are labeled from B1 to B8. B2 (histidine), B3 (lysine), B5 (lysine/arginine), B7 (lysine/arginine), and B8 (lysine) are involved in the binding of all four chemokines (Fig. 1). Interestingly, B1 (arginine) is involved only in CXCL1 binding. B4 (lysine) and B6 (lysine) are involved in CXCL1 and CXCL5 but not in CXCL7 and CXCL8 binding, and, in the latter, the corresponding residues are serine and glutamine, respectively. Basic residues that are unique to a given chemokine are in blue and underlined.
The observation that chemokine-specific residues are not only involved in GAG interactions but also play a major role in determining the geometry and topology is quite unexpected. We will discuss how conserved and unique residues dictate binding topology for chemokines CXCL1 and CXCL5 (Fig. 5). NMR studies of CXCL1 indicate heparin binds two non-overlapping surfaces that lie on opposite faces of the protein and spans the dimer interface. One surface (defined as α-domain) consists of conserved residues B2, B3, B4, B6, B7, and B8 from both monomers of the dimer, and the second surface (defined as β-domain) consists of conserved residues B1, B5, a lysine (K29) unique to CXCL1, and a quasi-conserved lysine (K49) also from both monomers of the dimer. 31 The binding surface in CXCL5 is distinctly different and consists of residues B2 to B8 that lie within the monomer. 32 Unlike CXCL1 that contains two distinct binding surfaces that span the dimer interface, CXCL5 contains two equivalent binding surfaces that lie within each monomer. The observation that there is no CXCL5-specific GAG-binding residue is striking. CXCL5 sequence shows a Phe corresponding to K29 and a glutamate corresponding to K49, which highlight how as few as two lysine residues at specific locations can dictate very different binding geometries in two related chemokines.
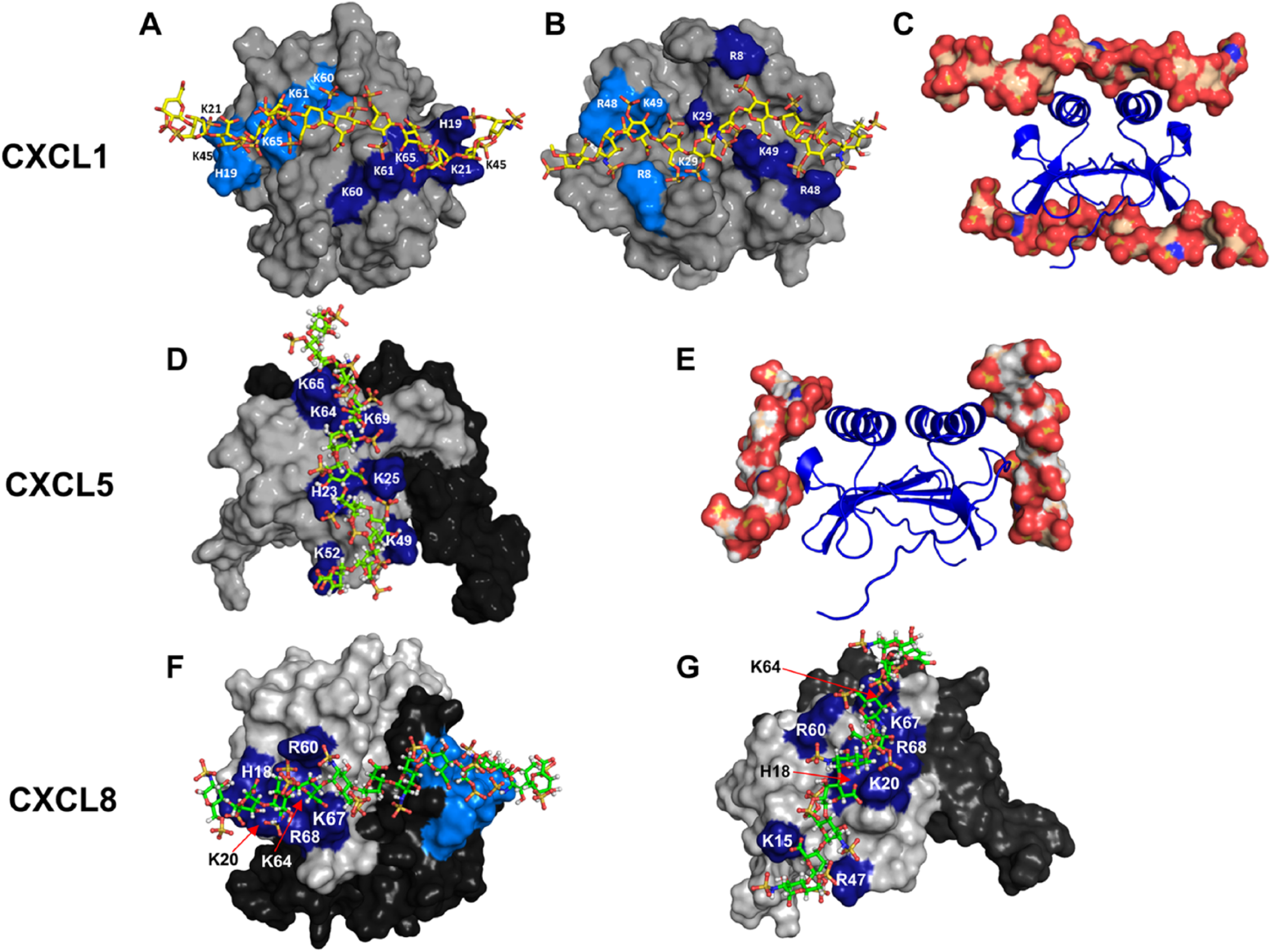
Models showing heparin binding in CXCL1, CXCL5, and CXCL8. Panels A and B show the non-overlapping heparin-binding surfaces (defined as α- and β-domains) in CXCL1. Heparin-binding residues from both monomers are highlighted in light and dark blue. Panel C shows heparin binding to both the domains. Panel D shows the heparin-binding surface in CXCL5. The second monomer of the dimer is shown in black for clarity. Panel E shows the binding of heparin to each of the two monomers. Panels F and G show two of the heparin-binding surfaces observed for CXCL8.
Several computational, biophysical, cellular, and mutational studies have characterized CXCL8 interactions.33,36 –38,44 –50 CXCL8 sequence lacks B4 and B6, and binding studies reveal B2, B3, B5, B7, and B8 are involved in binding. NMR studies reveal as many as four CXCL8-specific residues (K15, K54, K67, and R68) are also involved in binding. 33 For the dimer, modeling studies indicated that a single geometry cannot satisfy binding to all of the conserved and specific residues and that heparin binds to multiple surfaces. Analyses of the models indicate binding residues can be divided into two categories: core residues that are common to all surfaces and a second set of residues in the periphery of the core residues that define the different binding geometries. The core set consisted of conserved B2, B3, B7, B8, and CXCL8-specific K67 and R68, and peripheral set consisted of conserved B5 and CXCL8-specific K15 and K54. We show two of the several geometries observed in Fig. 5. We would like to point out that existence of several binding geometries for the dimer does not mean that all are equally favored, and in reality, it is very possible that one or two poses are the most favored. Additional experiments and high-resolution structures are essential to determine whether the binding interface in CXCL8 is plastic or unique. In the case of monomer, binding is mediated by all of the core residues as observed for the dimer and only one peripheral residue (K15), and the binding geometry essentially corresponds to the second model observed in the dimer (Fig. 5).
We have characterized the binding interactions of CXCL7 monomer and dimer to heparin using NMR spectroscopy.30,34 Binding interactions of the native monomer were characterized by exploiting the weak propensity of CXCL7 to form dimers. This is the only structural characterization of GAG interactions of a native monomer in the NAC family. Although the dimer levels were lower than the monomer under the experimental conditions, the binding interactions of the native dimer were successfully characterized by exploiting its higher heparin-binding affinity compared with the monomer. In the dimer, conserved B2, B3, B5, B7, and B8, and CXCL7-specific R54 mediate binding. The observation that conserved residues B4 and B6 are not involved in binding is different compared with CXCL1 and CXCL5; interestingly, CXCL8 lacks basic residues at these positions. Modeling studies reveal heparin can bind two different surfaces, one within a monomer and one across the dimer interface. However, the model spanning the dimer interface does not involve B5. Additional studies are required to determine whether both binding geometries are equally favored. In the case of monomer, dimer-binding residues (except B8), and in addition, quasi-conserved residue K9, mediate binding interactions. Modeling studies reveal multiple geometries that can be described by a set of core residues (B2, B3, and R54) common to all surfaces and peripheral residues (B5, B7, and K9) that are involved in specific geometries. These studies once again reveal how different combination of conserved and chemokine-specific residues can result in different binding surfaces for related chemokines.
Currently, nothing is known regarding GAG interactions for CXCL2, CXCL3, and CXCL6 (Fig. 1). CXCL2 and CXCL3 show high sequence homology to CXCL1 but are missing a basic residue at positions B5 and B6, respectively. CXCL6 is also distinct as it is missing B2. Studies summarized here on other NACs show that multiple binding surfaces can exist for a given chemokine, binding surfaces can vary between the monomer and dimer, and that the binding surfaces can vary significantly among related chemokines. Therefore, it is possible that GAG interactions of CXCL2, CXCL3, and CXCL6 are also distinctly different, and future studies will reveal whether this is the case.
Animal model studies have also shown that mouse CXCL1 (also known as KC) is more efficient compared with mouse CXCL2 (also known as MIP-2) in recruiting neutrophils to the lung and that KC traverses more rapidly from the lung to circulation, suggesting differences in GAG interactions determine chemokine levels at different compartments that in turn determine gradients and neutrophil levels. 38 KC and MIP-2 also exist as monomers and dimers, and sequences reveal seven of the eight conserved basic residues (except B6) are conserved in KC but only five (except B2, B5, and B6) are conserved in MIP-2 (Fig. 1). The molecular basis of KC binding to heparin has been characterized using NMR spectroscopy, which showed that heparin binds across the dimer interface. 51 KC sequence reveals none of human CXCL1-specific basic residues (K29 and K49) from the β-domain. NMR data also show no chemical shift perturbation of B1 arginine, suggesting that the binding geometries of mouse and human CXCL1 are different. Characterization of heparin binding to MIP-2 turned out to be challenging, and data exist only of a disaccharide. 39 This study identified some of the basic residues, and a more detailed characterization is essential to describe how GAG interactions determine KC and MIP-2’s distinct in vivo activities.
How do GAG interactions influence chemokine function and neutrophil recruitment? As discussed above, GAG interactions determine levels of free chemokine monomer and dimer, which is intimately coupled to the makeup of the chemotactic and haptotactic gradients. Solution-phase free chemokines determine the chemotactic gradients, and solid-phase GAG-bound chemokines determine the haptotactic gradients. It is very likely that the nature of these gradients vary in the vasculature, across the endothelium, and in the ECM, and as a function of time. It has been proposed that chemotactic gradients are unlikely to exist in the vasculature, as free chemokines will be washed away with blood flow and that haptotactic gradients drive neutrophil egress across the endothelium.49–52 By extension, it is believed that it is the GAG-bound and not the free chemokine that binds and activates the receptor especially in the vasculature. Intravital imaging studies have also been interpreted to indicate that GAG-bound chemokine is presented to the receptors on leukocytes. 53 However, there is no direct experimental evidence for a ternary complex and that GAG-bound chemokine can activate the receptors on leukocytes.
Chemokine binding and activation of CXCR2 involve two distinct sites.54,55 One site involves interactions between the chemokine N-loop/β3-strand and receptor N-terminal domain residues (defined as Site-I) and the second site involves interactions between the ligand N-terminal and a groove in CXCR2 defined by the extracellular/transmembrane residues (Site-II). Site-I functions as an initial docking site, and the structural basis of Site-I interactions has been studied by characterizing chemokine binding to receptor N-terminal domain peptides. Such studies for CXCL1, CXCL5, CXCL7, and CXCL8 have shown several residues that are involved in CXCR2 binding are also involved in GAG interactions.28,30–32,34,56 A schematic of the GAG and receptor-binding residues and the extent of overlap are shown (Fig. 6). Furthermore, heparin-bound CXCL1 and CXCL7 are unable to bind the CXCR2 N-terminal peptide.30,31 Heparin-bound CXCL8 and CXCL7 have also been shown to be impaired for receptor activity.30,45 These studies collectively suggest that it is the free chemokine and not GAG-bound chemokine that can access the receptor and, by extension, it is the chemotactic gradients that drive neutrophil egress from the vasculature to the target tissue. Considering the dimeric form binds GAGs with higher affinity, these observations also indicate GAG interactions regulate the levels of free monomer and dimer available for receptor interactions.
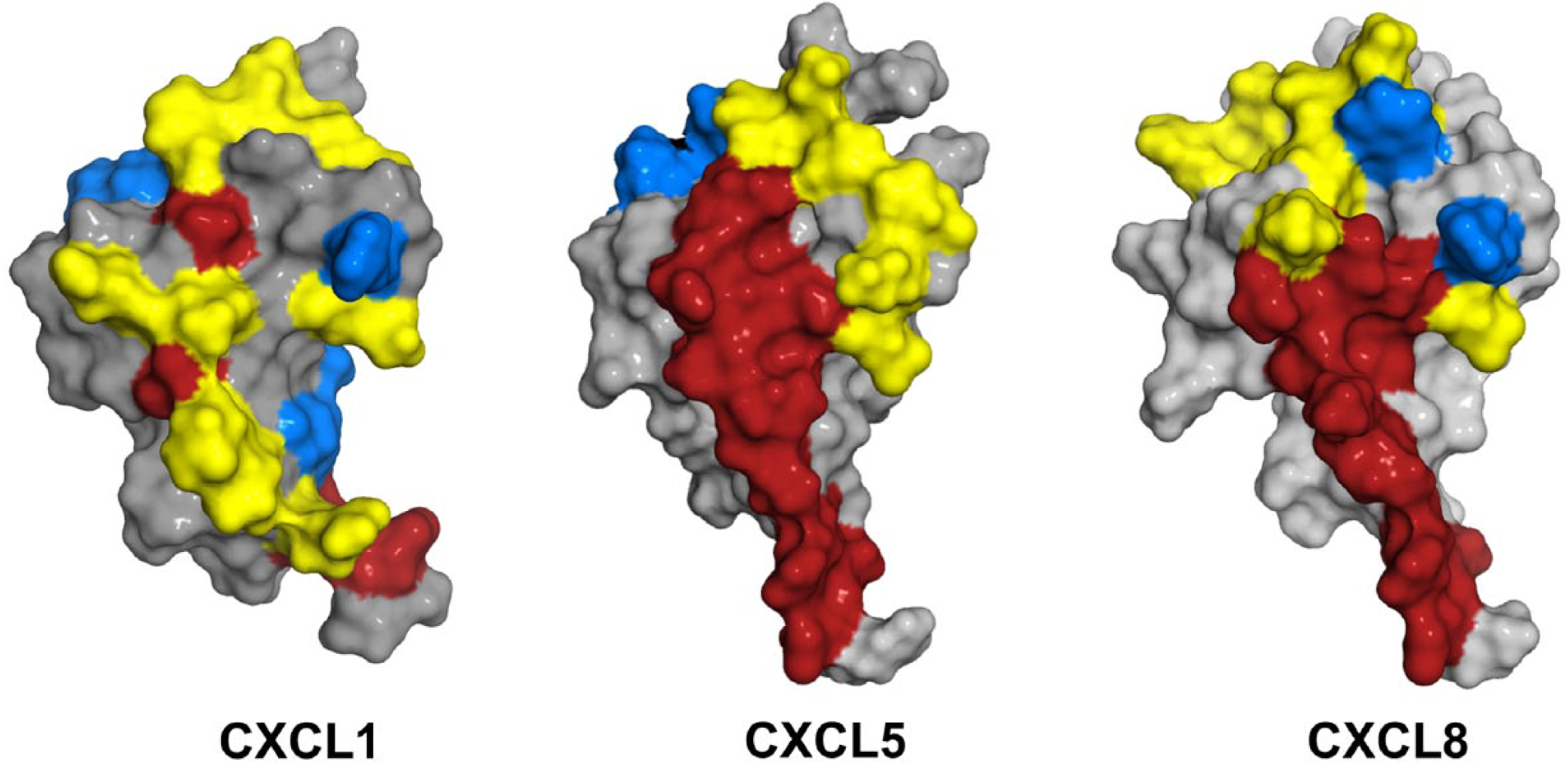
Structures of CXCL1, CXCL5, and CXCL8 showing GAG- and CXCR2-binding regions. CXCR2-binding domains are in red, heparin-binding domains are in blue, and residues that are common to both are in yellow. Abbreviation: GAG, glycosaminoglycan.
Considering in vitro studies used heparin oligosaccharides and receptor fragment, the question arises to what extent these observations capture the complexity of the in vivo interactions? The in vivo environment such as the vasculature, the proximity of the endothelium, and the architecture of the ECM are complex and crowded. Furthermore, NACs also bind chondroitin sulfate (CS) and dermatan sulfate (DS),36,57,58 and currently very little is known regarding how NACs bind to these GAGs. Structures of CS and DS, compared with HS, are different, and they are also less sulfated (Fig. 3). The basic building block of CS consists of
Chemokines were discovered in the late 1980s and that they are ligands for GPCRs and bind GAGs a few years later. One of the challenges in the field has been why humans express so many chemokines and is there a redundancy—a question very applicable to the seven NACs. Over the years, it has become apparent that the in vivo functions of chemokines are not redundant, 59 but the molecular basis underlying chemokine specificity remains unclear. Much has been learnt about the molecular basis of receptor interactions and signaling over the years, including a role for biased receptor signaling in defining the in vivo phenotype of closely related chemokines. 60 On the contrary, understanding and characterizing GAG interactions have been slow to come by due to absence of straightforward functional readouts and the difficulty in characterizing protein–GAG complexes unlike those of protein–protein and protein–DNA complexes. Furthermore, considering GAG-binding residues seem to be conserved and that binding occurs due to electrostatic interactions, it was thought that GAG interactions are promiscuous and that there is no specificity encoded in the chemokine. However, recent studies as summarized in this review make a compelling case that GAG interactions for NACs are specific, and that differences in these interactions most likely play an important role in fine-tuning the phenotype of each chemokine in a context-dependent manner.
Differences in GAG interactions also throw light on several observations, including selective expression of NACs by some cell types and/or under specific disease conditions, and, at the same time, co-expression of NACs in other conditions.61–65 Indeed, NACs have also been shown to form heterodimers, and that GAG interactions of heterodimers are distinctly different from those of homodimers. 66 Moreover, several of the NAC members have also been shown to play important roles in non-immune functions including angiogenesis and development, and clearly GAG interactions must play an important role considering these processes are both spatially and temporally regulated.67,68 In addition to gradient formation, GAG interactions have been implicated to regulate recruitment by increasing chemokines’ lifetime by preventing its proteolysis and from being washed away by blood flow in the vasculature. Chemokine activity is also modulated by bacterial proteases, but the cleavage site is not accessible in the GAG-bound form, indicating GAG interactions also restrict protease activity and extend chemokines’ lifetime and its function.16,17
Studies to date have provided important insights, but there is still much to be learnt. Some focus areas include structural basis of binding to other GAGs such as CS and DS, potential differences in chemokine binding between free and immobilized GAGs, structures of the chemokine–GAG complexes, and development of sensitive methods for detecting in vivo chemokine–GAG complexes and whether they can interact with the receptor to form ternary complexes. Distribution and levels of different GAGs can vary between tissues and between the endothelium and ECM.69–71 Considering GAGs bind hundreds of proteins,11,13,18,19 binding information of a given NAC to different GAGs must be encoded in the chemokine sequence. Therefore, it will be quite interesting to see what, if any, is the role of chemokine-specific residues in binding to CS and DS, and how the combination of conserved and chemokine-specific residues determines binding interactions. Ultimately, knowledge of the structures of the chemokine–GAG complexes is essential to fully describe the role of various basic residues and how each residue imparts affinity and specificity. Some success has been achieved in these arenas though not for the NAC family—A crystal structure of CCL2 bound to a heparin octasaccharide is now known and a recent NMR study reported binding of CCL5 to CS oligosaccharides.72,73 Knowledge from such studies will lead to a better understanding of how these versatile proteins by their GAG interactions in conjunction with receptor interactions fine-tune and define the phenotype for successful resolution of inflammation.
A hallmark of infection and tissue injury is the recruitment of neutrophils to the infected/injured site. In a healthy immune response, a sufficient number of neutrophils are recruited, and subsequent downstream processes of neutrophil apoptosis and efferocytosis result in successful resolution. However, under disease conditions, a dysregulation in chemokine levels (either few or more) results in altered GAG-bound and free monomer and dimer levels, which in turn result in altered gradients, leading to either impaired or robust neutrophil recruitment.74 –76 Too many neutrophils or sustained recruitment result in collateral tissue damage as in the case of acute lung injury and reperfusion ischemia, and impaired recruitment results in bacteremia and disease. Therefore, knowledge of the basic principles and the molecular mechanisms by which NACs bind GAGs also has direct impact on therapeutics. CXCL8 mutants that are inactive for receptor function but bind GAGs with higher affinity have been shown to impart protection in animal disease models. 77 More recently, hydrogels based on heparin and heparin derivatives were shown to outperform the standard-of-care product Promogran, by their ability to scavenge chemokines, and effectively reducing neutrophil activity and alleviating disease symptoms in humans. 78 These studies are very promising, indicating a detailed understanding of how individual basic residues impart affinity and specificity, and availability of high-resolution structures of chemokine–GAG complexes could lead to designing more effective hydrogels that could be used in a wide variety of clinical settings.
Footnotes
Competing Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
KR conceived and wrote the manuscript with input and suggestions from KMS, PRBJ, KVS, and AJB. All authors have read and approved the final manuscript.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Institutes of Health grants P01 HL107152 and R21 AI124681.