
Abstract
Cardiolipin (CL) is a unique dimeric phospholipid that exists almost exclusively in the inner mitochondrial membrane (IMM) in eukaryotic cells. Two chiral carbons and four fatty acyl chains in CL result in a flexible body allowing interactions with respiratory chain complexes and mitochondrial substrate carriers. Due to its high content of unsaturated fatty acids, CL is particularly prone to reactive oxygen species (ROS)-induced oxidative attacks. Under mild mitochondrial damage, CL is redistributed to the outer mitochondrial membrane (OMM) and serves as a recognition signal for dysfunctional mitochondria, which are rapidly sequestered by autophagosomes. However, peroxidation of CL is far greater in response to severe stress than under normal or mild-damage conditions. The accumulation of oxidized CL on the OMM results in recruitment of Bax and formation of the mitochondrial permeability transition pore (MPTP), which releases Cytochrome c (Cyt c) from mitochondria. Over the past decade, the significance of CL in the function of mitochondrial bioenergy has been explored. Moreover, approaches to analyzing CL have become more effective and accurate. In this review, we discuss the unique structural features of CL as well as the current understanding of CL-based molecular mechanisms of mitophagy and apoptosis.
Introduction to Cardiolipin
Cardiolipin (CL) was first isolated from beef heart in 1942 (Pangborn 1942). As an evolutionarily conserved phospholipid, CL is mostly confined to the inner leaflet of the inner mitochondrial membrane (IMM) in mammalian cells and plasma membranes of bacteria (Daum and Vance 1997; Schlame 2008). The structure of CL is characterized by a glycerol backbone connected to two phosphatidyl lipids. This structure is more flexible than regular phospholipids because CL possesses two chiral carbons and four fatty acyl chains that are usually polyunsaturated (Ardail et al. 1990). CL on the mitochondrial membranes can be induced to cause a negative membrane curvature, and this makes CL a perfect brick with which to build the inner folded cristae (Acehan et al. 2009; Schlame 2012; Xu et al. 2006a). Indeed, CL was reported to have an affinity with several proteins in the electron transfer chain and mitochondrial substrate carriers (Claypool 2009; Paradies et al. 2014). This profile may also partly explain how these respiratory chain proteins (complexes I, III, and IV, and ATP synthase) embed into the IMM and form relatively compact cristae. All of these proteins possess different sizes and shapes yet anchor perfectly in or through the IMM. Therefore they require close and diverse connections to phospholipids. But why is CL so special as compared with other phospholipids, such as phosphatidylethanolamine (PE) and phosphatidylserine (PS)?
The dimeric structure and polyunsaturated fatty acyl chains of CL may be responsible for its ability to switch between lamellar and negative curvature profiles. The biosynthesis of CL is a little more complex than that of PE and PS and involves two pathways: de novo synthesis and remodeling steps (Hatch 1998; Schlame et al. 2000) (Fig. 1). The de novo reactions originate from phosphatidic acid (PA). First, PA is transformed into cytidine diphosphate-diacylglycerol (CDP-DAG) in the presence of CDP-DAG synthase (Houtkooper and Vaz 2008). Then, CDP-DAG is converted to phosphatidylglycerol phosphate (PGP) by displacement of the cytidine monophosphate (CMP) moiety by sn-glycerol-3-phosphate through the activity of the PGP synthase. This is followed by a dephosphorylation reaction to produce PG (Chang et al. 1998; Stanacev et al. 1967). Finally, the combination of PG and CDP-DAG yields CL through the activity of cardiolipin synthase (CLS) (Chicco and Sparagna 2007; Lu et al. 2006). On the other hand, PE and PS possess relatively simple structures composed of a glycerol with two fatty acyl chains. Two chiral carbons on the two side glycerols of CL allow the four acyl chains to combine in different spatial locations. These distinct spatial structures may have important roles in the fluidity of cristae as well as in the fixation of proteins in mitochondrial membranes.
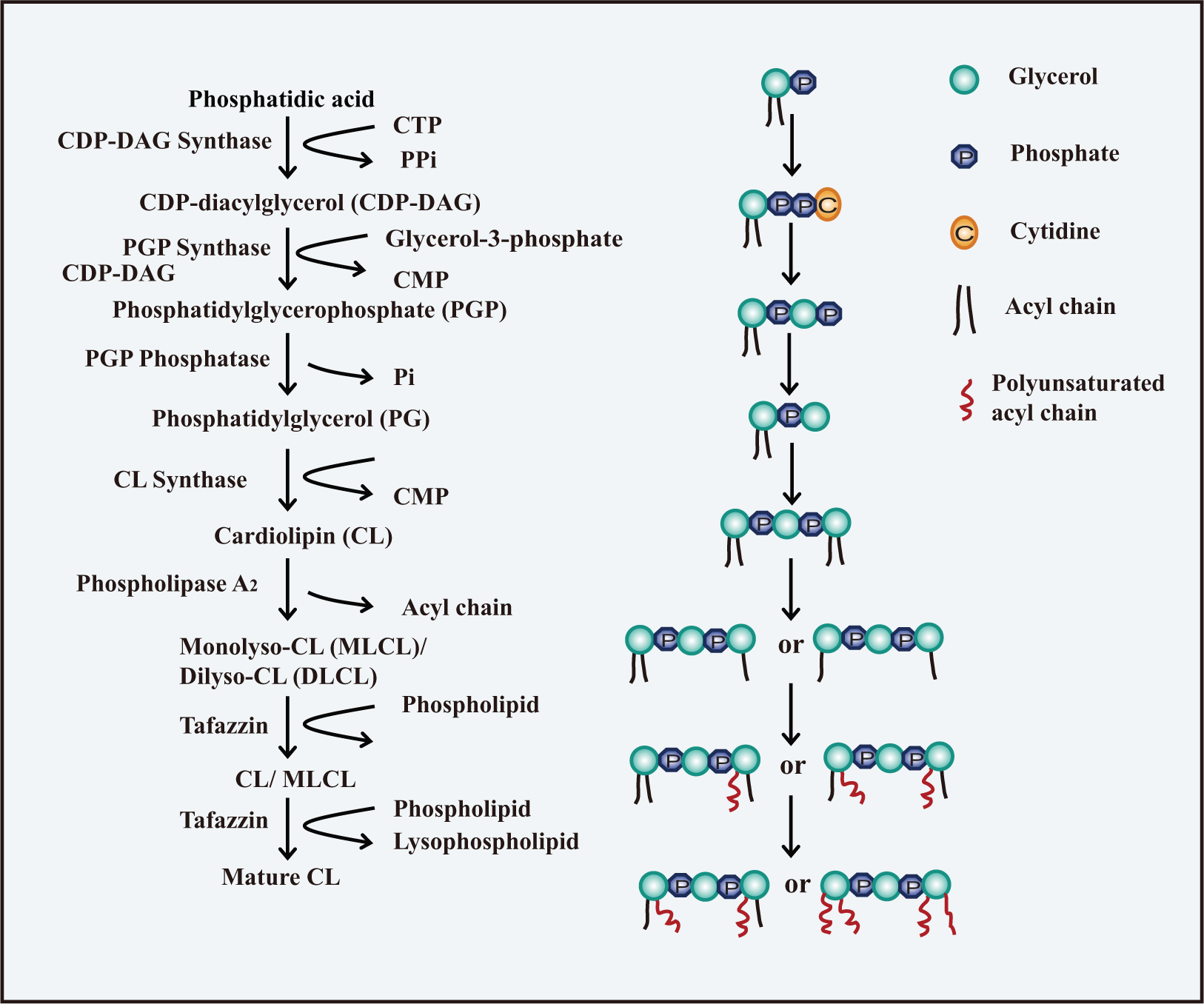
Biosynthesis of Cardiolipin (CL). The biosynthesis of CL involves two pathways: de novo synthesis and remodeling steps. The de novo synthesis of CL occurs through a series of four reactions originating from phosphatidic acid (PA). PA is first transformed into CDP-DAG in the presence of CDP-DAG synthase. CDP-DAG is then converted to PGP, followed by a dephosphorylation reaction to produce PG. The combination of PG and CDP-DAG yields CL. The remodeling process is a deacylation-reacylation cycle in the presence of PLA2 and tafazzin, resulting in the replacement of the fatty acyl chains in the newly synthesized CL with polyunsaturated chains.
CLS has no preference for certain acyl chains, indicating that post-synthetic remodeling is essential to produce specific polyunsaturated fatty acyl chains in CL. Remodeling is a cycle of deacylation and reacylation on the OMM or mitochondrion-associated endoplasmic reticulum (ER) (Eichberg 1974; Schlame and Rustow 1990; Schlattner et al. 2013). First, CL is hydrolyzed by phospholipase A2 (PLA2), generating monolysocardiolipin (MLCL) or dilyso-CL (DLCL) (Danos et al. 2008). A transacylase then takes an acyl chain from a phospholipid and attaches the acyl chain to the lyso-CLs. Three transacylases are proposed to be involved in the reacylation of lyso-CLs: monolyso-cardiolipin acyltransferase (MLCLAT), acyl-CoA: lysocardiolipin acyltransferase (ALCAT1) and tafazzin (Cao et al. 2004; Li et al. 2010; Ma et al. 1999; Taylor and Hatch 2003, 2009; Xu et al. 2006b). Tafazzin is accepted as the predominant transacylase that replaces acyl chains in CL to provide a functional mitochondrial membrane (Acehan et al. 2011; Gu et al. 2004; Xu et al. 2006a). However, ALCAT1 has been proposed to catalyze the deleterious remodeling of CL, leading to aberrant CL species that are commonly found in heart disease, diabetes, and obesity (Han et al. 2007; Li et al. 2010; Sparagna and Lesnefsky 2009). This may be associated with the finding that elevated reactive oxygen species (ROS), caused by over-expressed ALCAT1, leads to peroxidation of CL, although the mechanism is still unclear (Li et al. 2010). But what is the relationship between oxidation of CL and the CL remodeling reactions? The function of ALCAT1 may be different under normal or stressed conditions. Remodeling occurs under normal circumstances whereas stressed conditions result in enhanced ROS as well as oxidation and peroxidation of CL.
CL is responsible for adhering to and stabilizing the respiratory chain proteins, including complexes I, III, and IV, and ATP synthase (Camberg et al. 2007; Paradies et al. 2004; Petrosillo et al. 2003; 2013). Also, CL is necessary for aggregating and anchoring the oxidative phosphorylation proteins as well as being a reservoir for protons during ATP synthesis (Haines and Dencher 2002). Apart from its role in mitochondrial bioenergetics, CL has been reported to have a role in mitochondria fission (Montessuit et al. 2010). Mitochondria are dynamic organelles that constantly fuse and divide. Fission is necessary for degradation of injured mitochondria by mitophagy or for irreversible apoptosis (Lee et al. 2011; Park et al. 2012; Twig et al. 2008; Youle and Karbowski 2005). Furthermore, it has been reported that the distribution of CL is interchangeable between the IMM and OMM under autophagic or apoptotic stimuli. CL migrates from the IMM to the OMM when exposed to rotenone, staurosporine, and cyclosporine A (Chu et al. 2013; de Arriba et al. 2013; Fernandez et al. 2002). Externalized CL has more opportunities to interact with cytosolic proteins and thus affect cellular events like mitophagy and apoptosis. In this review, we emphasize the current understanding of CL-based molecular mechanisms of mitophagy and apoptosis, focusing on the crosstalk between CL and mitophagy/apoptosis-related proteins. In addition, we discuss briefly the CL-based interplay between mitophagy and apoptosis. The translocation of CL onto the OMM and the mechanism of CL oxidation are also highlighted in this review.
Properties of CL in Mitophagy
In 2005, Lemasters first defined the term mitophagy (Lemasters 2005). It refers to the selective removal of superfluous or damaged mitochondria by autophagosomes. This is a crucial mechanism in the control of mitochondrial quality (Ashrafi and Schwarz 2012). Recently, the distribution of CL was found to be changeable under mitophagic stress, and CL on the OMM interacts with LC3 and Beclin-1.
Translocation of CL in Mitochondria Membranes
Normally, CL constitutes about 25 mol% of mitochondrial membrane lipids (Daum 1985). In eukaryotic cells, over 65% of CL is predominantly confined to the inner leaflet of the IMM (Ardail et al. 1990; Krebs et al. 1979). However, Charleen T. Chu and colleagues found that both the content and species of CL in the OMM were increased in mitophagy-inducing cells. Mass spectra (MS) of CL exhibited clusters of similar masses, depending on the diversity of its fatty acyl chains. The mass spectrum of a control group exhibited seven IMM and four OMM CL clusters, whereas rotenone-treated samples exhibited seven clusters for both the IMM and OMM fractions. Notably, no increased amounts of CL peroxidation products were detected under those low-dose rotenone and STS-treated mitophagic conditions (Chu et al. 2013).
Phospholipid scramblase-3 (PLS3), Nm23-H4, mitochondrial creatine kinase (MtCK), and tBid help to reshape mitochondrial membranes by transferring CL (Fig. 2) (Kagan et al. 2006, 2014). PLS3 is probably responsible for the translocation of CL from the inner to the outer leaflet of the OMM (Chu et al. 2013; Liu et al. 2003; Van et al. 2007). tBid, a member of the BH3-only subgroup of the pro-death proteins, targets mitochondria by combining with CL on the OMM (Kim et al. 2004; Lutter et al. 2000; Sorice et al. 2004). PLA2 is activated by tBid, yielding MLCL and DLCL (Kagan et al. 2006). It also serves as a transmembrane transporter carrying lyso-CL and CL outwards. Moreover, tBid further activates PLS3 and forms a positive feedback loop that increases the amount of CL on the OMM (He et al. 2007; Kagan et al. 2006; Liu et al. 2008). MtCK and Nm23-H4 are both basic peripheral membrane proteins. Octameric MtCK binds to CL on the IMM and voltage-dependent anion channel (VDAC) protein on the OMM, appearing as a bridge across the mitochondrial membranes (Epand et al. 2007b). This may explain the means of CL translocation from the IMM to the OMM. MtCK does not function as a transporter when CL is mutated, indicating that CL is important for stabilizing MtCK (Epand et al. 2007a; Schlattner et al. 2009). In addition, Nm23-H4, a mitochondrial nucleoside diphosphate kinase, can also act as a bridge, attaching simultaneously to the IMM and OMM and allowing the transfer of CL (Epand et al. 2007a; Schlattner et al. 2013). The main transacylase, tafazzin, may also be involved in the migration of CL. Tafazzin exists in the outer leaflet of the IMM and the inner leaflet of the OMM; thus, it can be regarded as existing in the intermembrane space. It may only replace the acyl chains of certain non-bilayer CL in vitro (Schlame 2012; Schlame et al. 2012). The structure of tafazzin is sensitive to membrane tension that results from the arrangement of membrane phospholipids (Gawrisch 2012). It may alleviate membrane tension by starting the remodeling and migration of CL. Tafazzin may act as one of the sensors for membrane curvature and triggers the migration of CL to restore the mitochondrial membrane conformation. However, the exact and entire migration steps of CL still need to be further studied.
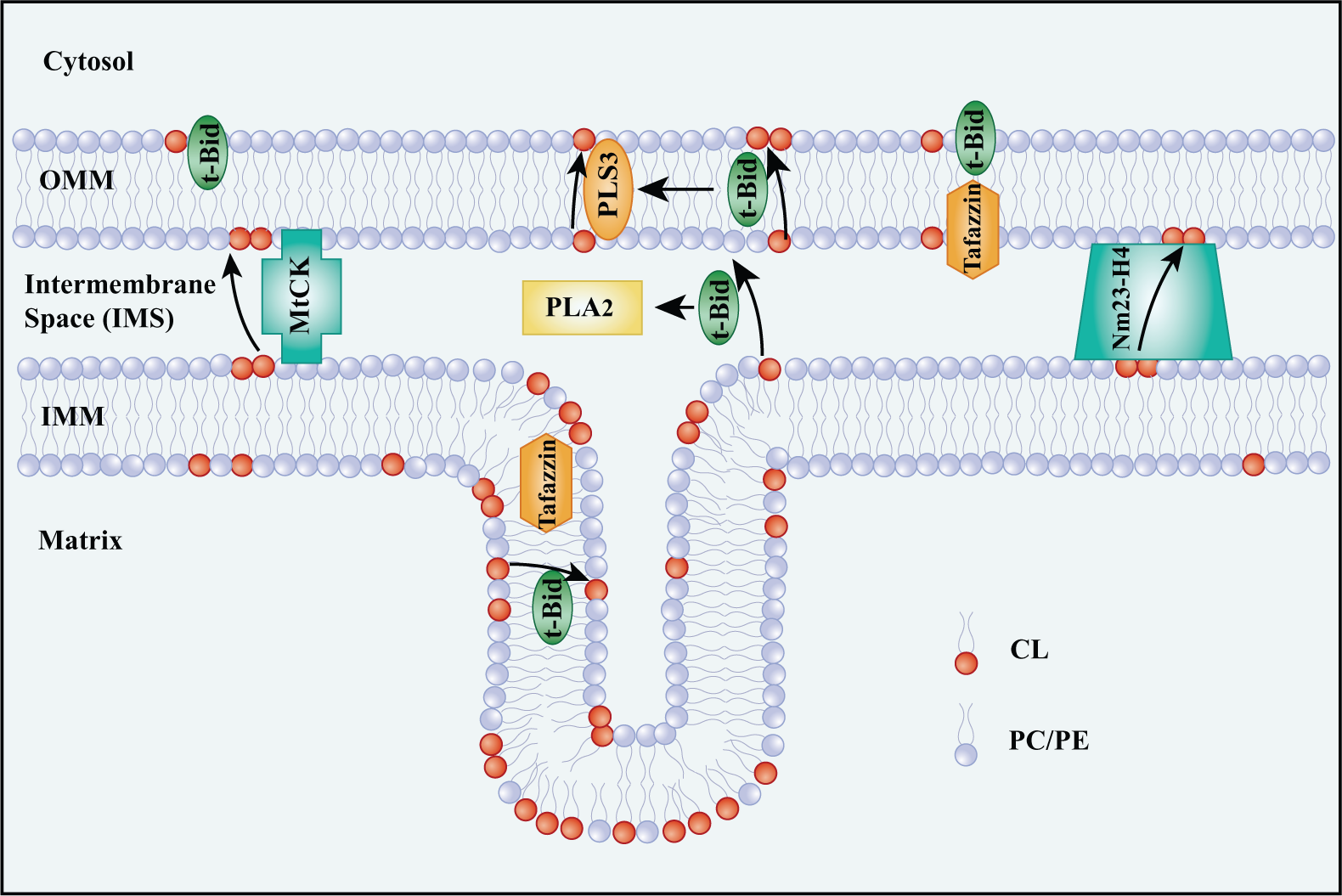
Migration of cardiolipin (CL) from the inner mitochondrial membrane (IMM) to the outer mitochondrial membrane (OMM). Several proteins are involved in the transfer of CL from the IMM to the OMM. Phospholipid scramblase-3 (PLS3) is mostly in charge of the transfer of CL from the inner to the outer leaflet of the OMM. tBid targets the mitochondria by binding to CL. tBid can swim through the mitochondrial membranes carrying CL and Lyso-CLs outward. In addition, tBid activates PLS3, which helps to increase CL on the surface of the OMM and recruit more tBid. Mitochondrial creatine kinase (MtCK) and Nm23-H4 bridge the IMM and OMM for CL transfer. Tafazzin may contribute to the migration of diversified CL by shuffling fatty acids between phospholipid species.
Interactions of CL and Autophagy-Related Proteins
CL Is Necessary for Mitochondrial Fission
Mitochondria are dynamic organelles, with a major axis of 5 µm in length (Cereghetti and Scorrano 2006). However, the diameter of an autophagosome is about 1 µm (Kraft et al. 2010). This size discrepancy presents an issue for mitophagy. Inhibition of mitochondrial fission decreases the level of mitophagy (Thomas and Jacobson 2012). Dynamin-related protein (DRP1), a large GTPase of the dynamin family, is activated and in charge of mitochondrial fission when DRP1 transfers to the OMM from the cytosol (Frank et al. 2001; Smirnova et al. 2001). IRGM, a human immunity-related GTPase in the IMM or matrix, affects mitochondrial depolarization and fission as well as subsequent mitophagy. IRGM has a role in fission only in the presence of DRP1. Notably, DRP1 and IRGM both show a strong affinity to CL in binding experiments using CL-agarose beads (Singh et al. 2010). Translocation of DRP1 from the cytosol to the mitochondria results in a ring-shaped DRP1 multimer, which wraps the constriction sites of the mitochondrial membrane like a twisted rope (Labrousse et al. 1999; Legesse-Miller et al. 2003). DRP1 on the OMM and IRGM on the IMM may cooperate to induce mitochondrial division, and these two proteins may be in charge of fission in both membranes. Fission events often generate uneven daughter mitochondria: one daughter exhibits an increased membrane potential with a high probability of subsequent fusion with other healthy mitochondria for recovery, whereas the second one has a decreased membrane potential and a high probability of degradation by peroxisomes or the mitophagy pathway (Twig et al. 2008) (Fig. 3).
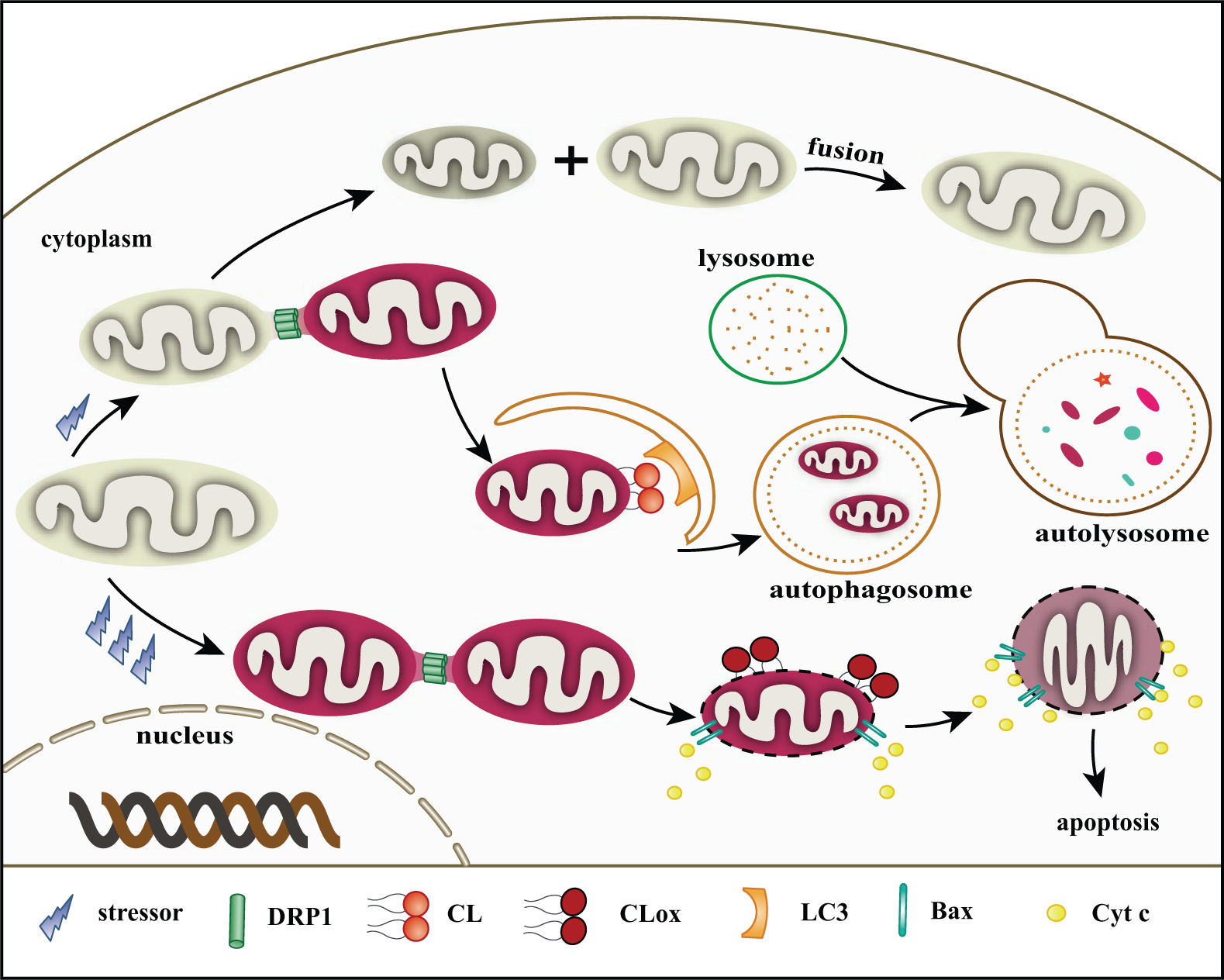
Roles of cardiolipin (CL) in mitophagy and apoptosis. (1) Under low and mild stress, dynamin-related protein (DRP1) localizes to mitochondria by binding to CL. Mitochondria are divided into two daughter units. The one with lower membrane potential is probably degraded via mitophagy. LC3 may target damaged mitochondria by combining with CL on the outer mitochondrial membrane (OMM). These proteins associate with CL, mediating the development of mitophagy. The one with higher membrane potential may be repaired by fusing with a healthy mitochondrion. (2) During apoptotic/high stress, abundant reactive oxygen species (ROS) are produced. CL is oxidized by Cytochrome c (Cyt c) under the help of hydrogen peroxide (H2O2) and transfers to the OMM. Mitochondria are also divided by DRP1 and other fission-related proteins. The mitochondrial membrane pore (MMP) is built via Bax oligomerization in the presence of tBid. Cyt c and other apoptotic factors then escape into the cytosol triggering a subsequent caspase cascade.
Roles of CL in Autophagic Cargo Recognition and the Formation of Autophagosomes
CL on the surface of the OMM may serve as a signaling molecule for mitophagy. But what does CL do to promote mitophagy? Since CL is unique to mitochondria, does it contribute to the selectivity of damaged mitochondria in mitophagy? In addition, since the mitochondrial membrane is considered to be one of the sources for the formation of autophagosomes (Hamasaki et al. 2013; Tooze and Yoshimori 2010), does CL have a role in their formation?
In 2009, Kagan and his colleagues hypothesized that the migration of CL in mitochondria may be a message for mitophagy (Kagan et al. 2009a, 2009b). Just like the externalization of PS on the plasma membrane serves as a “death signal” in apoptosis, CL on the OMM may be a signal for mitophagy. It has been shown that exposed CL on the surface of the OMM combines with LC3 and gives rise to the recognition of injured mitochondria (Chu et al. 2013). LC3, a homolog of autophagy-related gene 8 (Atg8) in yeast, is lipidated to LC3-Ⅱ via conjugation with PE upon induction of autophagy. LC3 is involved in the elongation of the autophagosomal membrane and is involved in autophagic cargo recognition (Tanida et al. 2008). It contains basic surface patches that can bind CL (Chu et al. 2013). In addition, knocking down the expression of CL synthase (CLS) or PLS3 leads to less CL on the OMM, and reduced mitophagy in primary neurons, SH-SY5Y, and HeLa cells (Chu et al. 2013). Regulation of mitophagy, however, could be much more complex. In fact, multiple additional players have been reported in the field and are attracting great attention. For instance, depolarization by a mitochondrial uncoupler increases PINK on the mitochondria followed by translocation of Parkin onto the OMM (Narendra and Youle 2011; Scarffe et al. 2014; Springer and Kahle 2011). Moreover, it has been very recently demonstrated that the recruitment of autophagic machinery to the endosome during infection involves a direct interaction between ubiquitin and Atg16L1, and this is independent of LC3-mediated mechanisms (Fujita et al. 2013). Clearly, multiple mechanisms exist, and these involve cargo recognition under different conditions. The LC3-CL pathway may be different from the PINK-Parkin pathway upstream of the damage mechanisms, since the LC3-CL pathway shows no depolarization in mitochondria.
CL may contribute to the formation of autophagosomes by associating with Beclin-1 and LC3. Beclin-1, a mammalian ortholog of Atg 6 in yeast, is also a subunit of the class III phosphatidylinositol 3-kinase (PtdIns3K) complex, which acts at the nucleation stage of autophagosome formation (Klionsky and Schulman 2014; Tooze and Yoshimori 2010). Beclin-1 is activated when phosphorylated on serine 14 (Russell et al. 2013). Interestingly, Beclin-1 preferentially interacts with membranes that are enriched with CL through an evolutionarily conserved domain (ECD) (Huang et al. 2012). Three aromatic amino acids on the tip of the ECD present a hydrophobic finger that may be the binding site (Huang et al. 2012). The ER and mitochondria provide membrane sources for the formation of the phagophore. Since Beclin-1 has a close relationship with mitochondria, it may be the linker that uses the mitochondrial membrane to build the pre-autophagosomal structure. Mitochondrial membranes may be also available for pre-autophagosomal structure elongation, since CL shows a close connection with LC3, which itself has a role in the growth of phagophores (Füllgrabe et al. 2013). Of note, it has been reported that phagophores are formed independently of the LC3-lipidation process in HeLa cells and mouse embryonic fibroblasts (MEFs) deficient in Atg5 or Atg3 (Kishi-Itakura et al. 2014; Uemura et al. 2014); this suggests that there is more than one mechanism involved in the growth of phagophores (Pangborn 1942) .
Oxidation of CL and the Formation of Mitochondrial Membrane Pores
During apoptosis, CL is not only externalized but also oxidized (Bayir et al. 2007; Fernandez et al. 2002; Kagan et al. 2004, 2005). The oxidized form (CLox) may help to form the mitochondrial membrane pore (MMP), which, in turn, sets Cytochrome c (Cyt c) free from the mitochondria (Kuwana et al. 2002). Cyt c in the cytosol binds with apoptotic protease activating factor-1 (Apaf-1), and further binds to pro-caspase-9 to create a protein complex known as the apoptosome. Activated caspase-9 triggers a series of caspase cascades (Li et al. 1997).
Oxidation/Peroxidation of CL under Apoptotic Stimuli
In 1999, Ushmorov and colleagues first reported that the loss of CL in APO-S Jurkat cells, as detected by a decrease in nonyl acridine orange (NAO) fluorescence, may be responsible for Cyt c release upon nitric oxide (NO)-induced apoptosis (Ushmorov et al. 1999). This is the earliest report of an association between CL and Cyt c in apoptosis. In fact, oxidation and peroxidation of CL are the reasons for decreased NAO fluorescence (Mileykovskaya et al. 2001), causing the release of Cyt c. Normally, CL binds about 15%–20% of the Cyt c in the IMM (Kagan et al. 2004). During apoptosis, CL migrates, potently enhancing the opportunities for CL and Cyt c to combine. The complex of Cyt c–CL turns Cyt c into a peroxidase, thereby catalyzing CL oxidation and peroxidation reactions in the presence of hydrogen peroxide (H2O2). H2O2 is an effective oxidizing equivalent feeding the peroxidation process (Kagan et al. 2005). However, CLox has no affinity for Cyt c and thus frees Cyt c from the complex (Orrenius and Zhivotovsky 2005).
CL and Cyt c interact on two sites on Cyt c (Kagan et al. 2009b; Rytömaa and Kinnunen 1994). The most important one is the C-site–a hydrophobic hole on the surface of Cyt c–where a tight combination occurs. One acyl chain of CL inserts into the C-site touching the inner covered heme group (Dickerson et al. 1971; Tuominen et al. 2002). Cyt c is then partially unfolded. This conformational change allows H2O2 access to the heme (Belikova et al. 2006; Choi and Swanson 1995; Tuominen et al. 2002). Nantes’s group proposed that the CL–Cyt c interaction sites were Lys22, Lys25, His26, Lys27, His33, and Lys87 (Kagan et al. 2009a; Kawai et al. 2005). A further study characterized the interaction spots Lys72 and Lys73 as well as the contiguous Lys99 and Lys100 based on an analysis of participants in close proximity to the binding domain (within a 5 Å distance) (Kagan et al. 2009a). A heavy (~350–400 mV) negative shift of the redox potential of Cyt c was detected when bound to CL, which indicated an increased capability for oxidation. The Fe∙∙∙S (Met80) bond was found disrupted upon CL binding, which gave H2O2 access to the heme (Kagan et al. 2009b; Kapralov et al. 2011). H2O2 takes two electrons from Cyt c and turns Cyt c into a reactive enzyme intermediate. Tyr67 of Cyt c turns it into a tyrosyl radical (Tyr•). Cyt c can then take a hydrogen proton and oxidize CL with an extra oxygen molecule (Kapralov et al. 2011).
CL Is Necessary for MMP Formation
Korytowski et al. (2011) showed that CLox species were significantly increased in mitochondria when exposed to apoptotic stress. This might sensitize the mitochondrial membrane for the recruitment of tBid and Bax. Both are quite necessary for MMP formation (Shamas-Din et al. 2013). Bax, a pro-apoptotic member of the Bcl-2 family, is soluble in the cytosol or combines loosely with mitochondria in an inactive manner. During apoptosis, it changes through a conformational transformation, followed by translocation and embedding into the OMM, where it eventually shapes the MMP via oligomerization (Annis et al. 2005; Antonsson et al. 2001; Kroemer et al. 2007; Schinzel et al. 2004; Youle and Strasser 2008). Cleavage of Bid by caspase-8 within a caspase-8–Bid complex produces truncated (t)Bid (Li et al. 1998). It has been suggested that tBid is necessary for Bax oligomerization, probably because it carries CL and Lyso-CL to the OMM.
Sani et al. (2009) established that the N-terminus of Bax (Bax-α1) was able to target mitochondria by combining with CL. In addition, CL-ablated cells showed a strengthened resistance to apoptotic stimuli and a decrease in the expression of Bax–tBid–CL complexes. When assembled into multimers, these complexes in the OMM help to shape the MMP (Lucken-Ardjomande et al. 2008; Sani et al. 2009; Youle and Strasser 2008). Liposomal studies have considered that CL is a prerequisite in Bax–tBid interactions. The bilayer destabilization caused by tBid is facilitated by increase in Ca2+ ion concentration in the mitochondria (Epand et al. 2002). This might partially explain why Ca2+ ions are the most prominent inducers of the MMP. The oxidized CL on the OMM may be appropriate for Bax targeting, whereas CL on the OMM in mitophagy does not combine with Bax and no tBid-Bax-CL complexes are formed. Thus, the oxidation of CL may be the determining factor for apoptosis.
It is generally accepted that Bax activation induces ROS accumulation during apoptosis (Kirkland et al. 2002; Manon 2004). Genetic ablation of Bax/Bak decreased superoxide production and CL (per)-oxidation as induced by actinomycin D. Moreover, in isolated mitochondria, recombinant Bax potentiated CL (per)-oxidation and Cyt c release under succinate stimulus (Jiang et al. 2008). The authors of that study suggested that two processes (generation of ROS and activation of Cyt c into a CL-specific peroxidase controlled by Bax/Bak) are essential for the oxidation of CL. It is possible that, in the early stage of apoptosis, CL is oxidized by Cyt c and externalized. CL, especially CLox on the OMM, recruits Bax. Bax on the OMM in turn promotes the (per)-oxidation of CL by further increasing ROS generation to recruit more Bax to form the MMP (Fig. 3). To the best of our knowledge, the detailed mechanism(s) regarding how Bax/Bak translocation causes ROS and how CL undergoes (per)-oxidation remain undetermined.
Both Bid and tBid have a high-affinity binding domain for CL (Ardail et al. 1990; Gonzalvez et al. 2005; Lutter et al. 2000; Tyurin et al. 2006). MS data have demonstrated that the α4-α6 domain of Bid is the targeting sequence (Kim et al. 2004). Lutter and colleagues (2000) explained that CL and tBid are not a direct ligand-receptor pair, but probably are connected by a peculiar structure. This explanation was raised following the observation that tBid neither bound directly to free CL in vitro nor to liposomes or membranes until the concentration of CL was equal to that of physiological levels (Lutter et al. 2000). Since CL is a linker bridging the gap between mitochondria and apoptosis, this may explain the interaction between CL and the Bcl-2 family (Mileykovskaya et al. 2001).
In addition, mitochondrial fission is necessary in apoptosis. Inhibition of DRP1 by RNA interference or a chemical inhibitor leads to elongated mitochondria and delays their fragmentation, resulting in decreased levels of apoptosis (Cassidy-Stone et al. 2008; Cereghetti et al. 2010; Frank et al. 2001). Moreover, DRP1 may be necessary for Bax oligomerization. It was shown that mutations in DRP1 heavily limited the combination of DRP1 with CL and inhibited Bax oligomerization. A later study showed that DRP1 facilitated Bax oligomerization by driving hemifusion/hemifission intermediate membrane structures (Basañez et al. 2002). At the time of fission, the IMM and OMM are close to each other in the contraction sites and present an obviously enhanced membrane tension. However, the formation of Bax oligomers could mitigate the tightness of the membranes and therefore cause leakage of mitochondrial contents into the cytosol through the newly formed MMP. But, a different opinion has been presented indicating that knocking down DRP1 cannot completely protect the cells against Bax-related apoptosis; therefore, DRP1 may be not indispensable for apoptosis (Estaquier and Arnoult 2007). This may explain why a certain membrane curvature of the mitochondrion may be needed for Bax targeting and Bax oligomerization. Other proteins related to mitochondrial dynamics, such as IRGM and FIS1, may also help to change the membrane conformation that is appropriate for Bax.
Outlook and Perspectives
CL is a unique dimeric phospholipid with four chains composed of different combinations of polyunsaturated fatty acyls, such as oleic acid, linoleic acid, and arachidonic acid. There have been many reports about CL and its roles in the functions of mitochondria. This review highlights the special structural features of CL as well as the properties of CL in mitophagy and apoptosis. It is the diversified combinations of four fatty acyl chains and the dimeric structure that make it flexible and appropriate for interactions with a number of proteins, such as respiratory chain complexes and mitochondrial substrate carriers. In addition, some CL transfers to the OMM during mitophagy and apoptosis, recruiting mitochondria fission proteins. Mitochondria fission occurs both in mitophagy and apoptosis. CL on the OMM also helps to recognize damaged mitochondria by autophagosomes via its combination with LC3 in mitophagy. CL may also have a role in the formation of phagophores through its association with Beclin-1 and LC3. In apoptosis, CL is oxidized by Cyt c at the time of externalization and both of these events are essential for MMP formation. The different characteristics of CL in apoptosis and mitophagy may result from the various associated oxidation reactions.
Distinct species of CL were considered to exist in different tissues, but it is unclear how CL transacylase chooses a certain polyunsaturated fatty acyl chain for a certain tissue. Do CL transporters distinguish CL from the CLox or do both forms share the same carrier(s)? Satisfying explanations need to be explored for these concrete questions. CL-based exploration of diseases remains a promising area for future investigation. CL oxidation has been considered to be an early step in apoptotic cell death. Inhibition of CL oxidation is thus a fascinating treatment strategy that will result in increased resistance to apoptosis. More and more pathophysiological situations, including heart ischemia/reperfusion, heart failure, diabetes, Barth syndrome, as well as aging and age-related cardiovascular and neurodegenerative disorders, are now realized to be related with common elements of abnormal CL content, remodeling, and fatty acyl chain composition. More knowledge about CL may provide us with new and effective ideas to prevent CL-related disorders.
Footnotes
Acknowledgements
This manuscript was edited for proper usage of scientific English by Dr. L. J. Sparvero.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by grant obtained from the