
Abstract
The present study is an initial analysis of whether p53 may function as guardian of the cardiomyocyte mitochondrial genome, with mitochondrial p53 localization proposed to be involved in both mitochondrial DNA (mtDNA) repair and apoptosis. Subcellular distribution, protein levels, and possible function(s) of p53 protein in the response of cardiomyocytes to adriamycin (ADR) were analyzed. Levels and subcellular localization of proteins were determined by Western blot and immunogold ultrastructural analysis techniques. Here we demonstrate that stress caused by ADR induced upregulation of p53 protein in cardio-myocyte mitochondria and nuclei between 3 and 24 hr. Increased expression of PUMA and Bax proteins, pro-apoptotic targets of p53, was documented following ADR treatment and was accompanied by increased levels of apoptotic markers, with elevation of cytosolic cytochrome c at 24 hr and subsequent caspase-3 cleavage at 3 days. Mitochondrial p53 levels correlated with mtDNA oxidative damage. Loss of p53 in knockout mouse heart resulted in a significant increase in mtDNA vulnerability to damage following ADR treatment. Our results suggest that mitochondrial p53 could participate in mtDNA repair as a first response to oxidative damage of cardiomyocyte mtDNA and demonstrate an increase of apoptotic markers as a result of mitochondrial/nuclear p53 localization.
A
It is now well established that the p53 tumor suppressor protein is a transcription factor that plays a central role in preserving nuclear genomic stability by induction of cell cycle arrest to allow DNA repair, with subsequent triggering of apoptosis if repair is not possible. In addition to its role as a transcription factor, p53 has been shown to act directly upon mitochondria via a transcription-independent pathway. It has been demonstrated that, at the onset of apoptosis following hypoxia or DNA damage, a small fraction of p53 translocates to mitochondria, where it interacts with Bcl-XL or Bcl-2, resulting in cytochrome c release and caspase-3 activation (Mihara et al. 2003; Park et al. 2005). However, several recent studies provide evidence that p53 protein can also localize to the mitochondria independent of apoptosis. A role of mitochondrial p53 protein in mito-chondrial base excision repair (mtBER) to maintain genomic stability of mitochondrial DNA (mtDNA) has been documented in mouse liver and cancer cell lines (de Souza-Pinto et al. 2004; Achanta et al. 2005; Chen et al. 2006). In addition, a novel role of p53 in regulation of mitochondrial respiration has been recently demonstrated in human cancer cell lines (Matoba et al. 2006). Therefore, the function of mitochondrial p53 localization in response to cellular stresses is still controversial, and it is unknown whether protein function is cell type- and/or stressor specific. Consequently, it is conceivable that p53 could have different functions in dividing vs non-dividing cells, or even in benign vs malignant cells. Most importantly, these observations suggest that p53 may not only be guardian of the nuclear genome, but of the mitochondrial genome as well.
It has been proposed that activation of the tumor suppressor protein p53, which in turn promotes apoptosis of tumor cells, is a key mechanism of action of antitumor drugs, including ADR (Lowe et al. 1994; Lotem et al. 1996). However, relatively few studies have focused on ADR-induced p53 activation and function in non-dividing and terminally differentiated cardiomyocytes. Recently, a role of p53 in ADR-induced cardiac injury has been documented. Liu et al. (2004) showed that a chemical inhibitor of p53, pifithrin-α, protects against ADR-induced acute cardiotoxicity via reduction of apoptosis. Moreover, targeted disruption of the p53 gene has been shown to attenuate ADR-induced cardiac toxicity (Shizukuda et al. 2005). These studies supported a significant function of p53 in cardiac toxicity associated with ADR. However, it is unknown whether differential subcellular localization of p53 has distinct functions in acute ADR-induced cardiotoxicity, especially because p53 may serve as guardian of the mitochondrial genome, being involved in mitochondrial DNA repair. In the present study we hypothesized that p53 plays a role in cardio-myocyte apoptosis associated with ADR-induced cardiotoxicity. We further postulated that mitochondrial p53 accumulation may be involved in maintaining mitochondrial genomic integrity or apoptosis induction, dependent at least partially on the extent of mtDNA damage and degree of mitochondrial injury.
To initially test our hypotheses, we used Western blot and immunogold ultrastructural analysis techniques with specific antibodies to examine the levels and subcellular localization of p53 protein, selected proteins that regulate apoptosis or mtDNA repair (PUMA, Bax, cytochrome c, active caspase-3, and DNA polymerase γ), and oxidized DNA [8-hydroxy-2′-deoxyguanosine (8-OHdG)] to better understand possible function(s) of p53 subcellular localization in acute ADR-induced cardiotoxicity. Our results document levels and subcellular localization of p53 immunoreactive protein in the cardiomyocyte response to ADR treatment and also indicate that mitochondrial p53 localization may play an additional role in protecting the cardiomyocyte mitochondrial genome in acute ADR-induced cardiotoxicity.
Materials and Methods
Chemicals, Reagents, and Antibodies
ADR was purchased from Pharmacia and Upjohn (Kalamazoo, MI). Anti-p53 polyclonal antibody (FL-393) and anti-Bax polyclonal antibody (N-20) were from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) monoclonal antibody (MAb) was obtained from Advanced Immuno Chemical (Long Beach, CA). Anti-8-OHdG MAb (N45.1) was from Nikken SEIL Corporation (JaICA; Shizuoka, Japan). Anti-cytochrome c MAb (clone 7H8.2C12) and anti-active caspase-3 MAb were purchased from PharMingen (San Diego, CA). Anti-PUMA polyclonal antibody was from Cell Signaling Technology (Danvers, MA). Anti-DNA polymerase gamma (DNA poly γ) polyclonal antibody (Ab-3) was from Lab Vision Corporation (Fremont, CA). All chemicals and reagents were obtained from Sigma Chemical Co. (St Louis, MO), unless otherwise indicated.
Animals and Treatments
All strains of mice used in this study were purchased from The Jackson Laboratory (Bar Harbor, ME) and housed at the animal facility of the University of Kentucky. In the first experimental protocol, males from inbred B6C3 mice (age 10–13 weeks and weighing 22–28 g) were used. In this protocol, mice were randomly injected IP with a single dose of either ADR at 20 mg/kg (n = 2 mice for each time point) or the same volume of saline (2.9% sodium chloride solution, n = 1 mouse for each time point) as a control. Mice were euthanized with an IP injection of 20 mg/kg pentobarbital (Abbott Laboratories; North Chicago, IL) at four different time points (0, 3, 6, and 24 hr), and heart tissues were subsequently collected and processed for further study. In a second study directed toward analyzing apoptosis markers at a later time point, mice as described above were injected IP with either saline (n = 1) or ADR (n = 2) at the same dose as above. After 3 days, mice were euthanized and heart tissues were then collected and processed for further study. For assessment of mtDNA oxidation levels, an additional experiment was performed using WT and p53 homozygous knockout mice [p53KO(-/-)] mice. Generation of p53KO(-/-) mice has been previously described (Jacks et al. 1994), and genotyping was confirmed by PCR analysis of tail DNA (unpublished data). WT and p53KO(-/-) mice in C57BL/6 background were injected IP with either saline (n = 3) or ADR 20 mg/kg (n = 3). Mice were sacrificed 6 hr following ADR treatment. Heart tissues were collected and then processed for immunogold ultrastructural analysis. Experimental protocols are summarized in Table 1.
Western Blot Analysis
Mouse heart tissues from either saline (n = 1) or ADR (n = 2) were homogenized with a lysis buffer containing 50 mM Tris (pH 7.4), 150 mM NaCl, 1% Nonidet P-40, and 1% Triton X-100. A protease inhibitor cocktail and a phosphatase inhibitor cocktail (Sigma Chemical Co.) were added according to the manufacturer's recommendations. Heart homogenates were incubated on ice for 30 min and were then centrifuged at 13,500 × g for 10 min at 4C. The supernatant was transferred, aliquoted, and stored at −80C. Protein concentrations were determined by Bradford protein assay (Bio-Rad Laboratories; Hercules, CA). Tissue homogenates (30–50 μg) were electrophoresed in 10% or 12% SDS polyacrylamide gels and then transferred onto nitrocellulose membranes. After blocking in 5% non-fat dry milk in TBS-Tween 20 for 1 hr at room temperature, the membranes were incubated with primary antibody (anti-p53 antibody diluted 1:200, anti-Bax antibody diluted 1:500, anti-PUMA antibody diluted 1:750, anti-GAPDH diluted 1:1000, and anti-active caspase-3 antibody diluted 1:400) overnight at 4C. Membranes were then incubated with appropriate secondary antibody conjugated to horseradish peroxidase (Bio-Rad Laboratories) diluted 1:3000–1:10,000 in TBS-T for 1.5 hr at room temperature. Protein bands were visualized on X-ray film using the enhanced chemiluminescence system (Pierce Biotechnology; Rockford, IL). Densitometric analysis was carried out using Kodak one-dimensional 3.6 software (Kodak; Rochester, NY). For each set of data, experiments were repeated two to three times, and representative images are shown.
Summary of experimental protocols used in this study

WT, wild type; p53KO(-/-), p53 homozygous knockout mice; ADR, adriamycin; 8-OHdG, [8-hydroxy-2′-deoxyguanosine].
Immunogold Labeling of Immunoreactive Protein
Heart tissues from the left ventricle were cut into 1-mm3 cubes, fixed, embedded, and processed for immunogold electron microscopy as described previously in detail (Oberley 2002), with minor modifications as described below in which the ultrasmall gold and silver enhancement technique (Humbel et al. 1995) was used to optimize the previously described immunogold labeling technique. Two embedded blocks from each heart for each mouse were sectioned and transferred to nickel grids. Grids were rinsed with TBS, blocked with 0.5% BSA-C (Aurion; Wageningen, The Netherlands), and then washed with PBS. Grids were incubated with primary antibodies (anti-p53 diluted 1:60, anti-cytochrome c diluted 1:40, anti-DNA poly γ diluted 1:40, and anti-8-OHdG diluted 1:100) at 4C overnight in a humidified chamber. Grids were washed and then incubated with gold-conjugated secondary antibody [ultrasmall gold-conjugated F(ab')2 fragments of goat anti-rabbit IgG diluted 1:100 (Aurion) or 15-nm gold-conjugated F(ab')2 fragments of goat anti-mouse IgG diluted 1:50 or goat anti-rabbit IgG diluted 1:75 (BB International; Cardiff, UK)] for 90 min at room temperature. Grids were then rinsed in TBS followed by double-distilled H2O. Experimental grids stained with anti-p53 polyclonal antibody and ultrasmall gold-conjugated secondary antibody were then incubated with R-gent SE-EM electron microscope-grade silver enhancement reagent (Aurion) for 20 min to enhance the visualization of gold particles. Grids were counterstained with uranyl acetate, observed, and photographed with an electron microscope (Hitachi H-600; Hitachi, Tokyo, Japan) operated at 75 kV.
In studies using the anti-8-OHdG antibody, the rinsing step, blocking step, and incubation with primary antibody were performed under vacuum to protect against false positive results from the reaction of atmospheric oxygen and DNA in dried tissues (Matsuo et al. 1995).
As a control, normal rabbit or mouse serum (1:1000; Dako, Carpinteria, CA) and antibody diluent (Scy Tek; Logan, UT) were used in place of primary antibody. These controls resulted in trace background labeling; ~0.01 gold beads/μm2 area were identified in nonspecific background labeling.
Quantification of Immunoreactive Protein
For relative quantification of an immunoreactive protein (p53, cytochrome c, DNA poly γ) or 8-OHdG in an experimental group vs a control group, all sections were stained simultaneously under the same conditions. Random sampling was achieved by scanning the grid at low magnification so that immunogold beads could not be seen, yet gross sample artifacts (folds in tissues, dust particles, etc.) could be avoided. Grids were scanned systematically from top to bottom and from left to right, and then photographs of entire cardiomyocyte cells were taken at ×8000 magnification every 8–10 grid fields.
Photographs of 30 cardiomyocyte cells were taken from each group. The area of each compartment (mitochondria, nucleus, and cytoplasm) was outlined and measured by image analysis software (Scion Image Beta 4.02; Scion Corporation, Frederick, MD). Gold beads within specific subcellular compartments were then counted per group. Mean density of gold beads/μm2 area was expressed as mean value ± SEM of 30 cardiomyocyte cells. Data were shown as the ratio of labeling density (ADR/saline). The procedure utilized for quantification has been used in several studies to estimate amount of protein of interest in specific subcellular compartments (Chaiswing et al. 2004; Lucocq et al. 2004; Mayhew et al. 2004).
For analysis of cytochrome c, data were also studied in a different fashion. Results from quantification of immuno-reactive protein, expressed as a mean value of 30 cardio-myocytes, will be misleading because the sampled population of cells is biochemically heterogeneous, consisting of normal, adapted, reversibly, and/or irreversibly injured cells. Data were therefore analyzed to determine the number of individual cardiomyocytes with significant mitochondrial release of cytochrome c. Labeling density (gold beads/μm2) of an individual cardiomyocyte was compared with the average density obtained from analysis of 30 cardiomyocytes. Individual cardiomyocytes showing both a significant decrease in levels of mitochondrial cytochrome c and an increase in cytosolic cytochrome c protein levels when compared with average values (mean ± SEM) from 30 cardiomyocytes in each subcellular compartment were considered to be cardiomyocytes with significant release of cytochrome c from mitochondria. In each case, significance was defined as a data point lesser or greater than 1 SEM from the average value. The number of cardiomyocytes fulfilling these criteria were counted, and data were expressed as the percentage of cardiomyocytes with release of mitochondrial cytochrome c and then a relative number obtained by determining the ratio of ADR/saline. The relative number data shown in Results contain a single number/group/time point; thus, statistical analysis was not performed.
Antibody Specificity
Specificity of anti-p53 antibody and anti-cytochrome c antibody used in these experiments was verified by testing specific binding of the antibody with purified protein. Purified p53 and cytochrome c protein were purchased from Santa Cruz Biotechnology and Sigma Chemical Co., respectively. Various concentrations of purified protein and 50 μg of mouse tissue homogenate (heart and kidneys) were electrophoresed in 12% SDS polyacrylamide gels, transferred onto nitrocellulose membranes, and then Western blot analysis was performed as described above. Results demonstrated a specific band of p53 at a molecular mass of 53 kDa and cytochrome c at a molecular mass of 15 kDa (data not shown). Specificity of p53 antibody was additionally verified using human colon carcinoma cell lines with WT (HCT116 p53+/+) and KO of p53 (HCT116 p53−/-). Results demonstrated a specific band of p53 in HCT116 p53+/+ cells, whereas no band was observed in HCT116 p53−/- cells (data not shown). For other antibodies, known positive control lysates were used for verification of antibody specificity.
Specificity of anti-8-OHdG MAb was tested by ELISA technique as previously described (Toyokuni et al. 1997). The antibody recognized both modified base and deoxyribose structure of 8-OHdG but did not cross-react with the original four deoxyribonucleosides or other DNA base-modified products such as 8-hydroxy-2′-deoxyadenosine and O6-methyl-2′-deoxyguanosine or urine components. In addition, this antibody has been used to determine oxidized DNA bases in specific sequences of single genes using a whole human genome approach (Akatsuka et al. 2006).
Statistical Analysis
Statistical evaluation was performed with SPSS11 for Windows (SPSS Inc.; Chicago, IL). For comparison between ADR-treated mice vs control mice, independent Student's t-test was utilized.
The differences of ratio values between time points (0 hr vs other time points) were compared using bootstrap analysis. Statistical evaluations were performed by the R project for statistical computing software (www.r-project.org). Standard errors for ratio data were calculated using propagation of error theory; p<0.01 was considered significant as indicated in Results and as shown in figures.
Results
ADR Induced an Increase of p53 Immunoreactive Protein in Various Cardiomyocyte Compartments
p53 protein is a primary mediator of the cellular stress response induced by genotoxic and oxidative stress-inducing agents. To determine whether ADR induced upregulation of p53 protein in cardiomyocytes, Western blot analysis of p53 protein was performed. As demonstrated in Figures 1A and 1B, p53 protein levels were increased as early as 3 hr in ADR-treated cardiomyocytes, and increased levels were maintained throughout the time course analysis following ADR treatment (3, 6, and 24 hr).
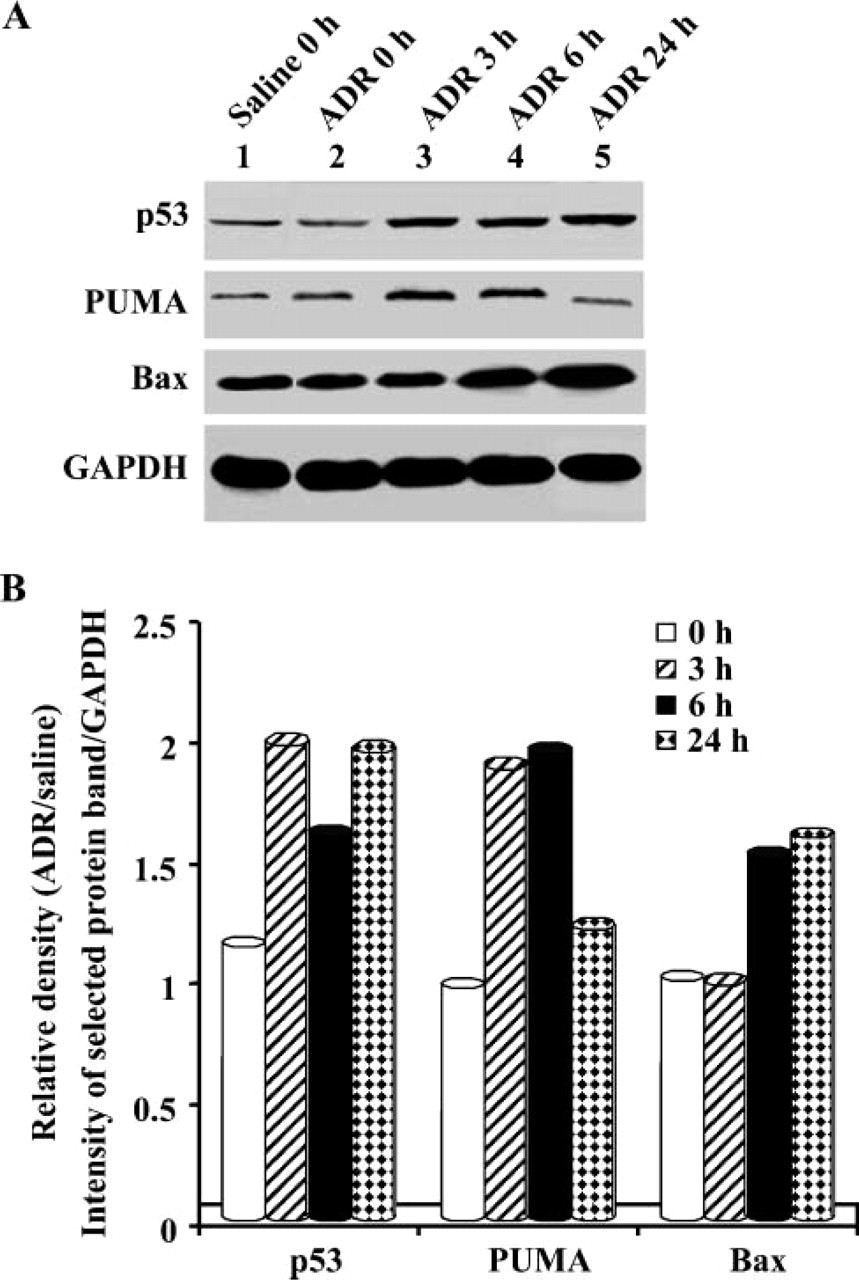
Effect of adriamycin (ADR) on the expression of p53, PUMA, and Bax immunoreactive protein. Mice were treated with either saline or ADR and then sacrificed at 0, 3, 6, and 24 hr following treatment, as indicated. (A) Western blot analysis was performed on 40 μg of heart tissue homogenate with antibody against p53, PUMA, and Bax protein. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a protein loading control. (B) Densitometric analysis of p53, PUMA, and Bax protein expression in cardiomyocytes following treatment. Density of selected protein bands was measured relative to density of GAPDH. Data were then expressed as the relative density of ADR at each time point to saline at 0 hr.
We further analyzed the level and localization of p53 protein in each subcellular compartment including nuclei, mitochondria, and cytoplasm (composed largely of myofilaments) using immunogold ultrastructural analysis. As shown in Figure 2A, gold beads representing p53 protein in cardiomyocytes were found in small numbers in all subcellular compartments of control mice (A' and C' in Figure 2A), whereas significantly increased labeling was observed over nuclei, mitochondria, and myofilaments after ADR treatment (B' and D' in Figure 2A).
Quantitative analysis of p53 protein gold labeling density ratio data (ADR/saline) in each subcellular compartment of cardiomyocytes is shown in Figure 2B. p53 protein levels were significantly increased in cardio-myocyte nuclei at every time point examined following ADR treatment, whereas p53 protein levels in cardiomyocyte mitochondria and cytoplasm were significantly elevated at 3 and 24 hr after treatment with ADR. These results indicated an increase in levels of p53 protein in all subcellular compartments of cardiomyocytes as part of the response to stress caused by ADR.
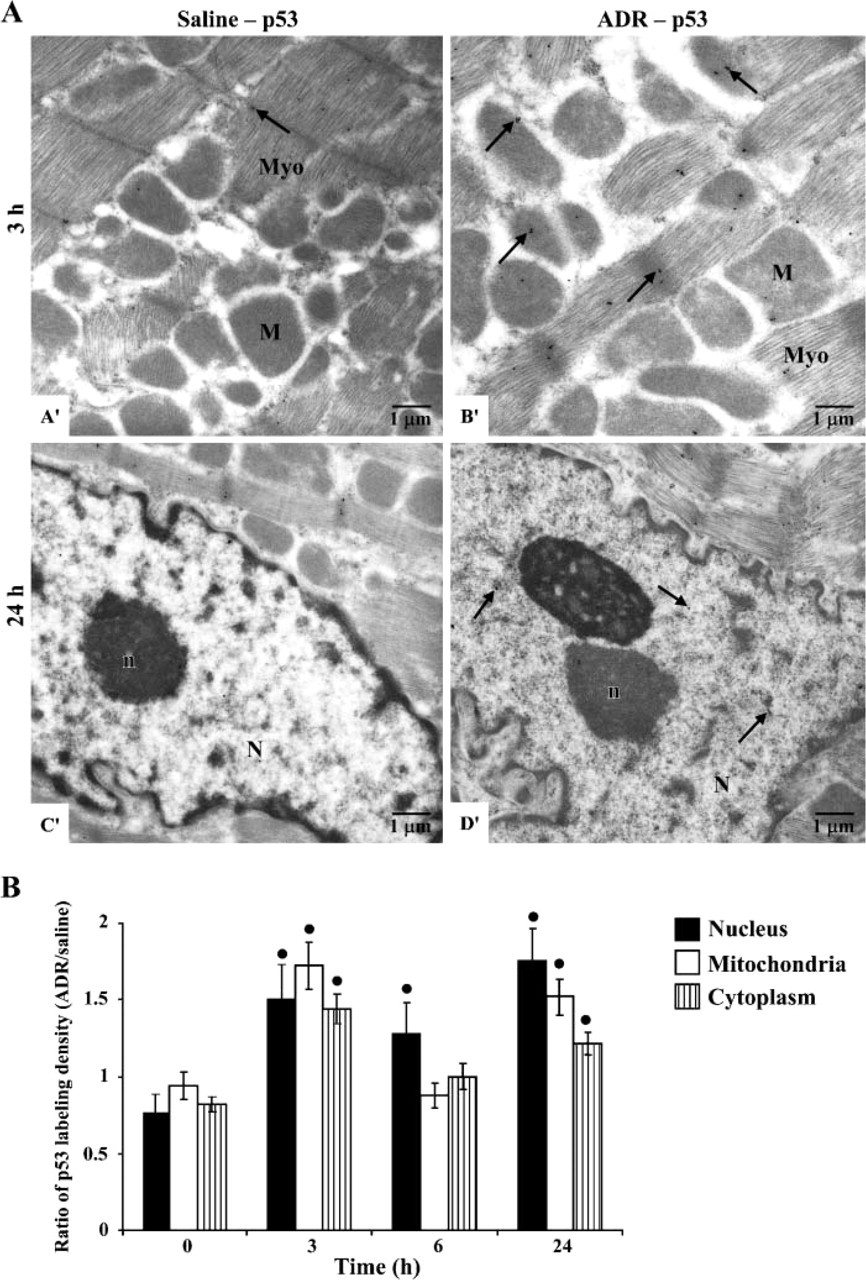
Effect of ADR on subcellular localization of p53 immunoreactive protein. Mice were treated with either saline or ADR and then sacrificed at 0, 3, 6, and 24 hr following treatment, as indicated. Cardiac tissues were prepared for immunogold ultrastructural analysis. (A) Representative immunogold electron microscope photographs using antibody against p53 protein in cardiomyocytes (×10,000). Electrondense beads indicate positive staining for p53 (arrows). Left ventricular tissues from mice treated with saline demonstrated low labeling of p53 protein (A', C') in all subcellular compartments. Mice treated with 20 mg/kg ADR showed significantly increased labeling of p53 protein (B', D') in the matrix and membrane of mitochondria (M), myofilaments (Myo), nucleolus (n), and nucleus (N). (B) Quantitative analysis of p53 immunoreactive protein in each subcellular compartment of mouse cardiomyocytes. p53 protein levels in mitochondria and nuclei were measured as density labeling (gold beads/μm2). Data shown present ratio of labeling density (ADR/saline). ∗p < 0.01 when compared with 0 hr.
Increased Expression of Pro-apoptotic Proteins in Cardiomyocytes Following ADR Administration
We further sought to examine whether increased p53 protein levels were involved in ADR-induced cardiac toxicity and apoptosis. A major molecular property of p53 is that of a transcription factor. Once activated, p53 in the nucleus binds to DNA in a sequence-specific manner to activate its downstream targets. Many proapoptotic proteins including PUMA and Bax have been shown to be targets of p53. To determine whether an increase of p53 protein following ADR treatment resulted in the induction of pro-apoptotic target genes, Western blot analysis of PUMA and Bax was performed. PUMA protein levels were upregulated at 3 and 6 hr, whereas an increase in Bax expression was observed at 6 and 24 hr following ADR treatment (Figures 1A and 1B).
Mitochondrial Cytochrome c Protein Release and Caspase-3 Activation in Cardiomyocytes Following ADR Treatment
Release of cytochrome c from the mitochondrial intermembrane space into the cytosol is a downstream event allowing evaluation of apoptotic cell death. To assess whether the identified subcellular increase of p53 levels and upregulation of pro-apoptotic proteins observed results in cytochrome c release from cardiomyocyte mitochondria, subcellular localization of cytochrome c in mitochondria and cytoplasm was assessed by immunogold ultrastructural analysis. The relative number of cardiomyocytes with release of mitochondrial cytochrome c was further analyzed as described in Materials and Methods. The relative number of cardiomyocytes with release of mitochondrial cytochrome c was increased ~5-fold at 24 hr after ADR treatment (Figure 3A).
Release of cytochrome c from mitochondria followed by activation of the intrinsic caspase cascade is a characteristic feature of the mitochondrial cell death pathway. To determine whether cytochrome c release from cardiomyocyte mitochondria following ADR treatment resulted in caspase activation and apoptosis, Western blot analysis of active caspase-3 was performed. Using antibody specific for the active form of caspase-3, we were able to detect active caspase-3 protein at 3 days following ADR treatment (Figure 3B). These results indicated that an apoptotic process occurred in cardiomyocytes following ADR treatment.
Acute ADR Treatment Resulted in Preferential Oxidation of Cardiomyocyte mtDNA Compared With Nuclear DNA
DNA damage and formation of reactive oxygen species (ROS) are a result of the cytotoxicity of ADR, and p53 is known to play a central role in maintaining genomic stability. To determine whether the observed increase in p53 protein levels was due to DNA damage induced by ADR, levels of 8-OHdG, a marker of oxidative DNA damage, in mitochondria and nuclei were measured by immunogold ultrastructural analysis utilizing a specific antibody. As shown in Figure 4A, gold beads representing 8-OHdG labeling were present in only very low numbers in mitochondria (A' in Figure 4) and nuclei of control cardiomyocytes examined (C' in Figure 4), whereas a significant increase in labeling was observed in mitochondria (B' in Figure 4) but not nuclei (D' in Figure 4) of ADR-treated cardiomyocytes. Nonspecific labeling was observed in cytoplasm.
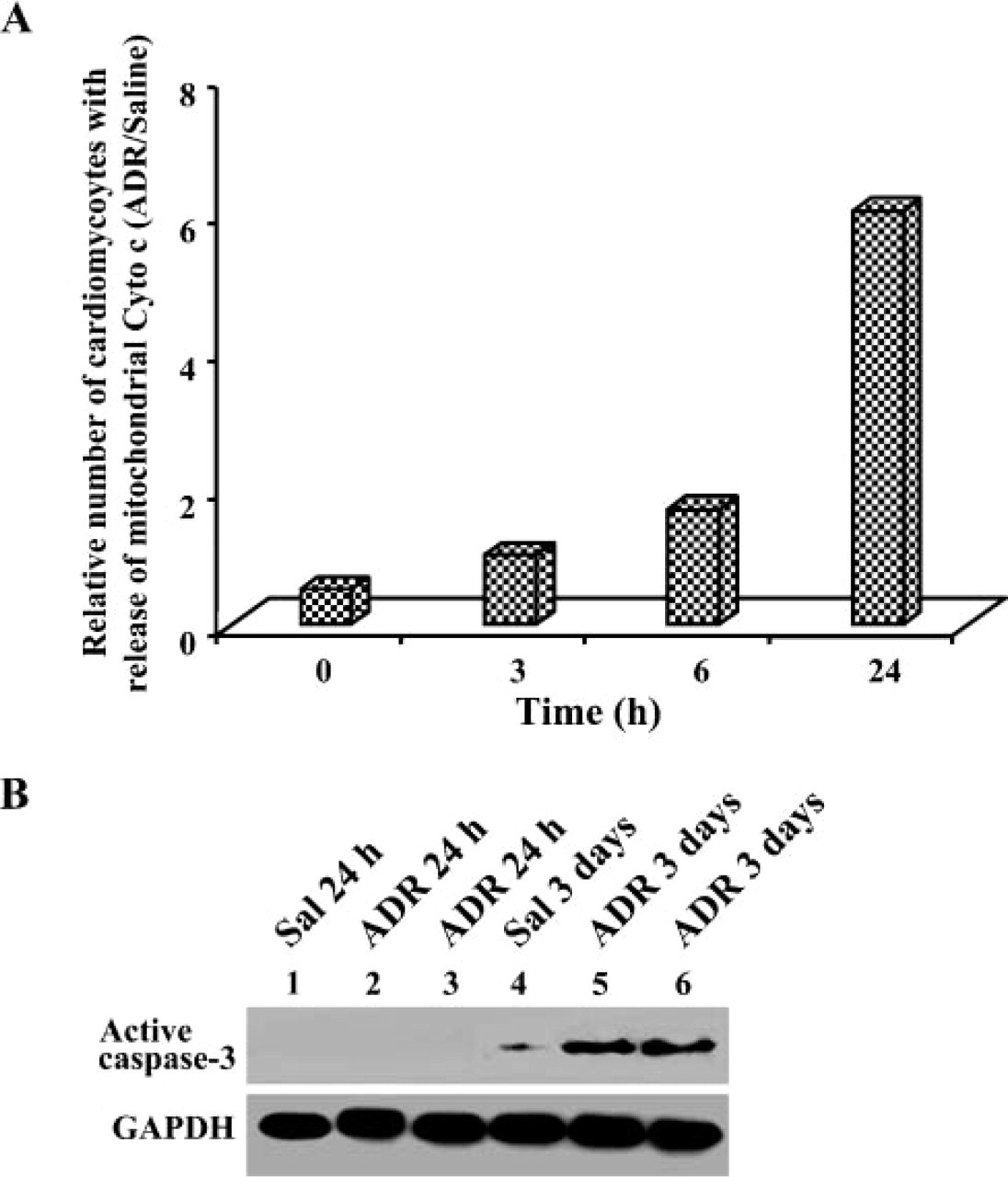
Evaluation of apoptotic markers in cardiomyocytes following ADR treatment. Mice were treated with either saline or ADR and then sacrificed at 0, 3, 6, and 24 hr following treatment, as indicated. Cardiac tissues were prepared for immunogold ultrastructural analysis. (A) Number of cardiomyocytes with significant release of mitochondrial cytochrome c after treatment with ADR. Data were analyzed as indicated in Materials and Methods. Data shown present relative number of cells with release of mitochondrial cytochrome c (ADR/saline). (B) Western blot analysis of active caspase-3. In addition to mice described above, additional mice were treated with either saline (Sal) or ADR (20 mg/kg) and then sacrificed at 3 days following treatment. Western blot analysis was performed on 50 μg of heart tissue homogenate with antibody against active caspase-3. GAPDH was used as a protein loading control. Representative image is shown analyzing active caspase-3 at 24 hr and 3 days following treatment.
Quantitative analysis of 8-OHdG gold-labeling density ratio data (ADR/saline) in each subcellular compartment demonstrated that 8-OHdG levels in mitochondria were significantly increased at 3 hr after ADR treatment, whereas a slight but not statistically significant increase of 8-OHdG was observed in cardiomyocyte mitochondria at 24 hr. In contrast, 8-OHdG levels in nuclei did not show any significant change after treatment with ADR (Figure 4B). These results indicated that within the first 24 hr ADR treatment resulted in mtDNA, not nuclear DNA (nDNA) damage. A decrease of 8-OHdG to steady-state levels at 6 hr from a peak at 3 hr suggests that mitochondrial DNA repair may have occurred.
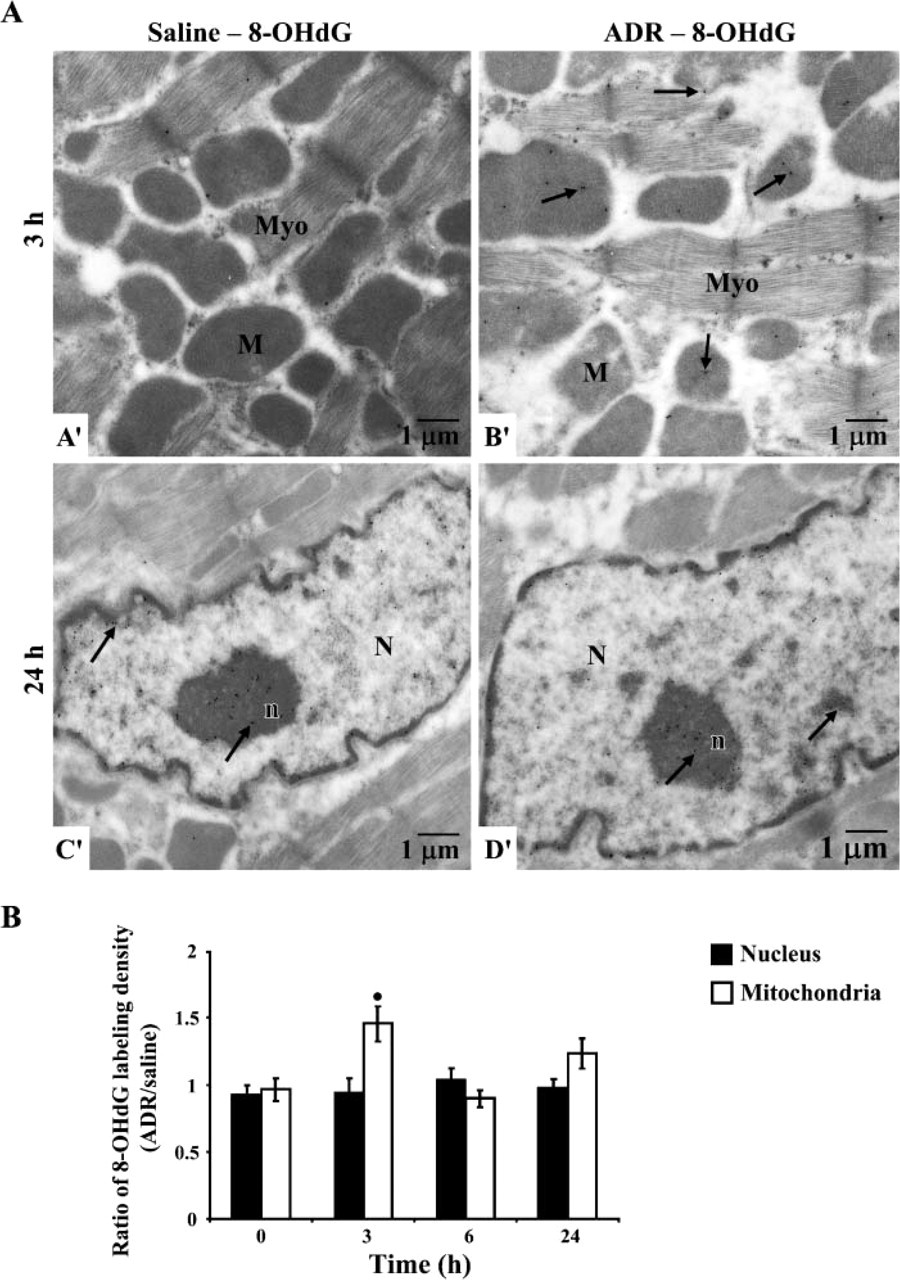
DNA oxidation in ADR-treated cardiomyocytes. Mice were treated with either saline or ADR and then sacrificed at 0, 3, 6, and 24 hr following treatment, as indicated. Cardiac tissues were prepared for immunogold ultrastructural analysis. (A) Representative immunogold electron microscope photographs using antibody against [8-hydroxy-2′-deoxyguanosine] (8-OHdG) in mouse cardiomyocytes (×10,000). Electron-dense beads indicate positive staining for 8-OHdG (arrows). Left ventricular tissues from mice treated with saline demonstrated low labeling of 8-OHdG in mitochondria (M) (A') and nucleus (N) (C'). Mice treated with ADR showed significant labeling of 8-OHdG in mitochondria (B') without increase in nuclei (D'). (B) Quantitative analysis of 8-OHdG in selected subcellular compartments of mouse cardiomyocytes. Levels of 8-OHdG in mitochondria and nuclei were measured as density labeling (gold beads/μm2). Data shown present ratio of labeling density (ADR/saline). ∗ p<0.01 when compared with 0 hr.
Distribution of p53 in Mitochondria Correlates With Levels of Damaged mtDNA
Levels of mitochondrial 8-OHdG observed following ADR treatment paralleled levels of mitochondrial p53. To determine whether mitochondrial p53 accumulation was attributable to mtDNA damage caused by ADR, correlation between levels of mitochondrial 8-OHdG and p53 protein in mitochondria was performed by linear regression analysis. As shown in Figure 5A, correlation analysis clearly demonstrated a direct relationship between levels of mitochondrial 8-OHdG and p53 protein in mitochondria throughout the time course of ADR treatment (R = 0.987, p = 0.013). These observations also support recent evidence, using confocal microscopy techniques, that stress occurring in mitochondria activates p53 mitochondrial migration (Zhao et al. 2005).
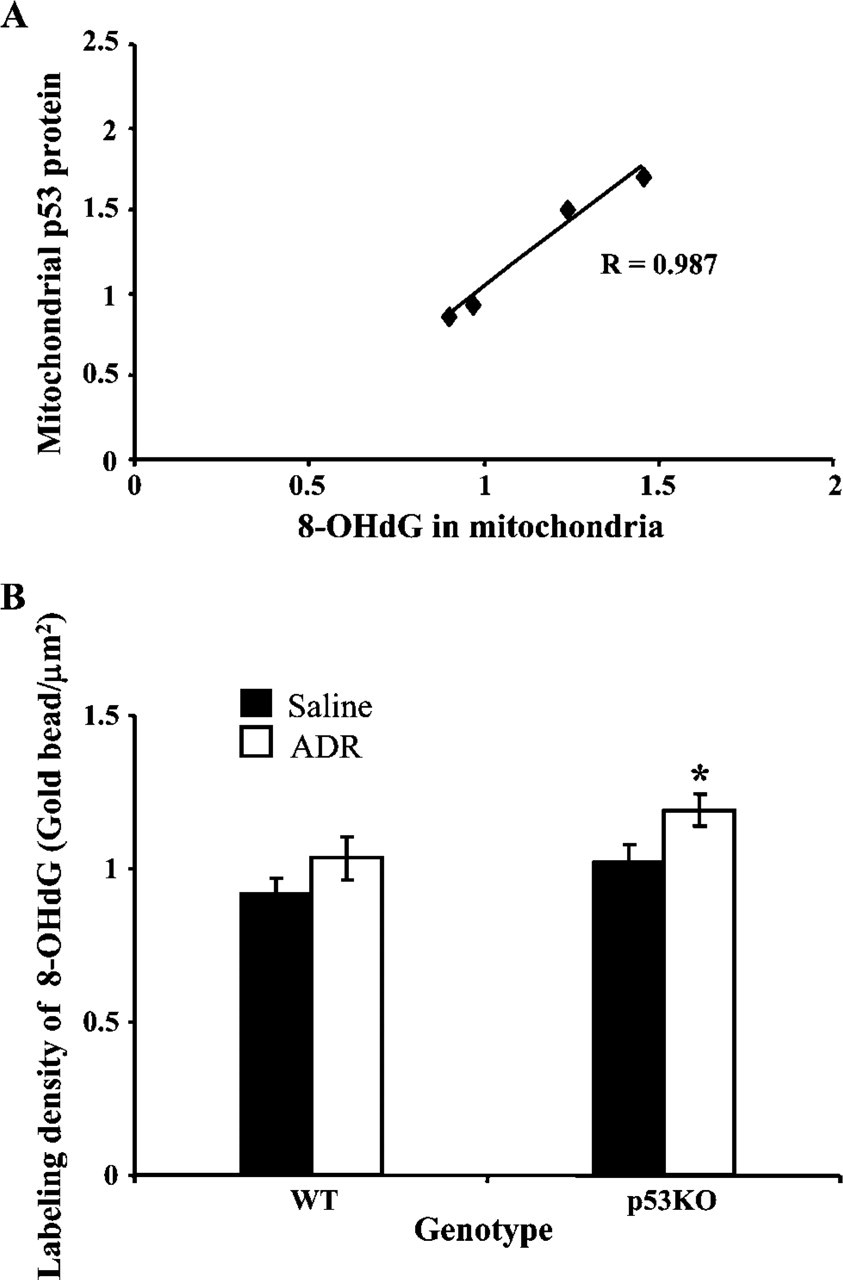
Analysis of mitochondrial 8-OHdG. (A) Correlation coefficient analysis of levels of 8-OHdG in mitochondria and mitochondrial p53 following ADR treatment. (B) mtDNA oxidation in p53 homozygous knockout mice [p53KO(-/-)] compared with wild-type (WT) mice. WT and p53KO(-/-) mice were treated with either saline or ADR (20 mg/kg) and then sacrificed at 6 hr following treatment. Cardiac tissues were prepared for immunogold ultrastructural analysis. 8-OHdG in mitochondria was measured and expressed as density labeling (gold beads/μm2). ∗ p<0.01 when compared with saline control.
p53KO(-/-) Mice Are Sensitive to mtDNA Oxidation Following ADR Administration
To determine the effect of p53 on cardiomyocyte mtDNA damage following ADR treatment, 8-OHdG immunogold ultrastructural analysis was performed on wild-type (WT) and p53KO(-/-) mice at 6 hr following ADR treatment. We selected this time point because Figure 4B showed that 8-OHdG in mitochondria returned to baseline levels at 6 hr in WT mice after an initial increase following ADR treatment. As shown in Figure 5B, levels of 8-OHdG were significantly increased in cardiomyocyte mitochondria of p53KO(-/-) mice treated with ADR when compared with control, whereas mitochondrial 8-OHdG levels were not significantly different in WT mice. These results indicated that mtDNA was more susceptible to oxidative stress caused by ADR in p53-deficient cardiomyocytes.
Discussion
ADR has been shown to cause cardiomyocyte apoptosis in several studies (Childs et al. 2002; Wu et al. 2002; Liu et al. 2004; Lien et al. 2006), a process mediated by the p53 tumor suppressor protein. Our present study utilized immunogold ultrastructural analysis techniques to determine levels of molecules of interest in particular subcellular compartments in vivo, allowing analysis of levels and subcellular localization of p53 protein in the acute ADR-induced cardiotoxicity model. Measuring levels and subcellular distribution of p53 protein as a function of time provided important evidence for novel p53 protein functions in the cardiomyocyte stress response to ADR. Our results suggested that mitochondrial p53 accumulation occurring as an early event may participate in mtDNA repair as a rapid adaptive response to oxidative stress in cardiomyocyte mitochondria, whereas nuclear p53 is more likely to play a major role in cardiac toxicity associated with ADR.
p53 protein is constantly produced but kept at a very low level in unstressed cells by MDM2 protein, targeting p53 for degradation by proteasomes. Western blot and immunogold ultrastructural analysis techniques demonstrated that treatment with ADR resulted in a rapid increase in levels of p53 immunoreactive protein. Additionally, data obtained from immunogold electron microscopy indicated an increase of p53 localization in all subcellular compartments of cardiomyocytes within the first 24 hr following ADR administration, without significant increase and alterations of p53 subcellular localization in endothelial cells. These results strongly suggest that p53 may be a major mediator in the cardiomyocyte stress response to ADR.
Apoptosis mediated by p53-dependent transcriptional activation of its target genes is well established. Although our present study demonstrated an increase in expression of PUMA and Bax protein (Figure 1), which are known to be p53 targets (Miyashita and Reed 1995; Nakano and Vousden 2001), it is uncertain whether upregulation of these pro-apoptotic proteins in our studies is p53 dependent because PUMA has been documented to induce apoptosis under certain conditions, independent of p53 (Jeffers et al. 2003). A previous study demonstrated that a chemical inhibitor of p53, pifithrin-α, prevented an increase in expression of Bax protein and protected against acute ADR-induced cardiomyocyte apoptosis (Liu et al. 2004), suggesting that transcription activation of p53 and Bax expression play a role in apoptosis induction of cardiomyocytes following ADR treatment. However, it has been reported that Bax alone was not sufficient for p53-induced apoptosis and required PUMA activation; PUMA is necessary for Bax conformational change and mitochondrial translocation (Liu et al. 2003). These combined observations suggest the possibility that these proapoptotic proteins may act in a coordinated fashion in the induction of cardiomyocyte apoptosis and may at least partially require p53 protein activation. Nevertheless, a study using p53 KO mice has shown improved cardiac functions with attenuation of cardiomyocyte apoptosis, supporting a significant function of p53 protein in ADR-induced cardiotoxicity.
Earlier studies suggested that p53 may have an apoptotic function separate from its regulation of gene expression (Caelles et al. 1994; Haupt et al. 1995). A more recent study has revealed a function of p53 outside the nucleus as a binding partner of anti-apoptotic members of the Bcl-2 family, including Bcl-2 and Bcl-XL (Erster and Moll 2005). In addition, Chipuk et al. (2005) demonstrated coordination of cytoplasmic p53 and the pro-apoptotic proteins PUMA and Bax in induction of mitochondrial permeabilization and apoptosis. Our present study demonstrated that p53 protein also accumulated in the cytoplasm of cardiomyocytes in response to ADR (Figure 2B). It is possible that cyto-plasmic p53 may be a part of apoptotic function by coupling with other p53 pro-apoptotic targets and amplifying the transcription-dependent apoptotic signals generated by nuclear p53 in cardiomyocytes. Alternatively, observation of cytoplasmic p53 may only relate to its transit during stress-induced subcellular redistribution. These possibilities need to be further analyzed.
The exact function of p53 in mitochondria remains unclear because mitochondrial p53 localization has been shown to be involved in both mitochondrialdriven apoptosis and maintainance of mitochondrial genomic stability (Marchenko et al. 2000; Mihara et al. 2003; de Souza-Pinto et al. 2004; Achanta et al. 2005; Chen et al. 2006). Our results demonstrated that p53 protein levels in mitochondria were significantly increased at 3 and 24 hr following ADR treatment (Figure 2B). However, the number of cardiomyocytes undergoing apoptosis was not increased until 24 hr, and activation of caspase-3 occurred at 3 days following ADR treatment (Figures 3A and 3B). These results raise questions regarding the function of p53 localization at early time points because it has been demonstrated that a fraction of p53 protein translocates to mitochondria at the onset of p53-dependent apoptosis (Marchenko et al. 2000). One possible explanation for data presented here is that the level of mitochondrial p53 induction was lower than the threshold for induction of apoptosis. Alternatively, it is possible that mitochondrial p53 accumulation observed at early time points may not be involved in the induction of apoptosis. We postulated that mitochondrial p53 may function similar to p53 in nuclei, playing a primary role in protecting mitochondrial genomic DNA, but leading to apoptosis induction if repair is impossible.
Documentation that p53 is present in mitochondria of normal unstressed cells (Mahyar-Roemer et al. 2004) and identification of a p53 binding motif within the mitochondrial 16S rDNA region (Heyne et al. 2004) implied that p53 may have a physiological role in cells. The observed increased p53 mitochondrial levels in cardiomyocytes following ADR treatment also suggest a role of mitochondrial p53 in response to cellular stress. A previous study demonstrated that oxidative stress occurs rapidly after ADR treatment (Chaiswing et al. 2004), and oxidative stress is known to be an activator of p53 (von Harsdorf et al. 1999), possibly due to oxidative DNA damage. Our results demonstrated that ADR treatment resulted in significant mtDNA oxidation at 3 hr following ADR treatment, but not nDNA oxidation (Figure 4B), suggesting that mtDNA is an early target for oxidative damage caused by ADR. It is not certain if the absence of detectable oxidative damage in nDNA is due to the actual absence of oxidation of DNA in the nucleus by ADR or the presence of abundant DNA repair enzymes in the nucleus, allowing rapid repair of oxidized nuclear DNA. mtDNA is more vulnerable to damage than nDNA due, in part, to its proximity to the respiratory chain, which is an important source of ROS production, lack of histone protection, and limited DNA repair capacity (Marcelino and Thilly 1999; Kang and Hamasaki 2002). Remarkably, the observed increase in 8-OHdG levels in cardiomyocyte mitochondria at 3 hr following ADR treatment occurred at the same time as mitochondrial p53 accumulation in mitochondria. In addition, regression analysis strongly supports the link between mtDNA damage and mitochondrial p53 localization (Figure 5A). These results suggest the possibility that p53 may have a primary role in maintenance of genomic stability. Consistent with this hypothesis is the fact that levels of oxidized DNA in mitochondria decreased to baseline levels at 6 hr following ADR treatment. Notably, we found a significant change in mitochondrial number and size between 0 and 6 hr, in which mitochondrial number was decreased at 3 hr, the same time as an increase of mitochondrial size was observed. These changes returned to basal state at 6 hr following ADR treatment (data not shown). Changes in number and size of mitochondria have been shown to be involved in mitochondrial turnover, a process related to mitochondrial degradation and renewal (Terman and Brunk 2004).
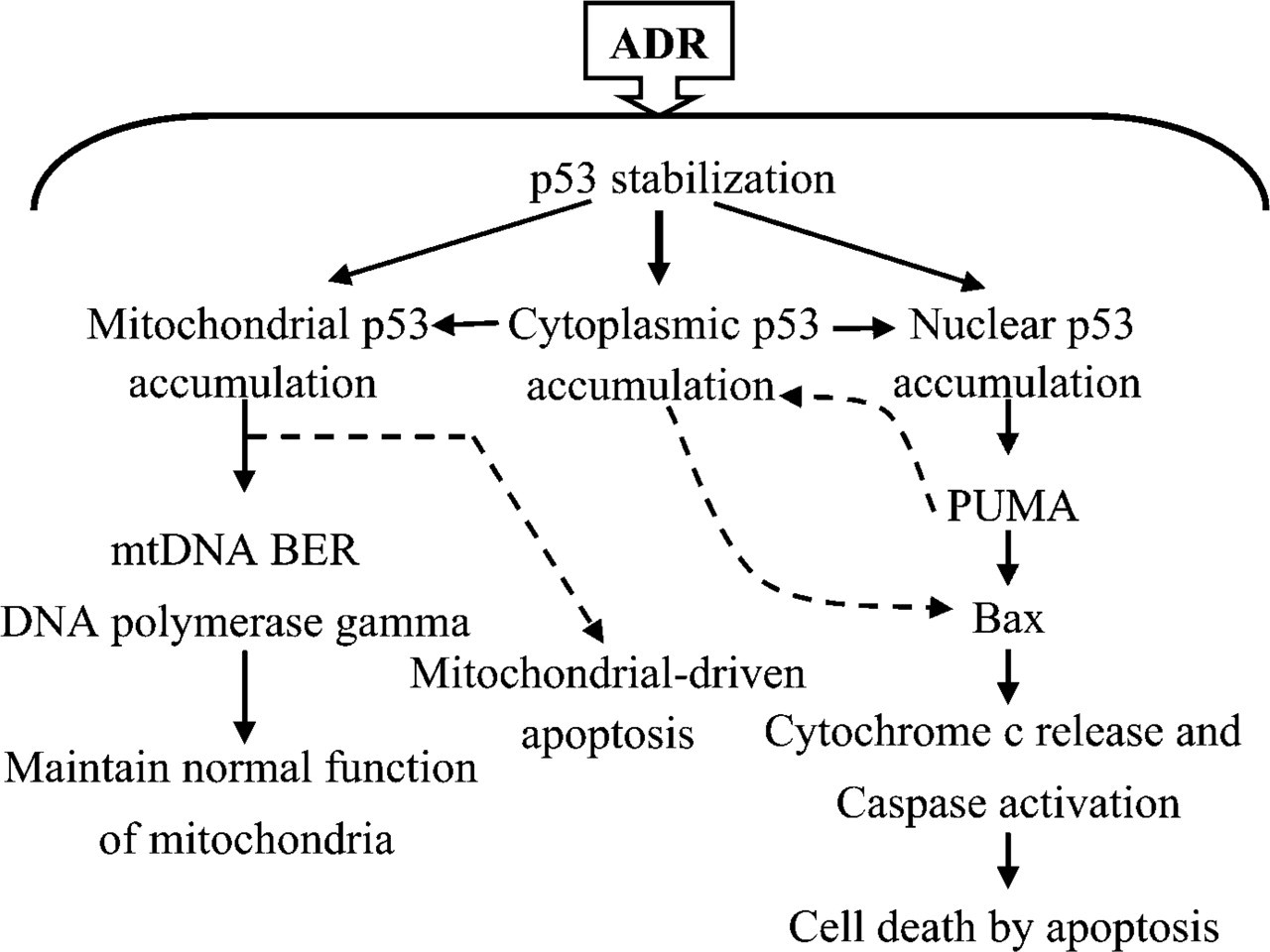
Schematic illustration showing the proposed role of p53 in ADR-induced cardiotoxicity. ADR treatment causes free radical production and stress resulting in p53 stabilization in response to stress. p53 localization to mitochondria is postulated to be triggered by mitochondrial DNA damage and may be involved in mitochondrial base excision repair. Expression of p53 in nucleus is hypothesized to regulate the expression of pro-apoptotic proteins such as PUMA and Bax, resulting in induction of apoptosis. p53 protein expression in the cytoplasm may relate to its transit during stress-induced redistribution or may be a part of apoptotic function that is coupled to other pro-apoptotic proteins in the cell.
The direct effect of p53 in protecting mitochondrial genomic stability was confirmed by examination of oxidized DNA (8-OHdG) levels in cardiomyocyte mitochondria from p53KO(-/-) compared with WT mice at 6 hr following ADR treatment (a time point at which repair processes presumably have already occurred). We found that mtDNA was more vulnerable to oxidative damage caused by ADR, in the absence of p53 (Figure 5B). These results are consistent with a previous study demonstrating that mtDNA was more susceptible to oxidative damage in p53-deficient cells after treatment with ethidium bromide (Achanta et al. 2005). These results support our hypothesis that p53 may play an additional role in cardiomyocyte mitochondria and is most likely involved in mtDNA repair. In addition, we found a slight elevation of DNA polymerase γ, a DNA polymerase enzyme in animal mitochondria, which is necessarily implicated in mtDNA base excision repair in cardiomyocyte mitochondria at 3 hr following ADR treatment (data not shown). Although we have not measured DNA poly γ activity, an increase of DNA poly γ activity has been documented in the hearts of ADR-treated rats in another study (Ogihara et al. 2002).
In summary, we documented the subcellular localization of p53 in acute ADR-induced cardiotoxicity and proposed that p53 may play an additional role in non-dividing cardiomyocytes in response to ADR, as opposed to responses documented in dividing cells (Figure 6). Cardiomyocytes are postmitotic and terminally differentiated cells that contain a large number of mitochondria for their high-energy demand and function. It is important for these specialized cells to maintain normal mitochondrial function because they are unable to regenerate to repair injury in response to a cell death signal. Persistent mtDNA damage would lead to respiratory chain dysfunction and cell death, a notion supported by a previous study in human hearts following ADR administration (Lebrecht et al. 2005). We propose that mtDNA, which is a target of oxidative stress caused by ADR, induces p53 localization to mitochondria and may participate in modulation of mtDNA repair as a protective adaptive response to maintain mitochondrial function in cardiomyocytes. However, the limited DNA repair capacity in mitochondria and p53-mediated induction of several pro-apoptotic proteins may ultimately result in ADR-induced cardio-toxicity and cardiomyocyte apoptosis. Therefore, the present study provides evidence for a novel role of p53 in cardiomyocyte response to ADR and presents new insights in understanding the mechanism(s) of ADR-induced cardiac toxicity.
Footnotes
Acknowledgements
This work was supported in part by grants from the National Institutes of Health (Grant #CA-94853 to DSC and TO) and from the Thailand Research Fund under the Royal Golden Jubilee program (to RN). The work was supported in part by resources and the use of facilities at the William S. Middleton Memorial Veterans Hospital, Madison WI.
We are grateful to Dr. Larry W. Oberley for critical review of this manuscript.