
Abstract
Focal adhesion kinase (FAK) is a protein tyrosine kinase that regulates cellular adhesion, motility, proliferation and survival in various types of cells. Interestingly, FAK is activated and/or overexpressed in advanced cancers, and promotes cancer progression and metastasis. For this reason, FAK became a potential therapeutic target in cancer, and small molecule FAK inhibitors have been developed and are being tested in clinical phase trials. These inhibitors have demonstrated to be effective by inducing tumor cell apoptosis in addition to reducing metastasis and angiogenesis. Furthermore, several genetic FAK mouse models have made advancements in understanding the specific role of FAK both in tumors and in the tumor environment. In this review, we discuss FAK inhibitors as well as genetic mouse models to provide mechanistic insights into FAK signaling and its potential in cancer therapy.
Introduction
Focal adhesion kinase (FAK) serves as a fundamental intracellular mediator of extracellular changes, such as extracellular matrix (ECM) remodeling, nutrient availability, and growth factors (Fletcher and Mullins 2010). FAK is a non-receptor protein tyrosine kinase that is activated by interactions with integrins, growth factor receptors, G protein-coupled receptors, and cytokine receptors (Fig. 1) (Lim et al. 2010; Mitra et al. 2005), and it is known to have a pivotal role in the regulation of cell adhesion, motility, proliferation, and survival in many cell types (Peng and Guan 2011).
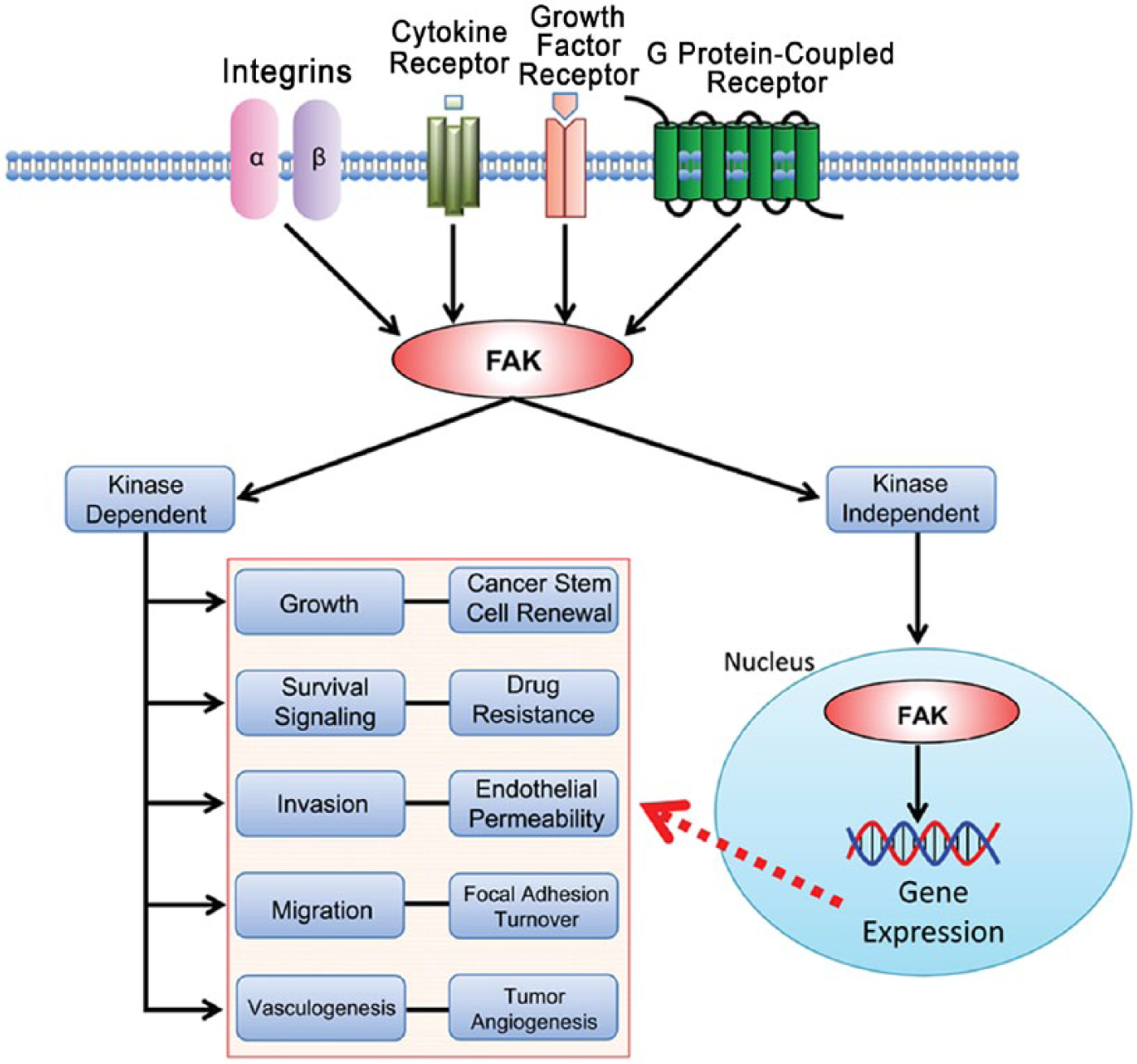
FAK signaling pathways in cancer. Integrins, cytokine receptors (CR), growth factor receptors (GFR), and G protein-coupled receptors (GPCR) are cell-surface transmembrane receptors that relay extracellular signals to FAK. FAK activation triggers subsequent signaling cascades in various cell processes: survival signaling, growth, angiogenesis, migration, and invasion. In cancers, FAK is overexpressed and activated and it is likely that the FAK signaling pathway is deregulated in cancer cells. FAK functions in both kinase-dependent and -independent manners. Kinase-inhibited FAK goes to the nucleus and potentially regulates gene expression to affect cancer progression.
The FAK gene is mapped on human chromosome 8q24.3 and ubiquitously expressed in most cells (Schaller et al. 1994; Zachary 1997). FAK itself does not act as an oncogene, but the FAK protein is overexpressed in many cancers (Table 1). FAK gene (gene symbol PTK2) amplification in the chromosome (Zhao and Guan 2009) or its upregulation by tumor-related transcription factors can contribute to FAK overexpression and activation in cancer (Cance and Golubovskaya 2008; Corsi et al. 2006). Activated FAK plays an important role as a key signal mediator in tumor progression and metastasis, suggesting that FAK is a potential target for cancer therapeutics (Fig. 1).
FAK Overexpression in Cancer.
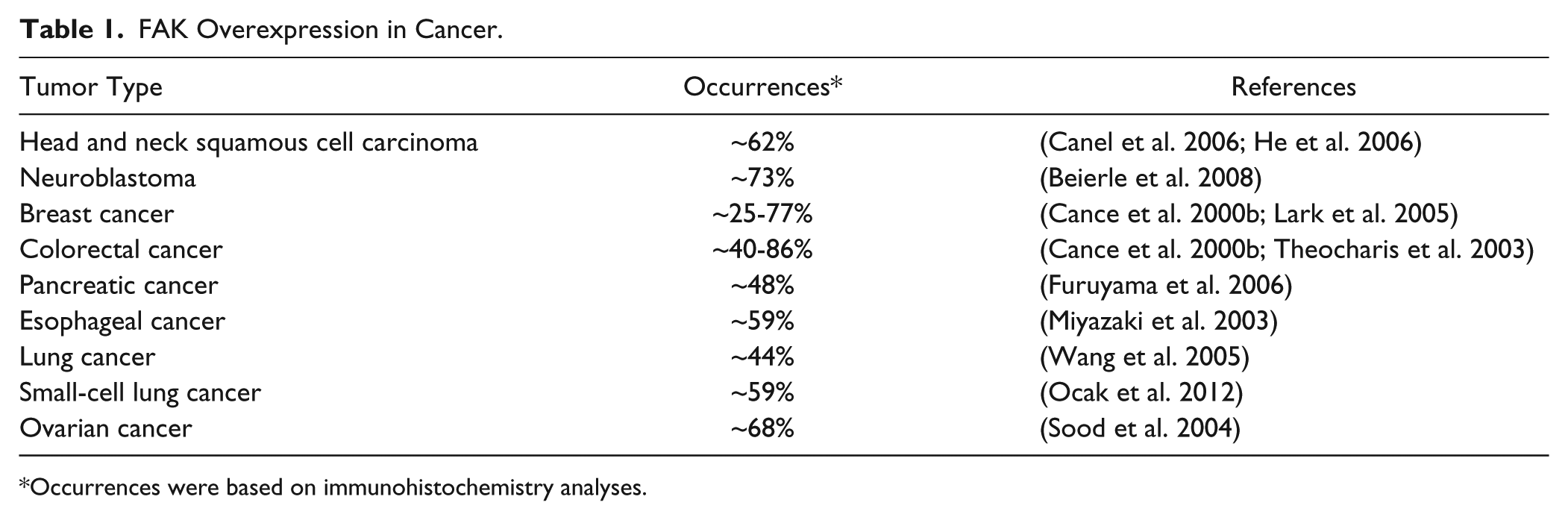
Occurrences were based on immunohistochemistry analyses.
In a recent study, FAK-Del33 (deletion of exon 33) was identified, and its mutation increased cell migration by promoting tyrosine 397 (Y397) autophosphorylation in human breast cancer cells (Fang et al. 2014); this suggested that genetic FAK mutations can play an important role in tumor progression and motility. Alternative splicing generates various FAK isoforms in cancers (Fang et al. 2014), which makes the study of FAK more complicated.
As higher FAK activity contributes to tumor proliferation and metastasis, efforts have been made by pharmaceutical companies to develop FAK inhibitors. Several small molecule FAK inhibitors have been tested in both preclinical and clinical phase trials.
In this review, we discuss therapeutic FAK inhibitors for cancer therapy and their recent status. Small molecule FAK inhibitors have been reported to affect both tumor cells and stroma. Therefore, this review also covers recent advances in FAK research with genetic animal models to elucidate unidentified functions of FAK along the various stages of cancer progression and in malignant phenotypes.
FAK Structure and Function
FAK functions can be separated into two main categories: kinase-dependent and kinase-independent. FAK kinase-dependent functions are often associated with integrin-related signaling at focal adhesions where FAK plays an important role in cellular migration and adhesion in both normal and cancer cells (Fig.1) (Cary et al. 1996; Parsons 2003). FAK also functions as a scaffold and participates in protein–protein interactions through its kinase-independent functions. Interestingly, FAK has been shown to localize to the nucleus and interact directly with p53 to promote cell proliferation and survival through p53 degradation (Golubovskaya et al. 2005; Lim et al. 2008b); this demonstrates a new function of FAK in the nucleus that occurs in a kinase-independent manner.
N-terminal Domain
FAK can be divided into three domains: N-terminal FERM (band
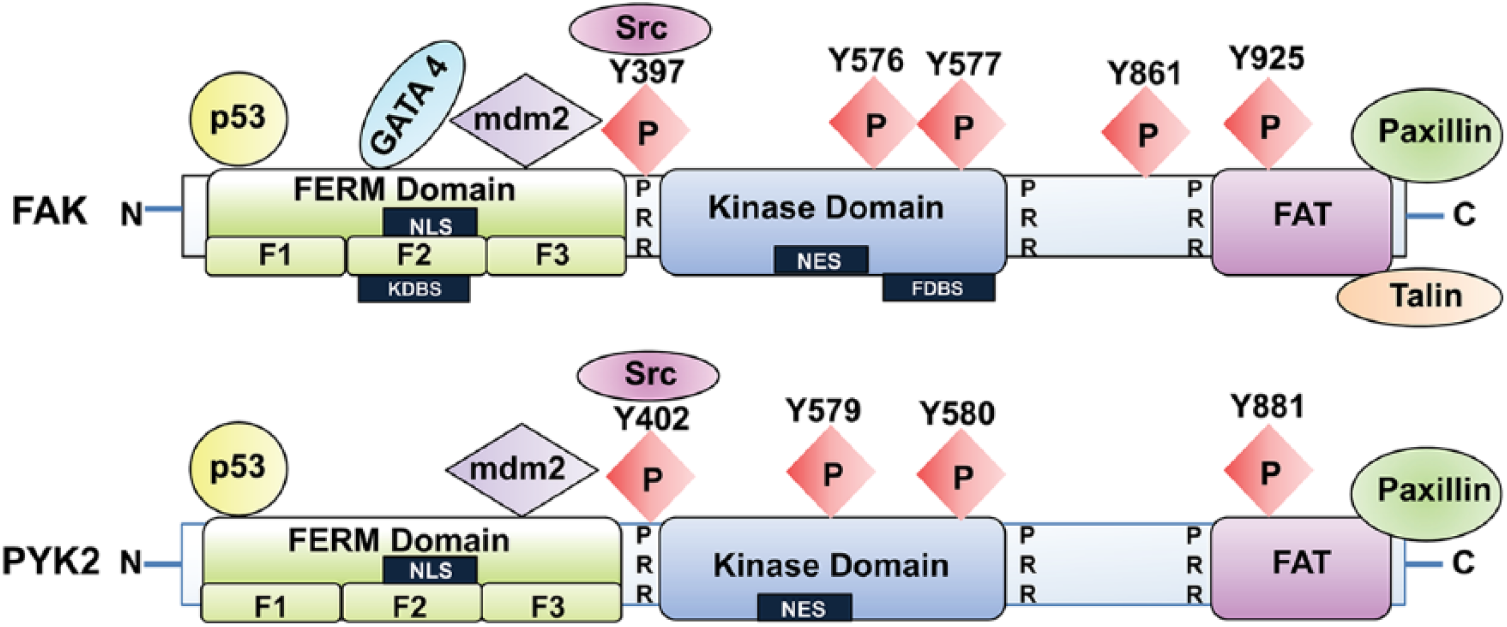
Schematic structure of FAK and Pyk2. FAK and Pyk2 contain N- and C-terminal domains and a central kinase domain. The N-terminal domain of FAK and Pyk2 contains the FERM (band
Kinase Domain
The FAK kinase domain resides in the middle of FAK (Parsons 2003) and contains the activation loop, tyrosine (Y) sites Y576 and Y577, which can be phosphorylated by Src; these sites ultimately regulate FAK kinase activity (Chan et al. 1999). Integrin, growth factor, and chemokine receptor stimulation result in the rapid autophosphorylation at Y397 (Calalb et al. 1995). The autophosphorylation site at Y397 and two other tyrosine phosphorylation sites at the activation loop, Y576 and Y577, are highly conserved within the catalytic kinase domain of protein tyrosine kinases (Owen et al. 1999). Phosphorylation-specific antibodies against phospho-Y397 and -Y576 (Life Technologies, #44-625G and 700013) are highly specific and present no cross-reactivity with Pyk2; these are therefore commonly used to monitor FAK activation during immunoblotting and immunohistochemistry.
Structural analysis of FAK FERM with the kinase domain revealed that the FAK FERM domain interacts and blocks the kinase domain, resulting in an auto-inhibited conformation (Lietha et al. 2007). The FERM domain binds directly to the kinase domain, centered at phenylalanine (F) 596 (Frame et al. 2010) (Fig. 2) and thus effectively blocks any catalytic activity. The structure further indicates that the intra-molecular interaction between FAK FERM and the kinase domain results in the “closed confirmation” of FAK, and the interaction of FERM with other activators can induce conformational changes that release the kinase domain from this closed conformation and subsequently leads to FAK activation (Cooper et al. 2003). Indeed, phosphatidyl inositol 4,5 bisphosphate (PIP2) binding to the FERM F2 basic cluster region opens the closed kinase domain and results in an active conformation of the protein (Barsukov et al. 2003).
C-terminal Domain
The C-terminal domain (CD) is characterized by two proline-rich sequences and the FAT (
FAK Functions in Cancer
FAK Gene Amplification
FAK protein is overexpressed in various cancers, including ovarian, cervical, kidney, lung, pancreatic, brain, colon, breast, and skin cancer (Golubovskaya et al. 2009) (Table 1). In addition, studies have confirmed a correlation between FAK overexpression levels and poorer clinical prognosis in several types of human tumors (Yom et al. 2011). One important mechanism of FAK overexpression is gene amplification, and increased FAK mRNA—which accounts for increased FAK expression—is also found in invasive and metastatic tumors, including head and neck, lung, colon, and breast cancer (Agochiya et al. 1999; Sulzmaier et al. 2014; Weiner et al. 1993).
Recently, several studies specifically examined FAK gene amplification in human tumors. FAK gene amplification was found to be a poor prognostic factor as well as an important indicator in gastric cancer (Park et al. 2010) and in invasive breast cancer (Yom et al. 2011). In contrast, correlations between FAK gene amplification and tumor progression in certain other human tumors have been inconsistent. Whereas overexpression of FAK in head and neck squamous cell carcinoma is independent of FAK gene amplification (Canel et al. 2006), there are still significant correlations between some invasive cancers and high FAK mRNA levels.
Cancer Initiation and Cell Survival
FAK expression has been linked to the initiation and survival of cancer cells and was first shown to prevent cell apoptosis in Madin-Darby canine kidney (MDCK) cells (Frisch et al. 1996). Genetic FAK deletion in skin keratinocytes has been shown to suppress dimethyl benzanthracene (DMBA)-induced skin cancer formation. In addition, deletion of FAK in keratinocytes induces cell apoptosis in vitro and in vivo (McLean et al. 2004a), and mammary tissue-specific FAK knockout with p53 deletion has been shown to reduce mammary tumor formation (van Miltenburg et al. 2014). FAK inhibition by FAK siRNA-mediated knockdown or overexpressing the FAK CD can decrease cell proliferation and tumor growth in breast cancer cells (Golubovskaya et al. 2009). Collectively, these studies suggest that FAK is critical in cancer cell survival.
Regulation of Cancer Stem Cells
Cancer stem cells (CSCs) have the ability to self-renew and to differentiate into cancer cells from a rare population of undifferentiated tumorigenic cells (Patel and Chen 2012). CSCs were first isolated from leukemia (Bonnet and Dick 1997) and, later, from many solid tumors, including brain, breast, prostate and pancreas cancers (Al-Hajj et al. 2003; Li et al. 2007; Li et al. 2009; Patrawala et al. 2006; Singh et al. 2003). CSCs generally contain specific cell surface markers, such as CD133, CD44, CD90, and CD24 (Anido et al. 2010; Singh et al. 2003) in addition to expressing specific transcription factors (Liu et al. 2013). FAK deletion in a murine breast cancer model led to a decrease in the number of mammary CSCs and a decrease in their self-renewal potential; this ultimately inhibited tumor progression (Luo et al. 2009a). Recent studies have also indicated that FAK is involved in the expression of several stem cell factors. FAK maintains the expression of critical transcription factors Slug (Snail family zinc finger 2) and Sox9, which were identified as important factors in maintaining mammary CSCs (Guo et al. 2012). In addition, NANOG, a key marker in stem cells, increases the level of FAK expression and activity in 293, SW480, and SW620 cancer cells (Ho et al. 2012). NANOG directly binds to the FAK promoter triggering FAK expression, and studies show that downregulating NANOG expression by siRNA can inhibit cancer cell growth, which can be reversed by FAK overexpression (Ho et al. 2012). These studies indicate that FAK expression may have an important role in the control of CSC function and activity.
Epithelial-to-Mesenchymal Transition (EMT)
EMT is a crucial process during embryogenesis, development, tissue remodeling and tumor progression. Over the past decade, numerous regulators have been identified as essential transcription factors in EMT, such as Snail, Slug, Twist, and Zeb (Chui 2013; Wang et al. 2013). EMT ultimately requires a decrease in epithelial markers (E-cadherin, α-catenin, and β-catenin), an increase in mesenchymal markers (vimentin, fibronectin, and N-cadherin) and the secretion of matrix metalloproteinases (MMPs). These changes in cell phenotype and genetic modulation promote a transition from benign tumor to invasive carcinoma.
Recent studies have identified evidence of FAK involvement in the EMT process. FAK has a functional role in TGF-β-mediated EMT by Src-dependent activation in hepatocytes (Cicchini et al. 2008). These studies revealed that FAK signaling is required for the transcriptional regulation of several mesenchymal markers and for the delocalization of E-cadherin. Additionally, a FAK inhibitor (1,2,4,5-benzenentetramine, 4HCl) repressed TGF-β-induced EMT in human squamous cell carcinoma (Saito et al. 2013). FAK signaling was required for Src-regulated E-cadherin expression in colon cancer cells, and inhibition of FAK activity reduced Src-mediated cell invasion (Avizienyte et al. 2002; Hauck et al. 2002a). More direct evidence of FAK involvement in EMT has been provided from a recent study of FAK-/- embryonic cells. FAK re-expression rescued the mesenchymal characteristics of FAK-/- embryonic cells to generate committed mouse embryonic fibroblasts via Snail1 gene expression and Snail1 protein stabilization (Li et al. 2011). Taken together, although the direct role of FAK is yet to be unveiled in EMT, the correlation between FAK and EMT may offer an important target in cancer metastasis and cancer therapeutics.
Invasion and Metastasis
FAK overexpression is also associated with the enhanced invasion and metastatic characteristics of EMT (Cance et al. 2000a). Integrin β1 and FAK signaling directly regulate the proliferation and invasion of metastatic cells in the lung (Shibue and Weinberg 2009). FAK phosphorylation is important in regulating E-cadherin expression by activating Src signaling pathways in colon cancers and, before small molecule FAK inhibitors were available, overexpression of the FAK CD was useful in inhibiting cancer cell invasion and metastasis (Hauck et al. 2002b). Furthermore, FAK promotes cellular membrane expression of MT1-MMP, a matrix metalloproteinase, which serves to degrade the ECM (Wu et al. 2005). A recent study from Schlaepfer’s group highlighted the importance of FAK in the tumor environment. FAK plays an important role in mediating VEGF (vascular endothelial growth factor)-induced vascular permeability by directly phosphorylating β-catenin at Y142 (Chen et al. 2012). In addition, vascular endothelial cadherin (VEC) phosphorylation, which is mediated by FAK, increases vascular permeability and cancer cell metastasis (Jean et al. 2014). Cancer cells release copious amounts of VEGF, which compromises barrier function by increasing vascular permeability and new blood vessel formation. FAK-mediated barrier function demonstrates how cancer cells take advantage of the compromised endothelial barrier and enter the vasculature in intravasation and extravasation processes.
Tumor Angiogenesis
FAK also plays an important role in tumor angiogenesis. FAK expression is increased in microvascular endothelial cells, which leads to enhanced angiogenesis (Haskell et al. 2003; Lechertier and Hodivala-Dilke 2012). VEGF stimulation increases the activation of FAK in endothelial cells (Chen et al. 2012). Increased FAK Y397 autophosphorylation is also correlated with increased endothelial cell migration (Cary et al. 1996). In addition, inhibition of FAK reduces VEGF expression from tumor cells and has been shown to decrease tumor angiogenesis in the 4T1 breast cancer model (Mitra and Schlaepfer 2006). Since FAK has an important role in the regulation of tumor angiogenesis, blocking FAK activity in endothelial cells should be considered in cancer prevention therapy.
Pyk2: FAK Family Protein
Pyk2 (proline-rich tyrosine kinase 2) is the only other kinase structurally and functionally related to FAK, also referred to as the second FAK family kinase. FAK and Pyk2 contain approximately 45% homologous amino acid sequences over the entire protein, and even up to 60% homology within the kinase domain (Fig. 2) (Lipinski and Loftus 2010). Despite these similarities, these two proteins are not equally expressed in all types of tissues. Pyk2 expression, in contrast to FAK, is limited to brain cells, fibroblasts, platelets, and hematopoietic cell types (Avraham et al. 2000; Orr and Murphy-Ullrich 2004). FAK is primarily activated through integrins, growth factor receptors, and cytokine receptors in contact with the ECM, whereas Pyk2 can be activated by integrins, and shares downstream targets with FAK, but it is usually activated in response to increases in intracellular calcium (Lev et al. 1995).
FAK is typically overexpressed in various cancers, and Pyk2 is also upregulated in several cancers (Wendt et al. 2013; Zhonghua et al. 2012). In breast cancer cells, Pyk2 overexpression was inversely related to E-cadherin level and necessary to initiate metastasis (Wendt et al. 2013). In benign prostatic hyperplasia, Pyk2 overexpression was observed and resulted in increased malignancy in prostate cancer (Stanzione et al. 2001). Interestingly, compensatory Pyk2 upregulation is often observed when FAK is deleted or inhibited. Pyk2 is upregulated in FAK-/- fibroblasts and endothelial cell-specific conditional FAK knock-out mice (Sieg et al. 1998; Weis et al. 2008). In adult mice lacking endothelial cell FAK, tumor angiogenesis can be promoted by compensatory Pyk2 upregulation (Weis et al. 2008), suggesting that Pyk2 plays an important role in tumor angiogenesis. Taken together, these two kinases may have overlapping signaling pathways in cancer progression, and it would be ideal to inhibit both FAK and Pyk2 activity.
FAK Inhibitors
As FAK expression and activity play important roles in tumor survival, motility, and angiogenesis in vitro and in vivo, FAK is recognized as a potential cancer target. Therefore, there have been numerous efforts to inhibit FAK signaling in cancer therapy. FAK inhibitors have changed significantly since the introduction of inhibiting FAK activity via antisense cDNA (Xu et al. 1996) and expression of the FAK CD (Xu et al. 2000). Major pharmaceutical companies have developed pharmacological FAK inhibitors, and some of these—mostly small molecule inhibitors and small inhibitory peptides—have made it to clinical trials (Table 2). The small molecule FAK inhibitors are ATP analogs in either pyrimidine- or pyridine-base form. FAK inhibitors can be mainly used to inhibit FAK activity in cancer cells by direct inhibition of FAK kinase activity. Our recent study demonstrated that treatment with the small molecule FAK inhibitor (PF-562,271) inhibited FAK activities as well as enhanced FAK nuclear localization in RPMI-7951 human melanoma (Fig. 3; unpublished data), suggesting that FAK inhibition also promotes FAK kinase-independent function (see also Fig. 1) via nuclear FAK localization, which may inhibit tumor growth and metastasis.
Pharmacological FAK Inhibitors.
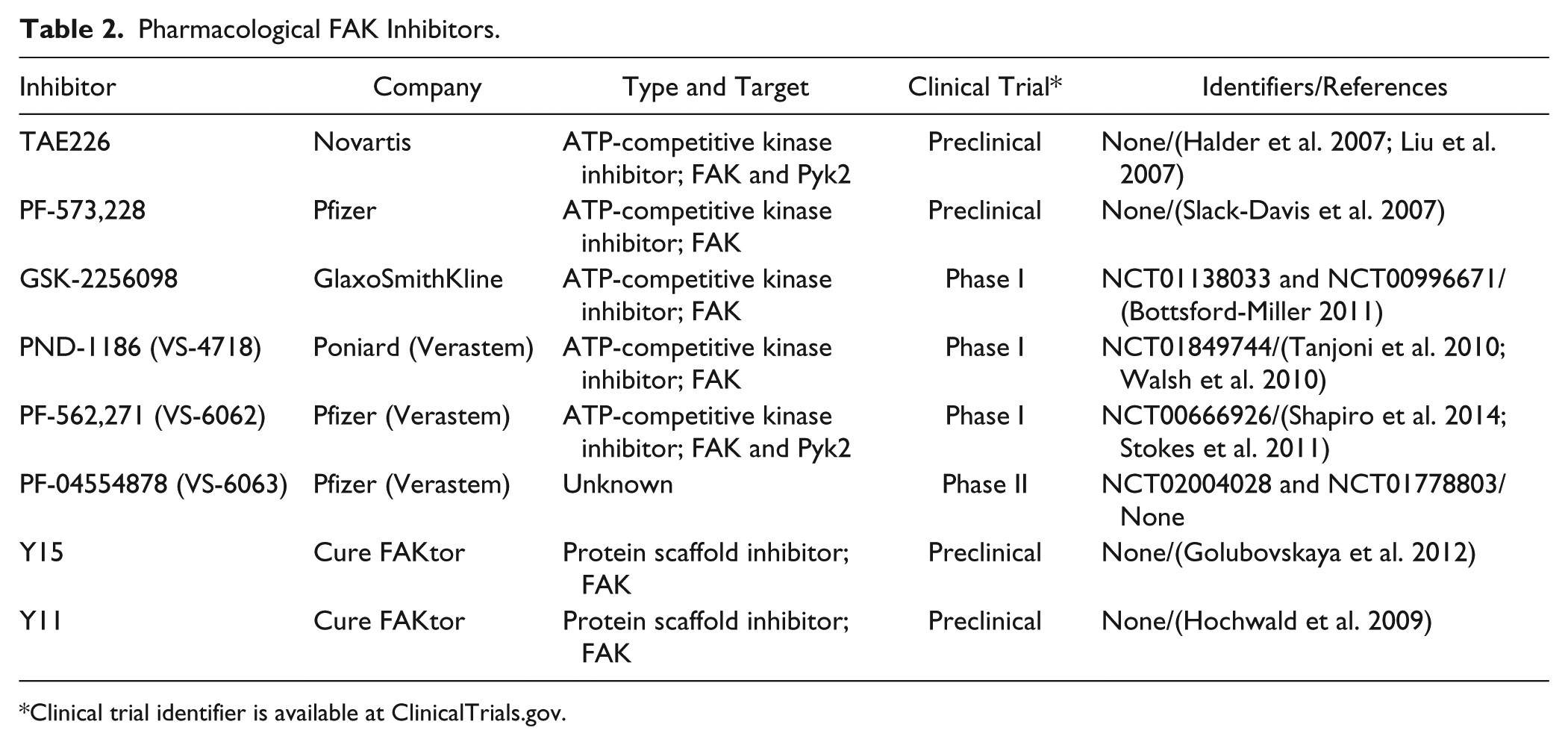
Clinical trial identifier is available at ClinicalTrials.gov.
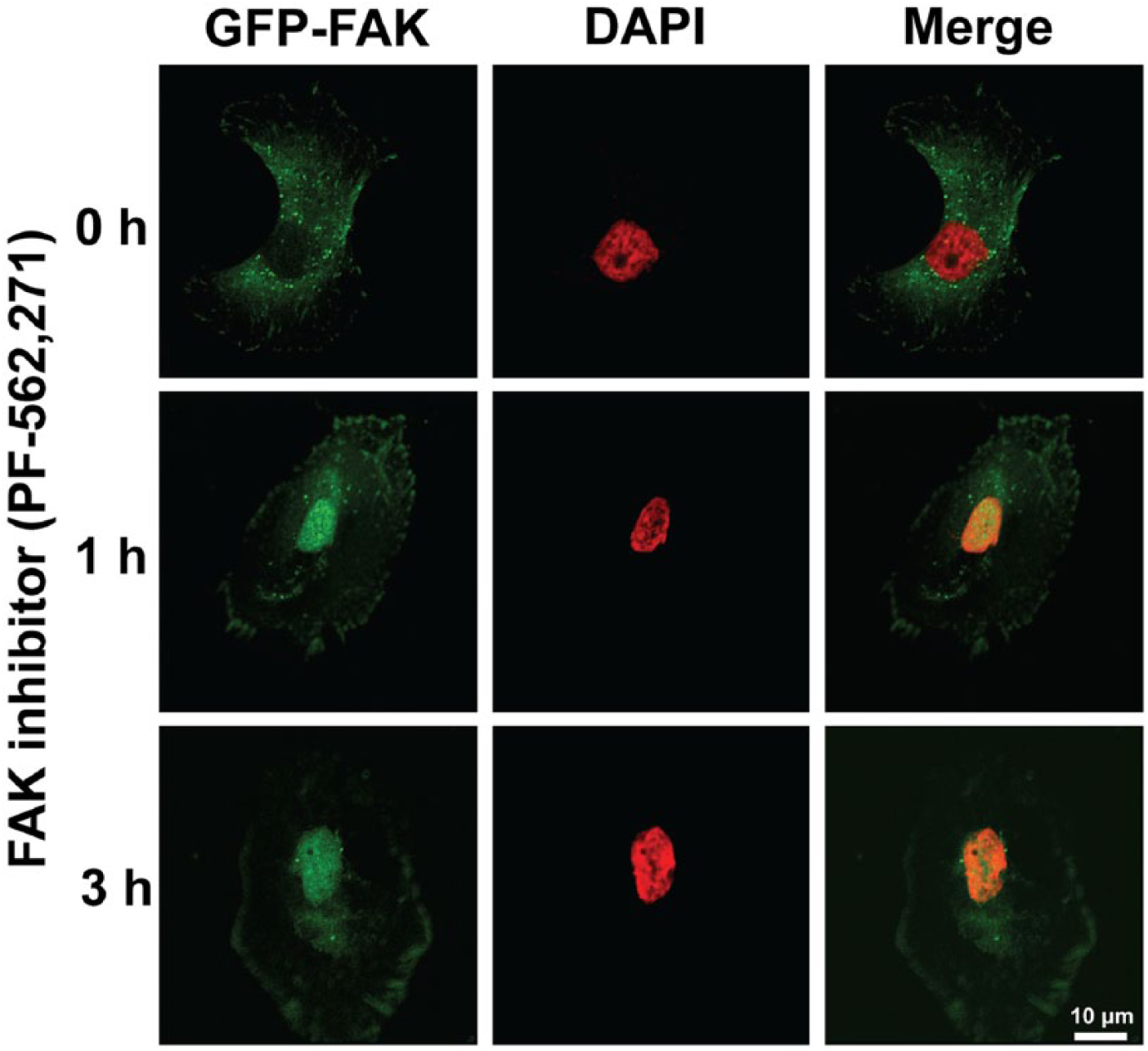
Pharmacological FAK inhibition promotes nuclear localization of FAK in RPMI-7951 human melanoma cells. GFP-FAK is stably expressed in RPMI-7951 cells. Cells were treated with PF-562,271 (1 μM) for 1–3 hr and cells were fixed. Images were collected with Nikon confocal microscope (A1R) and analyzed with NIS-Elements AR software. Nuclear localized FAK can be observed within 1 hr treatment of the FAK inhibitor. Green, FAK; Pseudo-colored Red, DAPI. Scale, 10 µm.
TAE-226
A majority of the available FAK inhibitors work as an ATP analog and target the ATP-binding domain surrounding lysine 454 in the FAK kinase domain. TAE-226 (Novartis) was the first effective FAK inhibitor used in vitro and in vivo. TAE-226 has been shown to inhibit FAK kinase activity as well as that of Pyk2 and other protein tyrosine kinases including insulin-like growth factor-1 receptor (Halder et al. 2007). TAE-226 blocks cell proliferation and invasion, and increases apoptosis in glioma and ovarian tumor models (Halder et al. 2007; Liu et al. 2007; Shi et al. 2007). Interestingly, TAE-226 in combination with docetaxel, a microtubule stabilizer, significantly decreases angiogenesis and invasion in ovarian carcinoma (Halder et al. 2007).
PF-573,228
PF-573,228 (Pfizer) is an ATP analog that acts to inhibit FAK kinase activity. This inhibitor is highly specific for FAK catalytic activity and other related kinases. Treatment with PF-573,228 at its IC50, 4.0 nM, blocks FAK kinase activity without affecting Pyk2 activity (Slack-Davis et al. 2007). Although PF-573,228 inhibits tumor cell migration in vitro, it cannot inhibit cell proliferation or induce apoptosis in fibroblasts and in prostate cancer tissues (Slack-Davis et al. 2007). These results suggested that PF-573,228 may have a different mode of action for cancer treatment and may effectively target FAK motility or metastasis in tumors.
GSK2256098
GSK2256098 (GlaxoSmithKline) is an orally available FAK inhibitor and was tested in a Phase I study (clinical trial identifier at ClinicalTrials.gov; NCT00996671). GSK2256098 treatment significantly reduced Y397 autophosphorylation of FAK at 1.0 μM (not for PYK2) and decreased migration and invasion in SKOV3 ovarian cancer cell line (Bottsford-Miller 2011). In addition, the 75 mg/kg dose was shown to significantly decrease tumor volume by 58% in a mouse xenograft model (Bottsford-Miller 2011). In this study, the double and triple combination of GSK2256098 with cytotoxic drugs including pazopanib, a multi-targeted receptor tyrosine kinase inhibitor, and docetaxel, a microtubule stabilizer, resulted in a 99% reduction in tumor weight.
VS-4718 (Previously PND-1186, Poniard)
VS-4718 (Verastem) effectively blocks both FAK and Pyk2 activity, and is currently in Phase I trials against various tumors (Clinical trial identifier; NCT01849744). Preclinical studies have demonstrated that VS-4718 has a high anti-tumor efficacy in a breast carcinoma mouse model (Tanjoni et al. 2010). Interestingly, VS- 4718 treatment was shown to reduce inflammatory cell infiltration and IL-6 expression, suggesting that FAK inhibition is associated with anti-inflammatory action in both tumor and stromal cells (Walsh et al. 2010). In addition, the oral administration of VS-4718 inhibited tumor growth through caspase-3 activation, and decreased tumor experimental metastasis in ovarian carcinoma models (Tancioni et al. 2014).
VS-6062 (Previously PF-562,271, Pfizer)
VS-6062 (Verastem) is capable of inhibiting both FAK and Pyk2 kinase activity (Roberts et al. 2008). VS-6062 is very effective both in vitro and in vivo by reducing tumor size and preventing metastasis (Stokes et al. 2011). In angiogenesis, compensatory Pyk2 upregulation in endothelial cells was observed upon inhibition or ablation of FAK (Weis et al. 2008). Therefore, the dual inhibition of FAK and Pyk2 is beneficial. In this respect, VS-6062 was shown to reduce human epithelial ovarian cancer angiogenesis and metastasis in nude mice (Stone et al. 2014). VS-6062 was actually the first FAK inhibitor tested in clinical Phase I trials (Infante et al. 2012), which established the recommended dosage of VS-6062, and overall showed that approximately 34% of patients experienced stable disease state at the end of the six-week treatment. However, VS-6062 showed non-linear pharmacokinetics and was thus discontinued (Infante et al. 2012).
VS-6063 (Previously PF-04554878, Pfizer)
VS-6063 (Verastem) was originally developed by Pfizer to overcome the nonlinear pharmacokinetics of VS-6062/PF-562,271. The results of a Phase I trial of VS-6063 indicated that stability was achieved in some patients with ovarian and colorectal tumors (clinical trial identifier; NCT017788033). VS-6063 is currently in Phase II study in patients with KRAS mutant non-small cell lung cancer (clinical trial identifier; NCT01951690). Although the chemical structure and specificity of VS-6063 are still not published, VS-6063 treatment has been shown to increase ovarian carcinoma cell sensitivity to taxane, suggesting that VS-6063 in combination with paclitaxel may be more effective as compared with monotherapy (clinical trial identifier; NCT017788033).
Y15 and Y11
There are drawbacks with using FAK inhibitors that function as ATP analogs to target the ATP binding pocket of the FAK kinase domain. Many tyrosine kinases share conserved sequences in their ATP binding sites, and it increases the probability that these FAK inhibitors could bind to and inhibit other kinases, diminishing the specificity of the ATP analogs to FAK (Golubovskaya et al. 2012). A new way to avoid this problem is to directly interfere with FAK autophosphorylation. This will not only inhibit FAK activity but also decrease the probability of inhibiting other tyrosine kinases.
Both 1,2,4,5-Benzenetetraamine tetrahydrocloride (FAK Inhibitor 14 or Y15) and 1-(2-Hydroxyethyl)-3,5,7-triaza-1-azaniatricyclo[3.3.1.13,7] decane bromide (Y11) are small molecule inhibitors of FAK but are not ATP analogs. These two molecules act by binding to and inhibiting the autophosphorylation at FAK Y397 (Golubovskaya et al. 2012; Golubovskaya et al. 2008). Y15 and Y11 have been shown to inhibit cell viability in breast, colon, melanoma, lung and pancreatic cancer cell lines (Golubovskaya et al. 2012; Golubovskaya et al. 2008; Hochwald et al. 2009). Y15 has an IC50 of about 1 μM, whereas Y11 has an IC50 of about 50 nM. Treatment with 1 μM Y15 or Y11 selectively inhibits FAK activity without interfering with the activity of other kinases (Golubovskaya et al. 2012; Golubovskaya et al. 2008). These two FAK inhibitors offer the potential for obtaining greater selectivity by targeting Y397 instead of the ATP binding site.
FAK Genetic Animal Models
The first genetic FAK mouse generated was a global ablation model (Ilic et al. 1995). These studies revealed that FAK deletion caused embryonic lethality at embryonic day E8.5 due to defects in mesodermal development, specifically cardiovascular development, suggesting that FAK is essential in embryonic vasculogenesis. Another mouse model with homologous knock-in mutations at FAK lysine 454 to arginine 454 (termed kinase-dead, KD), the ATP-binding residue, demonstrated that mice were lethal at E9.5 because of defects in vasculogenesis and chorio-allantois defects (Lim et al. 2010). Furthermore, another mutant FAK mouse model was developed with the deletion of exon 15, which contains autophosphorylation site Y397 (Corsi et al. 2009). These mice developed normally at E12.5, but died at E14.5-E16.5, with multiple developmental defects including hemorrhages, edema, delayed artery formation, vascular remodeling defects, and multiple organ abnormalities. This suggests that FAK has important kinase roles independent of autophosphorylation activity in early development. To avoid an embryonic lethal phenotype of various global FAK mutations in mice, several mouse models have utilized the inducible Cre-Lox recombinase genetic system to induce conditional FAK knockouts during development or in adult mice (Table 3).
FAKflox/flox Knockout and FAK Kinase-Dead (KD) Knock-in Mouse Models.
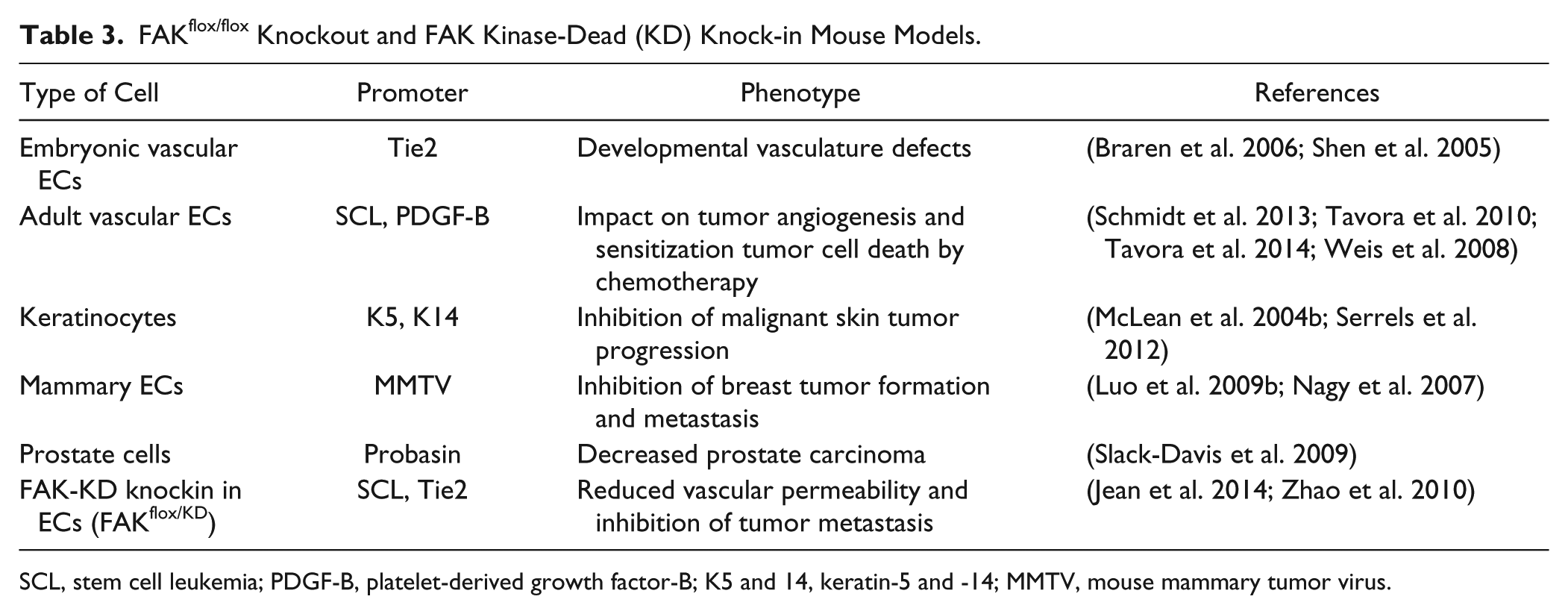
SCL, stem cell leukemia; PDGF-B, platelet-derived growth factor-B; K5 and 14, keratin-5 and -14; MMTV, mouse mammary tumor virus.
Endothelial Cells
Two independent groups generated Tie2-Cre driven endothelial cell knockout mice and showed that embryos died at E11.5 with developmental vasculature defects (Braren et al. 2006; Shen et al. 2005). A tamoxifen-inducible endothelial cell-specific knockout mouse (SCL-Cre-ER(T)) study revealed that FAK knockout in adult mice endothelial cells was not lethal. Surprisingly, there were no major tumor angiogenesis defects due to compensatory Pyk2 expression upon the loss of FAK (Weis et al. 2008). This result suggests the need for a dual protein tyrosine kinase inhibitor that targets FAK and Pyk2 so as to provide a total anti-angiogenic effect. To avoid Pyk2 upregulation in genetic FAK knockout mice, endothelial cell-specific, kinase-dead mice were generated by crossing FAKflox/flox SCL-Cre-ER(T) and FAK WT/KD mice. These mice exhibited defects in VEGF-mediated endothelial permeability by deregulation of adherens junctions (Chen et al. 2010; Jean et al. 2014).
Endothelial cell-specific FAK deletion with a tamoxifen-inducible platelet-derived growth factor-B (PDGF-B) Cre mouse demonstrated that, in contrast to the results shown from previous EC FAK knockout mouse models, FAK deletion in adult mice inhibited tumor growth and reduced angiogenesis (Tavora et al. 2010). With the same mouse model, cancer cells were injected subcutaneously and allowed to grow for just over 7 days. Mice were then injected with tamoxifen and given a tamoxifen-containing diet to delete endothelial cell FAK. These mice then underwent chemotherapy with doxorubicin or irradiation. Surprisingly, endothelial cell FAK knockout sensitized the tumors to chemotherapy (Tavora et al. 2014), suggesting that EC FAK may play a novel role in protecting tumors from chemotherapy, and that targeting EC FAK may enhance current cancer therapeutics.
Keratinocytes
Mice with a floxed FAK kinase domain under the control of tamoxifen-inducible keratin-14 Cre (K14 CreER(T)) were generated. Skin tumors were induced chemically and the loss of FAK inhibited tumor formation (McLean et al. 2004a). FAK-deleted squamous skin carcinoma cells (SCCs) were obtained from K14 CreER(T) FAKflox/flox mice and FAK re-expression in SCCs restored tumor-forming activity. But, importantly, PF-562,271 treatment abolished tumor proliferation. This result suggests that kinase activity of FAK is required for skin tumor progression in this system (Serrels et al. 2012).
Mammary Epithelial Cells
To evaluate the importance of FAK in mammary tumorigenesis, several groups have generated MMTV (mouse mammary tumor virus)-Cre mice, interbred from conditional FAK flox mice (Lahlou et al. 2007; Luo et al. 2009a; Provenzano et al. 2008). The first FAK-deleted mice using this system resulted in an inhibition of the benign-to-malignant transformation of tumors as well as inhibition of mammary tumor cell proliferation. This provided evidence that FAK plays an important role in initiating mammary tumorigenesis (Lahlou et al. 2007). Another conditional FAK knockout in mammary epithelium limited breast tumor formation and metastasis (Provenzano et al. 2008). In MMTV-Cre mice with a conditional deletion of FAK exon 3, it was demonstrated that FAK deletion in the mammary epithelium limits tumor formation via its effect on mammary cancer stem and progenitor cells (Luo et al. 2013).
Prostate Cells
Transgenic adenocarcinoma of the mouse prostate is a well-studied mouse model of prostate cancer under the control of the androgen-responsive minimal rat probasin promoter (Greenberg et al. 1995; Kaplan-Lefko et al. 2003). In this mouse model, the loss of FAK using probastin-Cre expression or a pharmacological FAK inhibitor, PF-562,271, did not affect the progression of the adenocarcinoma. However, continued FAK expression and activity is needed for androgen-independent formation of neuroendocrine carcinomas (Slack-Davis et al. 2009).
Nuclear FAK in Cancer
In addition to the canonical roles of FAK in the regulation of development, migration, proliferation, tumor angiogenesis, and metastasis, several recent studies have identified novel roles for FAK in the nucleus (Fig. 1). One of these studies identified that nuclear FAK promotes cell proliferation and survival by reducing p53-mediated cell cycle arrest under stress conditions (Lim et al. 2008b). Genetic FAK inhibition (kinase-dead knock-in cells) or pharmacological FAK inhibition increased FAK accumulation in the nucleus, which prevented inflammatory VCAM-1 expression by promoting the degradation of a transcription factor, specifically GATA4, which is required for VCAM-1 expression (Lim et al. 2012). Collectively, there are several conditions that can stimulate FAK nuclear localization: 1) stress signals, 2) cell de-adhesion, 3) FAK inhibition (Lim et al. 2008b; Lim et al. 2012). However, the mechanisms by which FAK shuttles from the membrane to the nucleus remain undefined (Lim 2013; Lim et al. 2008b).
In human melanoma cells, we found that FAK inhibition with PF-562,271 not only inhibits FAK activity, but also enhances FAK nuclear localization (Fig. 3).
Taken together, these studies have provided evidence of how nuclear FAK could regulate cancer cell proliferation and motility. Current evidence indicates that nuclear FAK may provide a potential mechanism for cancer therapeutics.
FAK Regulation of Epigenetics
Recently, it was reported that nuclear FAK binds to methyl CpG-binding protein 2 (MBD2) in myoblast cells, and their binding enhanced heterochromatin remodeling to promote gene expression (Luo et al. 2009c). The nuclear FAK-MBD2 complex decreased histone deacetylase complex 1 (HDAC1) and the methyl CpG site in the myogenin promoter, resulting in myogenin expression (Luo et al. 2009c). In addition to the known role of nuclear FAK, which controls transcription factor stability via ubiquitination, these studies suggest that FAK may play an important role in the regulation of gene expression through interactions with molecules responsible for DNA modification.
Conclusions
Preclinical and clinical trials of small molecule FAK inhibitors have demonstrated that FAK can be an effective cancer target in various cancers. Interestingly, based on genetic FAK mouse studies in cancer progression, we now know that FAK plays critical roles in promoting invasive properties of tumor cells as well as the tumor environment, which controls tumor metastasis. Therefore, FAK inhibitors have beneficial effects in terms of blocking tumor progression in both tumor cells and tumor stroma.
An emerging role of FAK signaling is the dual ability of FAK inhibition to inhibit FAK activity and promote nuclear FAK to potentially regulate gene expression. As FAK moves to the nucleus upon FAK inhibition via genetic or pharmacological means, it will be extremely intriguing to explore as yet unknown functions of nuclear FAK and reveal unidentified targets of FAK inhibitors for cancer therapy.
Footnotes
Acknowledgements
We thank Dr. Erin Ahn for critical reading of the manuscript.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work is supported by 2013-2015