
Abstract
Organ-specific cell types are maintained by tissue homeostasis and may vary in nature and/or frequency in pathological situations. Moreover, within a cell lineage, some sub-populations, defined by combinations of cell-surface markers, may have specific functions. Dendritic cells are the epitome of such a population as they may be subdivided into discrete sub-groups with defined functions in specific compartments of various organs. Technically, to study the distribution of DC sub-populations, it involves performing multiparametric immunofluorescence on well-conserved organ structures. However, immunodetection may be impacted by protein cross-linking and antigenic epitope masking by the use of 10% neutral-buffered formalin. To circumvent this and to preserve a good morphological tissue structure, we evaluated alternative fixatives such as Periodate Lysine Paraformaldehyde or Tris Zinc fixatives in combination with other embedding techniques. The cryosection protocols were adapted for optimal antigen detection but offered a poor morphological preservation. We therefore developed a new methodology based on Tris Zinc fixative, gelatin-sucrose embedding and freezing. Using multiple DC markers, we demonstrate that this treatment is an optimal protocol for cell-surface marker detection on high-quality tissue sections.
Introduction
Any histological evaluation necessitates organ fixation and embedding. One of the most commonly used techniques is based on 10% neutral-buffered formalin fixative, corresponding to 4% formaldehyde (4% FA) and associated with paraffin embedding. This technique allows the identification of all the previously described tissue structures but proteins are often altered by formaldehyde-induced methyl bridges precluding antigen recognition by specific antibodies. To circumvent this hurdle, antigen retrieval methods (chemical, enzymatic, physical) may be used. If this step is unsuccessful, alternative fixatives based on lower percentages of formaldehyde (0.25% in Periodate Lysine Paraformaldehyde (PLP)) or on other chemicals (Tris Zinc) have been developed. Paraffin embedding may represent an additional constraint for antigen recognition due to its fusion temperature (55–58C). Direct organ freezing offers better antigen preservation but poor tissue structure. Organ inclusion in embedding media may improve this latter point. Balancing tissue preservation and antigen recognition necessary to localize a cell sub-population, as defined by multiple antigen detection, can easily become a very complex task.
Immunofluorescence is well adapted for the detection of multiple antigens on the same tissue section. To define a protocol adapted for multiparametric immunofluorescent staining combined with good tissue preservation, dendritic cells (DCs) were chosen as model because of their diversity in terms of sub-populations and tissue distribution. Three of the main DC reservoirs were used for protocol set-up on steady-state resident DCs.
Materials & Methods
Animals
Specific pathogen-free healthy female C57Bl/6 mice were obtained from Charles River Laboratories (Les Oncins, France). All animal procedures followed the CEE directive 86/609 of 24th November 1986 on the harmonization of laws, regulations or administrative provisions relating to the protection of animals used for experimental or other scientific purposes. It is also conducted in compliance with the French law, décret n° 87.848 of 19th October 1987 modified by the décret n° 2001-464 of 29th May 2001 and the décret n° 2001-486 of the 6th June 2001. The 6-week-old mice were sacrificed, and the skin, spleens and the draining axillary and inguinal lymph nodes were collected and placed into different fixatives.
Fixation
Three methods were compared. Organs were placed in 4% formaldehyde (4% FA) in PBS overnight at 4C. Alternatively, organs were placed in periodate lysine paraformaldehyde (PLP) fixative as described by McLean and Nakane (1974) and Gendelman et al. (1983). This fixative consists of 0.25% formaldehyde in a 5% dextrose solution containing 0.2 M of
Embedding
Paraffin embedding was performed for organs fixed in 4% FA, PLP or TZ. After fixation, tissues were rinsed twice for 30 min in PBS and were dehydrated in graded ethanol baths (30% ethanol, 70% ethanol, 90% ethanol, water) followed by three baths of 100% ethanol for 1 hr each. After three baths of Microclearing (Microm Microtech, France), tissues were incubated in paraffin overnight at 58C. Prepared paraffin tissue blocks were stored at room temperature.
Fresh-frozen tissues were directly placed in Optimal Cutting Temperature compound (OCT, Tissue Tek®, Sakura, France) and placed on dry ice-cooled isopentane. Once solid, samples were then stored at -80C.
Combined Protocol with Tris Zinc Fixative, Sucrose and Gelatin Embedding
The Tris Zinc fixative was modified by adding 15% sucrose. Organs were fixed for 24 hr at 4C and then rinsed in 15% sucrose/PBS thrice, each time for 1 hr. Organs were incubated in a 7.5% gelatin/15% sucrose/PBS solution for 1 hr at 37C and blocks were finally prepared in the same medium. Other gelatin solutions (15% or 30%) were tested. Gelatin solutions containing the specimens were solidified in a Petri dish at room temperature. Upon reaching a suitable consistency, gelatin cubes, each with the specimen in the center, were cut out and frozen in isopentane cooled on liquid nitrogen vapors. Once solidified, samples were stored at -80C.
Sectioning
For all paraffin-embedded samples, 5-µm-thick serial sections were cut with a 2045 Jung Multicut microtome (Leica Microsystems GmbH; Wetzlar, Germany). Paraffin sections were mounted onto Superfrost Plus slides (05305190, Labonord; France), dried at 40–50C and stored at room temperature (for 4% FA and TZ-fixed samples) or -20C (for PLP-fixed samples).
Frozen samples were processed with a Microm HM560 cryostat (Microm Microtech France, Francheville, France). O.C.T. frozen sections were mounted onto gelatin and chrome alum coated-slides, dried at room temperature, and then fixed in acetone for 10 min at -20C before storage at -80C. Gelatin-frozen sections were mounted on Superfrost Plus Gold slides (F/1650-54, Microm Microtech France), dried and stored at -20C.
Staining Procedures
Hematoxylin and Eosin Staining
Paraffin sections were dewaxed by immersion in two baths of xylene for 5 min each followed by two baths of 100% ethanol, 70% ethanol and 30% ethanol, again for 5 min each, and then rehydrated in PBS. Cryosections were equilibrated at -20C and rehydrated before staining. Sections were stained for 3 min in Harris’ Hematoxylin (HHS-16, Sigma-Aldrich; St Louis, MO) followed by a 3-min wash in water. Sections were eosin-stained (312740-1000, RAL Diagnostics; France) for 30 sec followed by a 30-sec wash in water. Finally, sections were dehydrated and mounted in Eukitt® (053-47505, Labonord).
Immunohistochemistry
Antigen unmasking procedures were used for FFPE tissue sections by exposing them to citrate buffer at pH 6.0 with heating at 95C. Milder techniques were also used for FFPE tissue sections, such as incubation with thepepsin protease for 15 min at 37C. For PLP-fixed organs, 0.0025% trypsin in 0.1% CaCl2 buffer was used for 5 min at room temperature.
Primary, secondary and tertiary antibodies, as well as the detection systems, are listed in Table 1. The primary antibody concentration used for immunofluorescence was determined by serial dilution of each antibody tested alone on the three studied organs. For the MHC Class II detection, it was necessary to adjust the primary antibody concentration between the immunohistochemical protocol (0.05 µg/ml) and the immunofluorescent protocol (0.5 µg/ml) as the amplification system was different between these two methods.
Antibodies and Detection Systems Used for Dendritic Cell Detection by Immunohistochemistry.
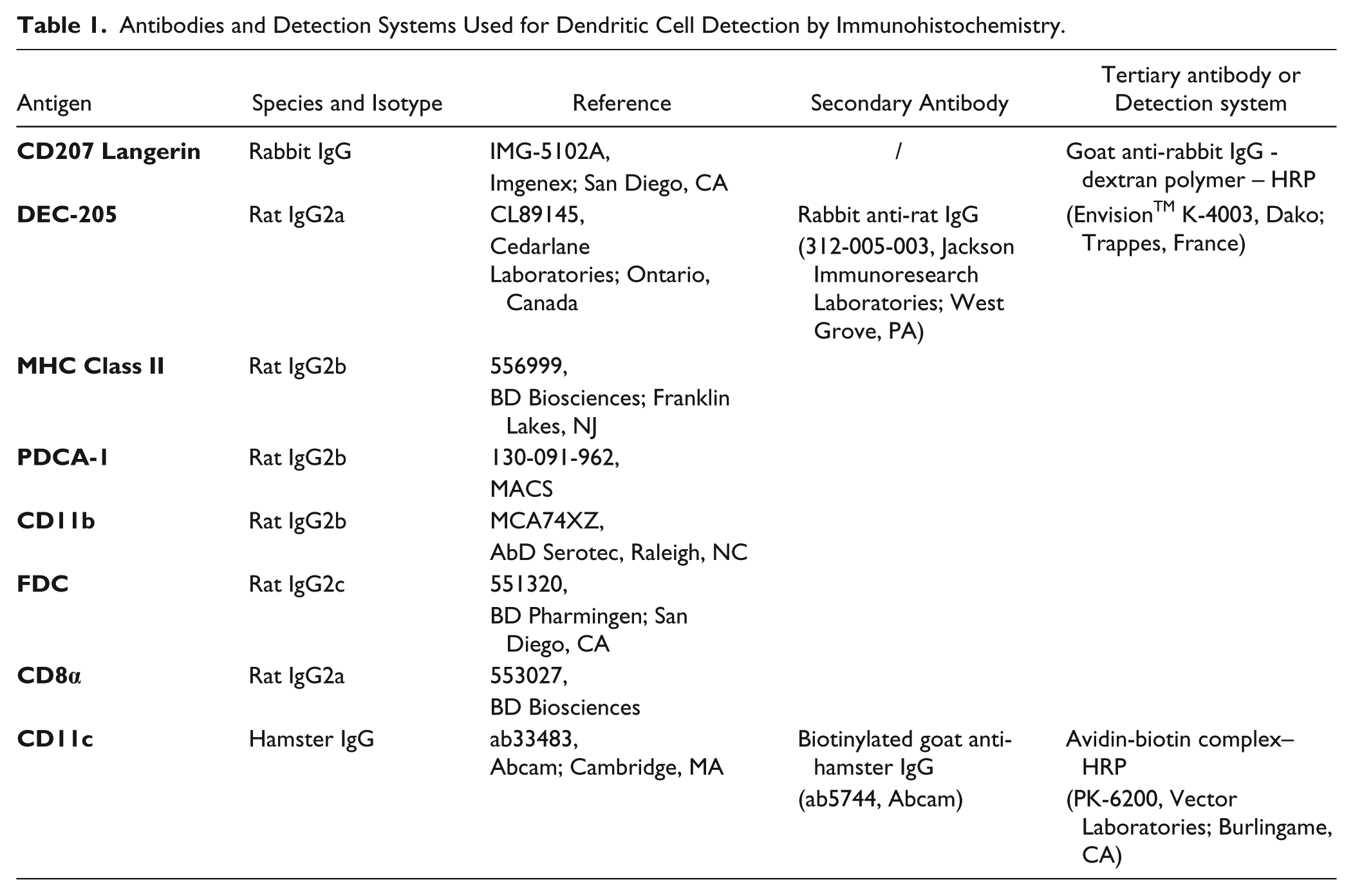
Protocol
Paraffin sections were dewaxed, rehydrated and unmasked, as previously described in Laboratory Methods in Histotechnology (Prophet 1992). Frozen sections were re-equilibrated at -20C and washed in PBS twice for 5 min.
Endogenous peroxidase activity was blocked by incubation in 3% H2O2 in PBS for 20 min at room temperature. Sections were rinsed twice in PBS and, for biotinylated secondary antibodies, endogenous biotin was blocked by the use of a specific kit (X0590, Dako; Trappes, France). Non-specific protein interactions were prevented by incubation in PBS containing 10% goat serum (G6767, Sigma-Aldrich) for 30 min. Diluted primary antibodies were incubated for 1.5 hr at room temperature or overnight at 4C. Following extensive washes in PBS, secondary antibodies (rabbit anti-rat IgG or biotinylated goat anti-hamster IgG) were incubated for 30 min at room temperature. After extensive washes in PBS, the amplification systems (goat anti-rabbit-dextran polymer-HRP or avidin-biotin complex-HRP) were applied for 30 min at room temperature. HRP activity was visualized with diaminobenzidine (DAB, K-3468, Dako, Trappes, France). Following color development, the reaction was stopped by washing the slides in distilled water. The tissue sections were counterstained with hematoxylin, washed in water and mounted in Mowiol® (Calbiochem, Merck, Darmstadt, Germany). Staining specificity was verified by the absence of a reaction when an isotype-matched unspecific antibody was used.
For immunofluorescence, the identical immunohistochemical protocol was used, and a fluorochrome-coupled tyramide was added for 10 min at room temperature, therebyexploiting the capability of tyramide to precipitate close to HRP (Werner et al. 1996). For double stainings, we adapted the secondary antibodies and the amplification systems to avoid cross-reactivities between antibodies used (Shindler and Roth 1996; Toth and Mezey 2007). Adapted protocols are summarized in Supplemental Table 1. The nucleic acid counterstaining was performed with a 0.5 µg/ml 4,6-diamidino-2-phenylindole (DAPI) solution (Invitrogen, Life Technologies; Grand Island, NY) for 15 min. Slides were mounted in Mowiol® (Calbiochem, Merck) and stored at 4C to avoid background autofluorescence (Staughton et al. 2001). The combinations are summarized in Table 2.
Antibody Combinations.
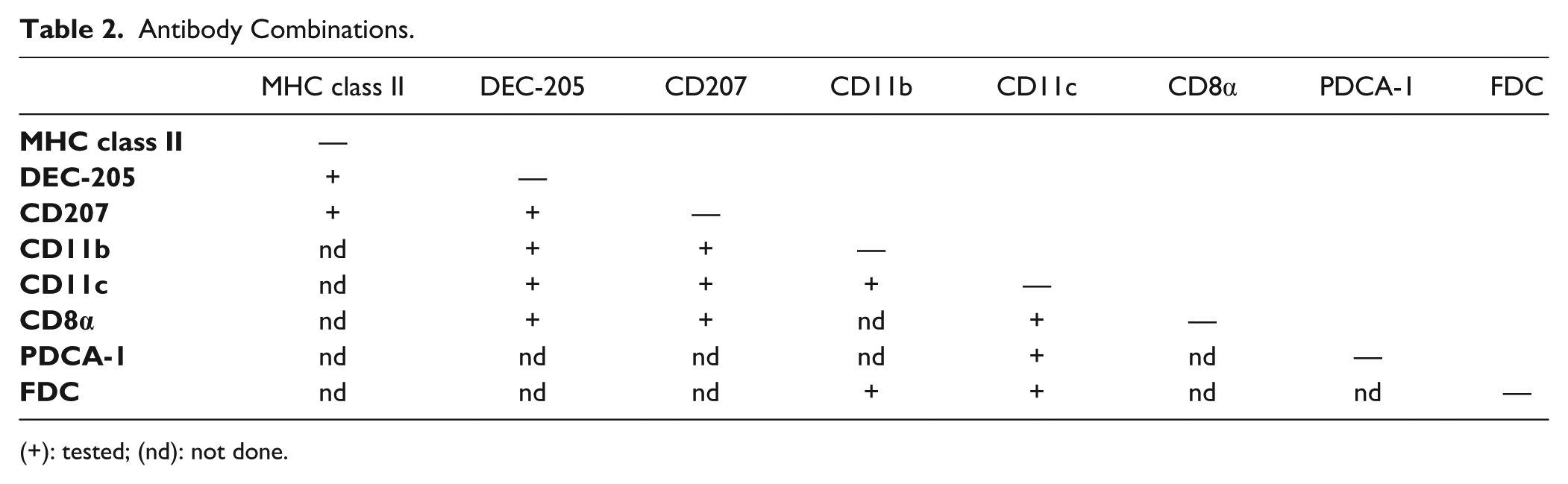
(+): tested; (nd): not done.
Immunostaining Evaluation
Stainings obtained with the chromogenic immunohistochemical protocols were observed and analysed with a bright-field optical microscope Nikon 90i (Nikon, France). The tissue integrity was evaluated by nuclei counterstaining, the presence of the different tissue compartments and their aspects according to the Comparative anatomy and histology atlas (Treuting and Dintzis 2012). The specificy of staining was confirmed when the slide incubated with the specific antibody but not the isotype-matched, non-specific antibody stained the defined cells. The staining quality was determined by comparing the signal/noise ratio between the specific and the non-specific antibody. This latter parameter was used to choose the optimal tissue treatment.
Immunofluorescence was evaluated by using excitation and emission filters adapted for the fluorochromes observed: DAPI (excitation, 357nm; emission, 461nm), FITC or Alexa Fluor 488® (excitation, 488; emission, 525), and Cy3 or Alexa Fluor 555® (excitation, 548nm; emission, 561nm). Images were acquired for the three colors on the same field. The optimal exposure time for fluorescence image acquisition was determined as the maximum time at which no background (unspecific staining) or tissue autofluorescence was detected on the slide incubated with the fluorochrome-matched isotype control antibody without there being overexposed signal on the slide incubated with the specific antibody. The free software, ImageJ (http://imagej.nih.gov/ij/; NIH, Bethesda, MD) and associated plugin (RGB Profiler) were used to ascertain the co-localization of combined markers.
Results
Single Staining of Different DC Markers
To determine an immunohistological procedure common to all antibodies, we first tested each antibody individually using a chromogenic protocol. The antibodies were validated for fixation, embedding and unmasking techniques, which allowed a good preservation of the tissue structure, as evaluated by a Hematoxylin counterstaining. All the selected markers were detected by at least one chromogenic immunohistological protocol in the studied organs. Detailed results of the chromogenic single staining patterns obtained on spleen, lymph node and skin sections are shown in Supplemental Tables 2, 3 and 4, respectively. A summary of the results correlating the capacity to detect DC markers as a function of the fixative used are shown in Table 3 and matched with the location and subtypes described in the literature (Table 4).
Summary of the Chromogenic Single Staining in Spleen, Lymph Node and Skin Sections.
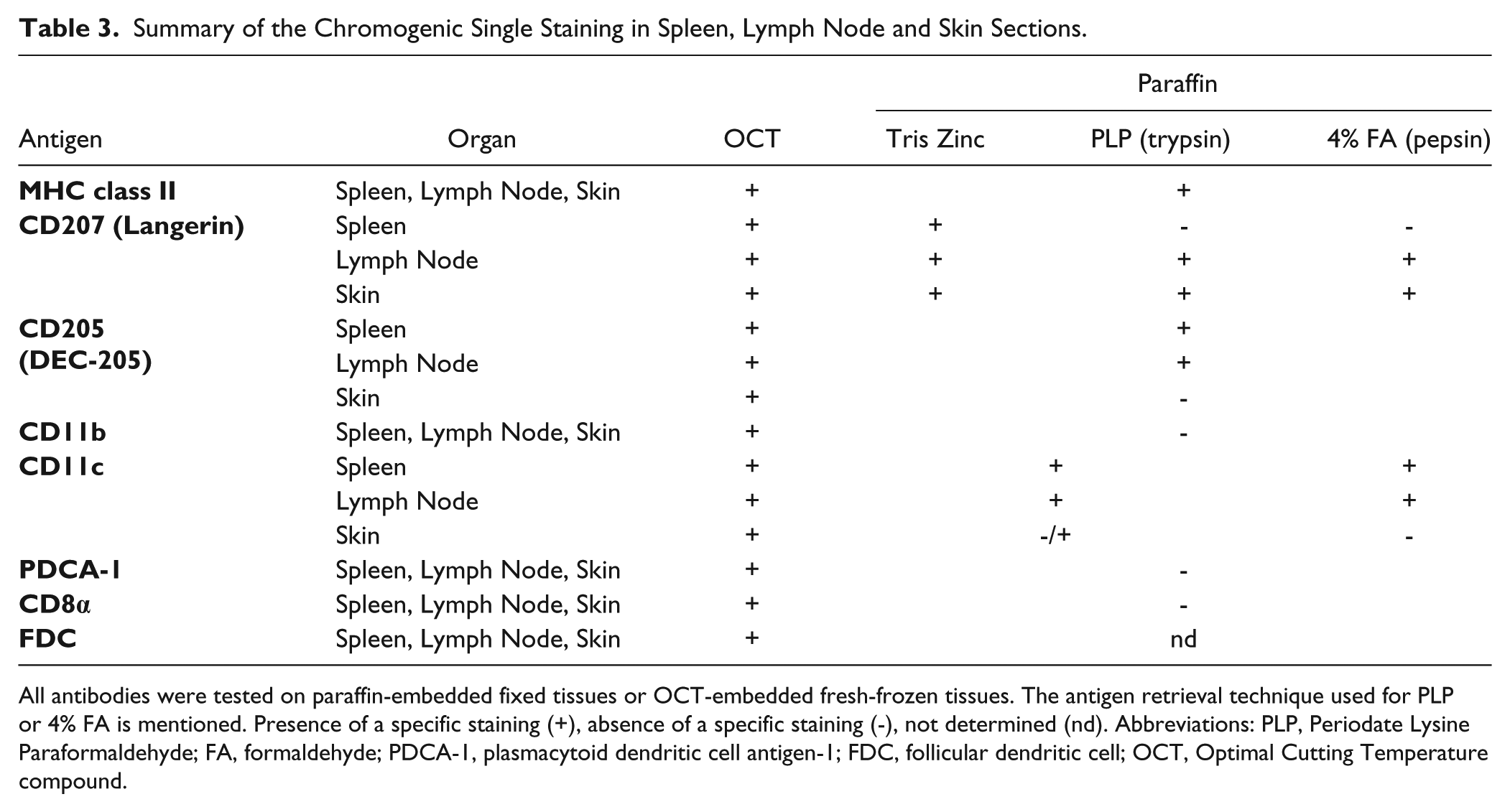
All antibodies were tested on paraffin-embedded fixed tissues or OCT-embedded fresh-frozen tissues. The antigen retrieval technique used for PLP or 4% FA is mentioned. Presence of a specific staining (+), absence of a specific staining (-), not determined (nd). Abbreviations: PLP, Periodate Lysine Paraformaldehyde; FA, formaldehyde; PDCA-1, plasmacytoid dendritic cell antigen-1; FDC, follicular dendritic cell; OCT, Optimal Cutting Temperature compound.
Summary of Main Dendritic Cell (DC) Subtypes, Location and Associated Markers from Literature.
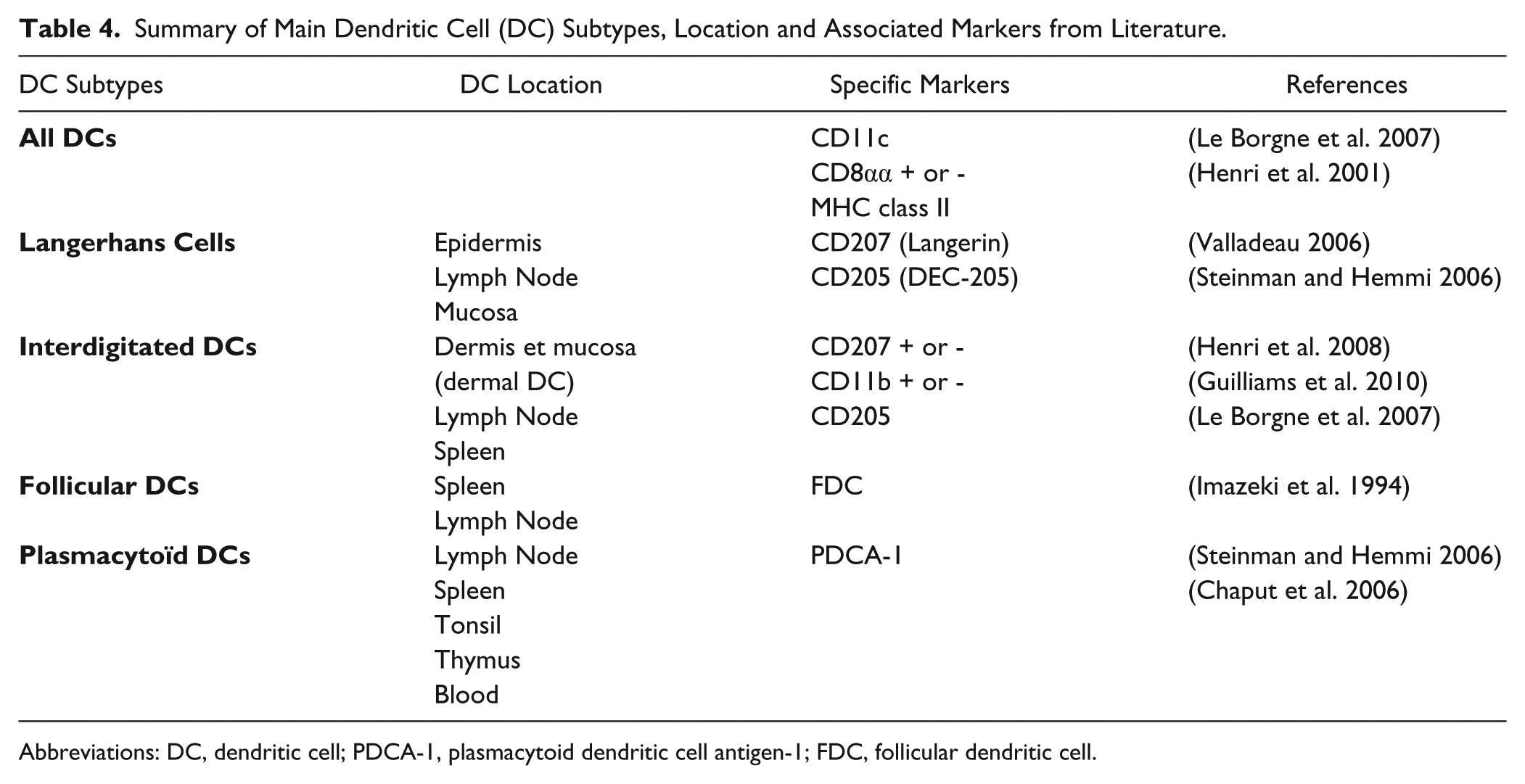
Abbreviations: DC, dendritic cell; PDCA-1, plasmacytoid dendritic cell antigen-1; FDC, follicular dendritic cell.
Some differences in the signal detection and/or staining intensity between tissues were also noticed among the fixative techniques. For example, CD205 (DEC-205) in skin was detected only in fresh-frozen tissue sections whereas it was observed on both frozen and paraffin sections of the spleen and lymph node (Table 3). CD207 (Langerin) was detected on skin sections for all conditions of fixatives and embedding procedures (Fig. 1A–1D). This observation was valid on lymph node sections whereas few langerin-positive cells were detected in the spleen, and only in frozen tissue sections and Tris Zinc-fixed-paraffin embedded tissue sections (Table 3). CD11b (Fig. 1E–1G), CD8α and PDCA-1 were not detected under paraffin-embedded conditions but they were on acetone-fixed sections (as shown for CD11b, Fig. 1H). The tissue structure was evaluated after hematoxylin nuclear counterstaining. The three paraffin-embedding conditions showed a good preservation of tissue structure, as seen by the strong, well-defined nuclear staining (Fig. 1A–1C, 1E–1G) compared with the diffuse and weak nuclear staining in the frozen sections (Fig. 1D and 1H); this difference confirmed the poor quality of the latter support. However, the three fixatives used before paraffin embedding gave varying results in terms of marker detection, and precluded antibody combinations for multiple stainings protocols. Overall, acetone-fixed sections of fresh-frozen tissue are the best support for multistaining immunofluorescent protocols of well-defined DC subpopulations.
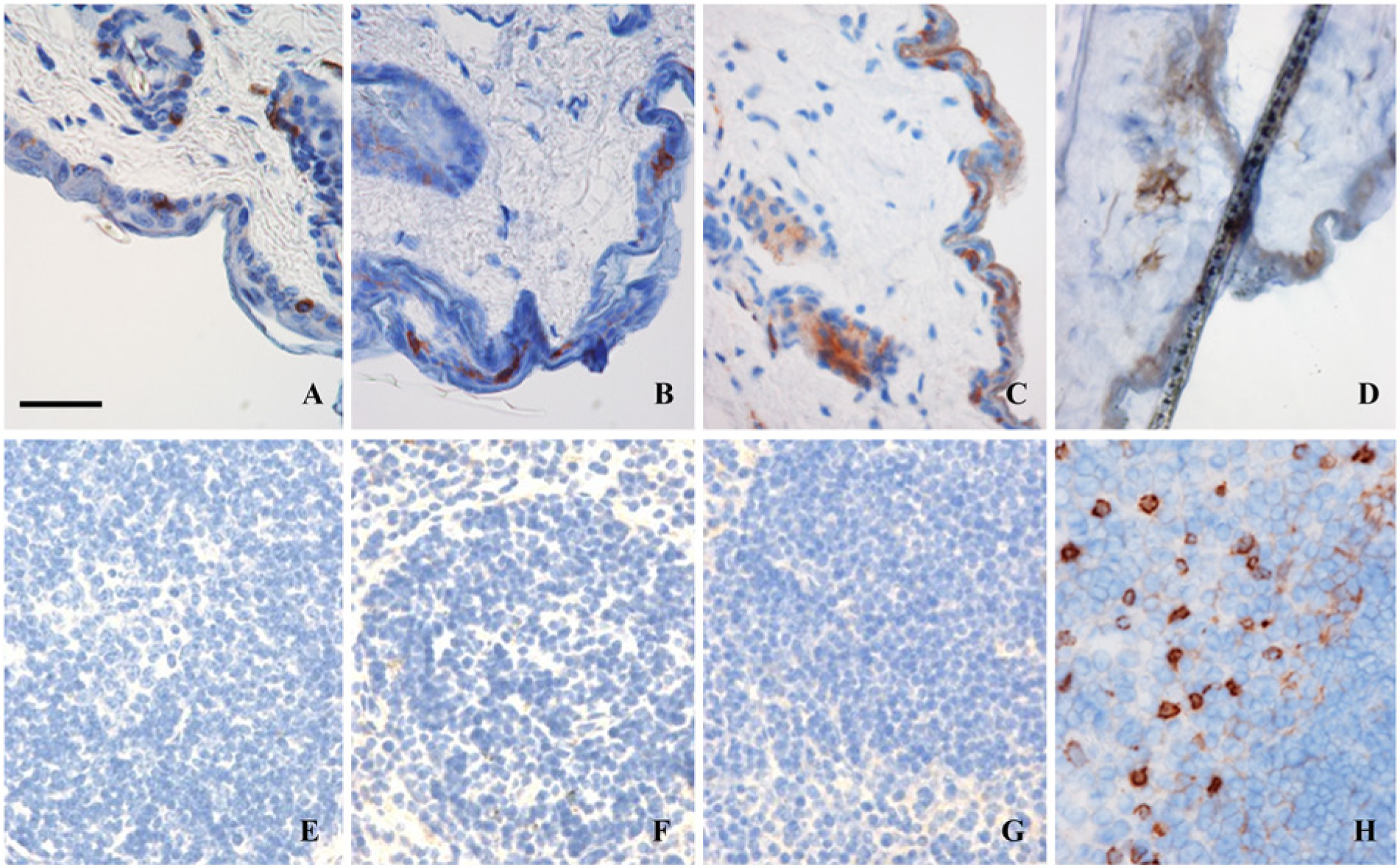
Two examples of immunohistochemical staining in the skin (A–D) and spleen (E–H): CD207 and CD11b. CD207 (Langerin) is detected in all fixative and embedding conditions: Paraffin-embedded skin after (A) 4% FA, (B) periodate lysine paraformaldehyde (PLP) and (C) Tris Zinc fixation methods. (D) Acetone-fixed skin cryosections. The CD11b marker is not detected on paraffin-embedded splenic tissue after (E) 4% FA, (F) PLP or (G) Tris Zinc fixations, and is detected only on spleen cryosections (H). Scale bar = 50µm.
To this end, multiparametric immunofluorescence was obtained by a direct translation of the chromogenic protocols to fluorescent protocols by the use of tyramide. Both methods yielded similar numbers of positive cells and a similar cell distribution in the studied tissues (Fig. 2).
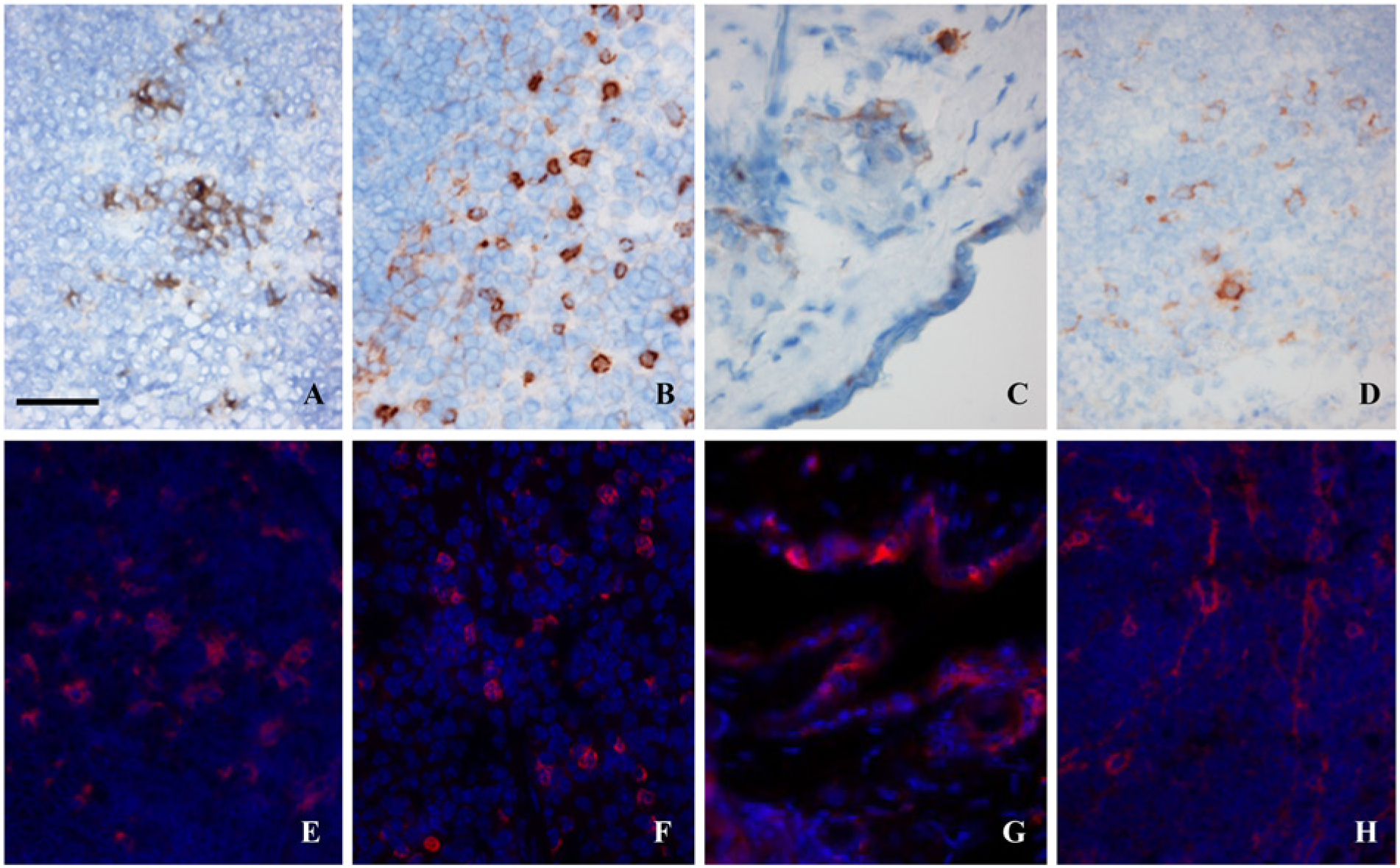
The translation of chromogenic protocol to fluorescent protocol did not affect the detection of all DC markers. For example, langerin-positive cells were detectable in lymph node (A, E), CD11b in spleen (B, F), CD205 in skin (C, G) and PDCA-1 in spleen (D, H). Scale bar = 50 µm.
DC Subset Detection after Antibody Combinations
This work was performed only on acetone-fixed sections of fresh-frozen tissues. For double staining, some secondary antibodies and amplification systems had to be changed from those used for the chromogenic staining and immunofluorescent single staining to avoid cross-reactivities between antibodies. To this end, other amplification systems were first tested in immunofluorescent single staining protocols (Supplemental Table 1) and selected for their ability to give the same results as described above. The use of the EnvisionTM detection kit associated with TSA was replaced by a biotinylated secondary antibody associated with a fluorochrome-labeled streptavidin for MHC Class II and CD11b detection. CD8α was detected with a HRP-labeled secondary antibody associated with a tyramide signal amplification system (TSA™). CD205 was detected with a biotinylated secondary antibody associated with an avidin-biotin complex and TSA™. No change was possible for CD207, CD11c, PDCA1 and FDC.
Once the conditions for the fluorescent antibodies were defined, the combinations described in Table 2 were applied to tissue sections. The results reflected the complexity of the various DC subsets. For example, CD11c is not a pan-dendritic cell marker as shown by the presence of the follicular DC marker (FDC+ CD11c-; Fig. 3A, cell C1) in the white pulp of the spleen, or by the presence of plasmacytoid DC marker, PDCA1+, which can be CD11c+ (Fig. 3B, cell C2) or CD11c- (Fig. 3B, cell C1) in the follicle and paracortex of the lymph node (Fig. 3). Once again, in these multiparametric staining conditions, the frozen sections showed a diffuse and weak nuclear staining, confirming the poor quality of the support.
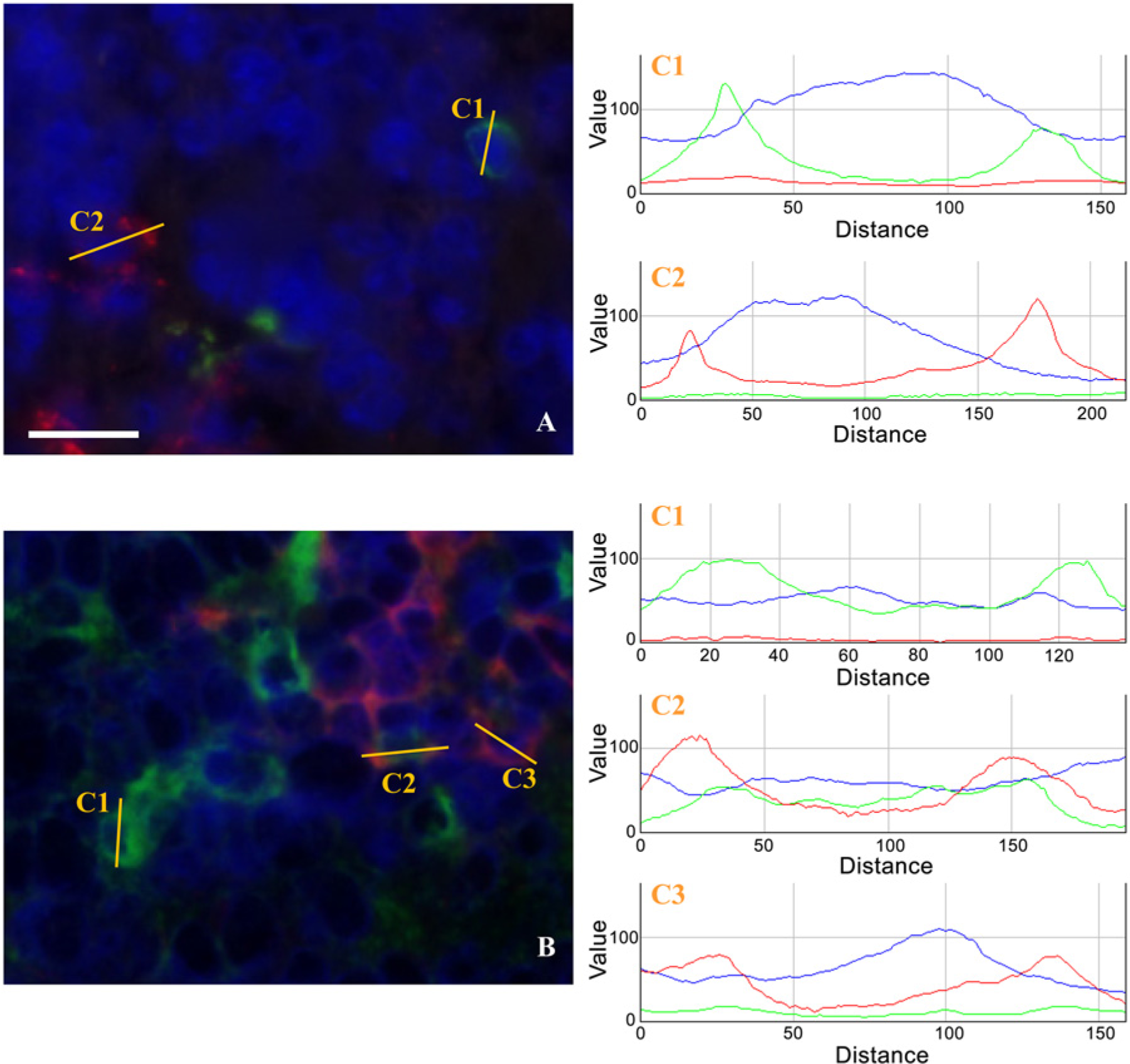
Double staining. Follicular dendritic cells stained positively using their specific follicular dendritic cell (FDC) marker (green) but were negative for the CD11c marker (red) (A) in the white pulp of the spleen. Cell 1 (C1) is an FDC that is also CD11c-. Cell 2 (C2) is a CD11c+ FDC- dendritic cell. Plasmacytoid dendritic cells stained positively using their specific marker, PDCA-1 (green) (B). These cells could be either negative (C1) or positive (red, C2) for the CD11c marker. The dendritic cell 3 (C3) is positive for CD11c and negative for PDCA-1. Scale bar = 50 µm.
To obtain better tissue preservation for the easier localization of these DC subsets, we developed an alternative fixation and embedding protocol based on a soft fixative, sucrose and gelatin.The first read-out was the tissue structure observed after Hematoxylin and Eosin staining in FFPE tissues (Fig. 4A), Tris Zinc-fixed, paraffin-embedded tissues (Fig. 4B), fresh-frozen OCT-embedded tissues (Fig. 4C) and Tris Zinc-fixed gelatin-embedded frozen tissues (Fig. 4D). Tissue fixation before embedding in a modified water-soluble embedding medium enhanced the preservation of layer components, especially in skin tissue samples, as demonstrated by the clear observation of nuclei in the epidermis or the presence of muscle striations (Fig. 4D, arrows). In the OCT condition, we observed cell disruption with bad nuclei preservation in non-compact organs such as the spleen and lymph node but well-preserved tissues using the gelatin condition (data not shown). The 7.5% gelatin/15% sucrose combination was chosen to detect combinations of DC markers. All of the selected DC markers were detected on cryosections from tissues fixed in Tris Zinc/15% sucrose and embedded in 7.5% gelatin/15% sucrose (Fig. 5), in the same manner as previously described. These conditions of fixation and embedding were used for the antibody combinations described in Table 2. The results are presented in Supplemental Tables 5, 6 and 7, and reflect the complexity of the DC subsets.
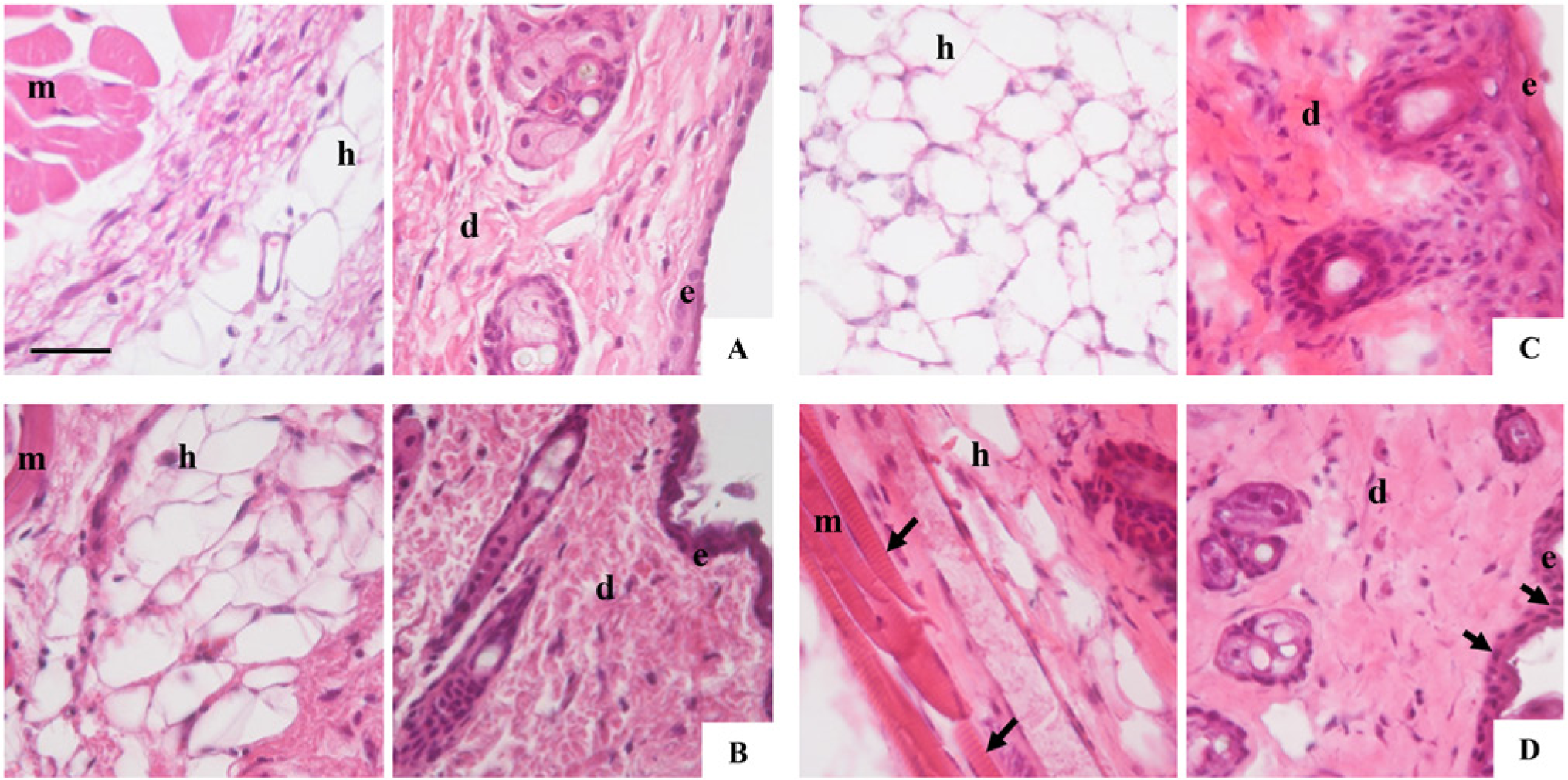
Organ structure in different fixation and embedding conditions. Hematoxylin and Eosin staining of skin sections that were (A) 4% formaldehyde (FA)-fixed and paraffin-embedded, (B) Tris Zinc-fixed and paraffin-embedded, (C) fresh-frozen and O.C.T.-embedded, or (D) Tris Zinc/15% sucrose-fixed and 7.5% gelatin/15% sucrose embedded. Left panels show hypodermis (h) and muscle layer (m). Right panels show dermis (d) and epidermis (e). (D) Arrows show epidermal nuclei in the right panel and muscle striations in the left panel. Scale bar = 50 µm.
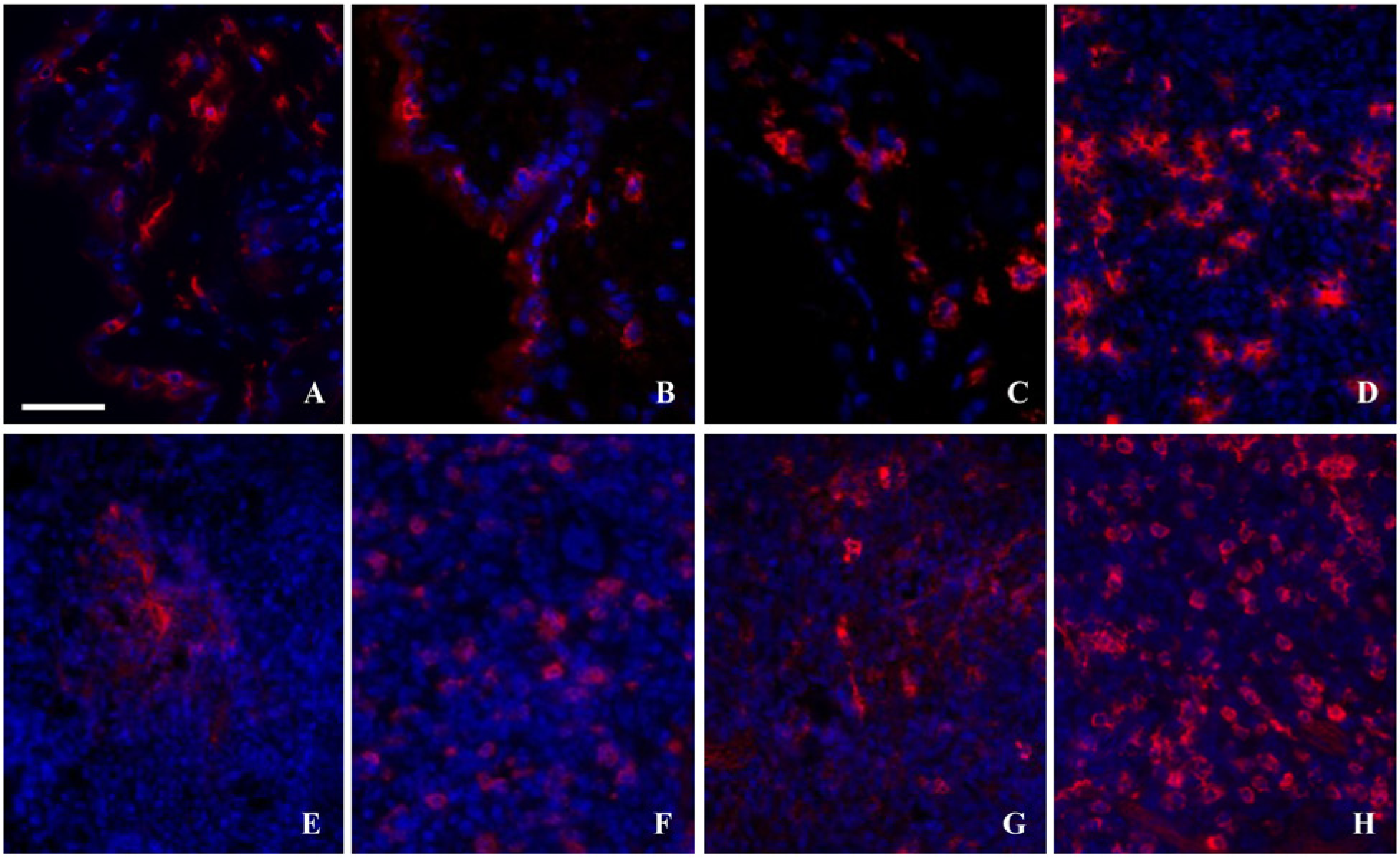
Dendritic cell (DC) markers on Tris Zinc/15% sucrose-fixed, 7.5% gelatin/15% sucrose-embedded sections: (A) MHC class II, (B) DEC205, and (C) CD11c are detected in skin. Langerin (D) andfollicular dendritic cells (FDCs) (E) are observed in lymph node and CD11b (F), PDCA-1, (G), and CD8α (H) are found in the spleen. Scale bar = 50 µm.
Discussion
In order to achieve a combination of tissue fixation and embedding that is satisfactory for immunofluorescence of multiple markers, it was interesting to work on DCs because of the complexity of their subsets. Many in vitro methods, such as direct isolation of DCs from sites of immune induction (Waithman et al. 2007), DC targeting by mAb such as CD205 (Bonifaz et al. 2002; Bonifaz et al. 2004; Hawiger et al. 2001) or driven expression of the simian diphtheria toxin receptor (DTR) under the control of the CD11c promoter (Jung et al. 2002; Saito et al. 2001) have been used to study DCs and their involvment in immune responses. But an exhaustive in vivo detection of DC subsets had yet to be published and we decided to establish protocols for an in situ observation of the different DC subsets. To this aim, we used and compared different methods available for fixation associated with embedding of tissue samples for the detection of all of the selected DC markers on well-preserved tissues.
Ten percent neutral-buffered formalin fixative associated with paraffin embedding is one of the most commonly used techniques in histology. Although these techniques allow for the excellent preservation of tissue structures, the tri-dimensional structure of proteins is often altered by formaldehyde by the formation of reactive hydroxyl-methyl bridges at sites of reactive hydrogens (Sompuram et al. 2004) and reactions with other chemical compounds, such as calcium (Shi et al. 2001). The optimal preservation of the antigen immunoreactivity is also dependent on the formaldehyde concentration and decreasing it from 4% (FA 4%) to 0.25% (PLP) allows for a better immunostaining for numerous markers (Whiteland et al. 1995). We confirmed this point for cutaneous CD11c-positive cells. The use of formaldehyde may be a problem when a native protein has been used for antibody generation, as the antibody could be directed against a conformational epitope. Antibodies generated with a peptide or a denaturated protein, such as an immunogen, may increase the chances of successful staining on formaldehyde-fixed tissues (Bordeaux et al. 2010).
An alternative method is the use of soft fixative based on Tris Zinc (Beckstead 1994). The reaction between the Zn2+ ions and the negative groups maintains the tri-dimensional structure of molecules like nucleic acids and proteins. This fixative, composed of safe, non-environmentally damaging chemicals, allows for the detection of Langerin-positive cells in splenic tissue whereas formaldehyde-based fixatives do not. But embedding in paraffin, for which the fusion point is 58C, is also a destructive treatment for thermolabile proteins. No or weaker CD11b, CD8α, DEC205 and CD11c staining in the skin is detected after Tris Zinc fixation and paraffin embedding as compared to that obtained using cryopreserved, non-fixed tissues.
The condition allowing the detection of all of the studied markers, including thermolabile or chemically cross-linked proteins, which are not detected after chemical tissue fixation and/or paraffin embedding, is the technique using fresh tissue freezing from which sections are fixed in acetone. However, the limitation of this method is the bad preservation of tissue structure because OCT is only an inclusion medium and does not penetrate inside the cells as paraffin does. Water is not evacuated and forms crystals inside the cells during the freezing process; this contributes to the tissue disruption (Callis 2004). In order to circumvent these problems and to achieve a satisfactory compromise between good tissue structure and antigen detection, an alternative protocol was derived from a technique used in electronic microscopy (Fernandez-Moran and Finean 1957). The original protocol describes the use of a tissue fixation adequate for electronic microscopy, followed by successive baths in gelatin solutions until the concentration of gelatin reached 30%. The duration of the protocol makes the tissue fixation necessary before inclusion. The choice of a soft fixative, such as Tris Zinc, was dictated by the arguments listed above. The addition of 15% sucrose allowed for better tissue penetration during the protocol and, in particular, acts as a cryoprotectant, preventing ice crystal formation during freezing (Barthel and Raymond 1990). Moreover, the use of gelatin at the physiological temperature of 37C does not impact on thermolabile antigens. This technique has been derived for multiple applications, including the study of odontoblasts (Byers and Sugaya 1995) and the obtention of ultrathin cryosections of polarized cells (Oorschot et al. 2002). Different embedding solutions containing variable percentages of gelatin were compared with structure and staining quality as readouts. Due to the difficulty in preparing a 30% gelatin/15% sucrose solution, a comparison of the structure preservation was done with a 15% gelatin/15% sucrose solution and a 7.5% gelatin/15% sucrose solution. No clear difference was observed between gelatin/sucrose solutions. For our purpose, all the selected markers have been detected on cryosections from tissues fixed in Tris Zinc/15% sucrose and embedded in 7.5% gelatin/15% sucrose and tissue structures are well preserved as compared to that obtained with O.C.T. Single stainings as well as multiple stainings reflect the complexity of DC subsets in terms of the localization and detection of markers (Douillard et al. 2005). This technique, initially developed for electron microscopy, was easily adapted and applied for light microscopy. Encouraging is the fact that these improvements in section quality are achieved with materials already at hand, and they do not require training in a new technique. Therefore, the system is less flexible in that tissue samples cannot be left in fixative for prolonged periods before processing.
In summary, the ability to maintain antigen integrity while providing excellent morphology makes the Tris Zinc fixation and 7.5% gelatin/15% sucrose embedding method we describe suitable for immunohistochemical localization of immune cells.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.