
Abstract
In the current era of genomic medicine, diseases are identified as manifestations of anomalous patterns of gene expression. Cancer is the principal example among such maladies. Although remarkable progress has been achieved in the understanding of the molecular mechanisms involved in the genesis and progression of cancer, its epigenetic regulation, particularly histone deacetylation, demands further studies. Histone deacetylases (HDACs) are one of the key players in the gene expression regulation network in cancer because of their repressive role on tumor suppressor genes. Higher expression and function of deacetylases disrupt the finely tuned acetylation homeostasis in both histone and non-histone target proteins. This brings about alterations in the genes implicated in the regulation of cell proliferation, differentiation, apoptosis and other cellular processes. Moreover, the reversible nature of epigenetic modulation by HDACs makes them attractive targets for cancer remedy. This review summarizes the current knowledge of HDACs in tumorigenesis and tumor progression as well as their contribution to the hallmarks of cancer. The present report also describes briefly various assays to detect histone deacetylase activity and discusses the potential role of histone deacetylase inhibitors as emerging epigenetic drugs to cure cancer.
Keywords
Introduction
Heritable alterations in gene expression, without any deviation in the primary sequence of DNA, are the crux of epigenetic modifications. DNA methylation and the covalent modification of histones encompass major epigenetic features on chromatin, which, by altering nucleosomal organization, leads to gene activation or repression (Vaissiere et al. 2008). An extensive literature documents an elaborate collection of reversible, dynamic post-translational modifications (PTMs) namely, acetylation, methylation, phosphorylation, ADP-ribosylation, ubiquitination, biotinylation and SUMOylation occurring extensively in the histone-tail and occasionally in the core histones. Of all the PTMs, histone acetylation has been the most extensively studied and appreciated (Grunstein, 1997). Histone acetylation involves the addition of an acetyl moiety to the ϵ-amino terminal of lysine residues within the tail domain of the core histone, mostly in H3 and H4. This neutralizes the positive charge: lessening the electrostatic interaction between DNA and histones, thereby forming relaxed chromatin. For example, H4-K16 acetylation disrupts the formation of higher-order chromatin structures (Shogren-Knaak et al. 2006). The relaxed and open chromatins thus generated are easily accessible to transcriptional machinery. Moreover, acetylated histones act as binding platforms for bromodomain proteins, which are transcriptional activators. Thus, histone acetylation aids in transcriptional activation and maintenance of euchromatin (Shogren-Knaak et al. 2006). In contrast, the removal of acetyl groups from the acetylated histones, “histone deacetylation”, forms the basis of inactive chromatin environment. This implies transcriptional inactivation due to inaccessibility of transcriptional proteins on a gene promoter (Figure 1). This establishes histone acetylation/deacetylation as a chief epigenetic regulator of gene transcription and chromatin topology. Two antagonistic sets of enzymes, histone acetyl transferases (HATs) and histone deacetylases (HDACs) reversibly catalyze the acetylation and deacetylation processes, respectively (Smith and Denu, 2009) (For detail mechanism, see Figure 2c).
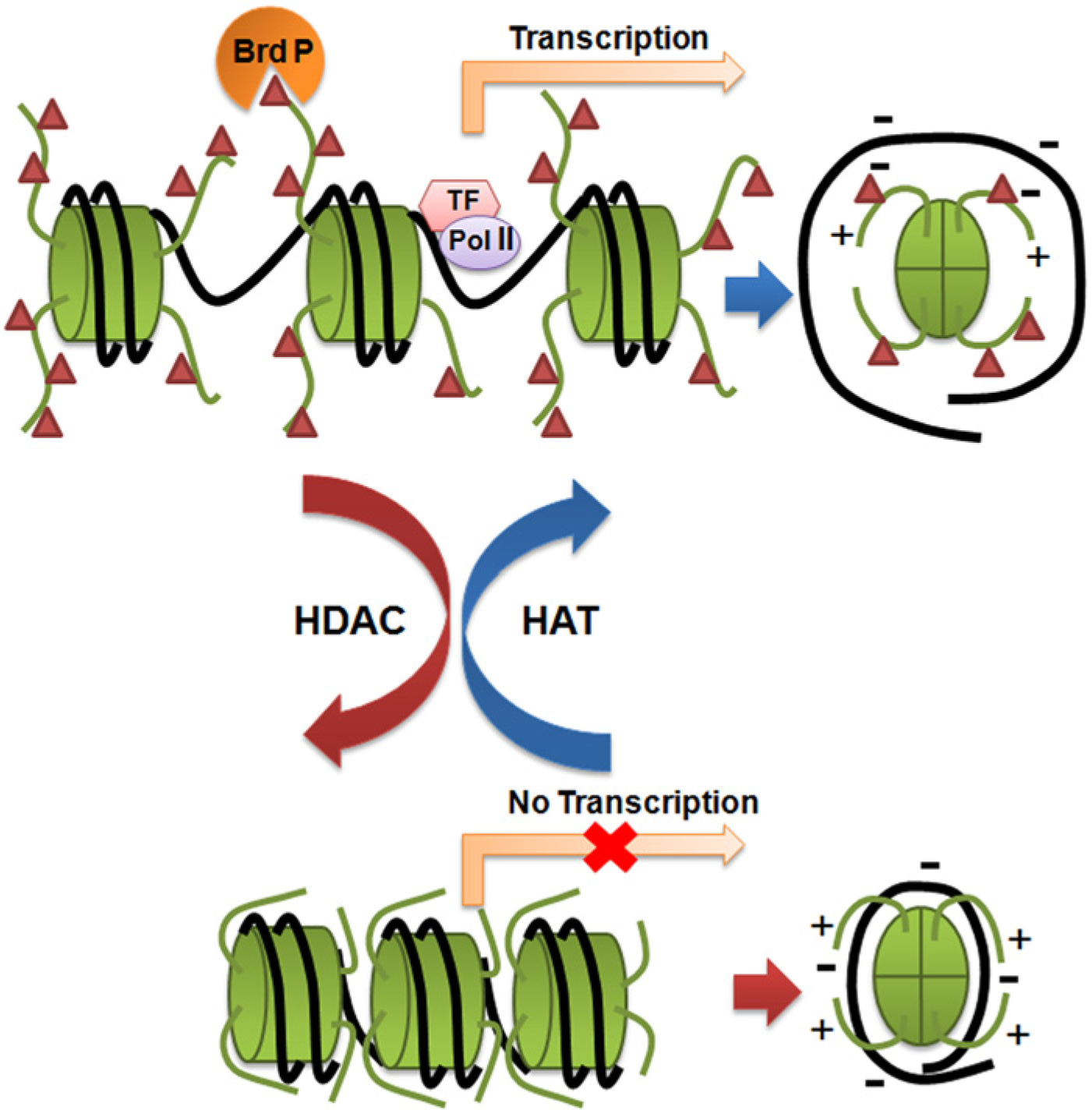
Alteration in chromatin topology catalyzed by histone acetylases (HATs) and histone deacetylases (HDACs). Top portion shows open, transcriptionally active chromatin with exposed binding sites for transcriptional machinery and acetylated histone tails acting as docking site for Brd P (Bromodomain-containing transcriptional factors). Bottom portion depicts the compact and transcriptionally inactive chromatin with no available binding sites for transcriptional machinery. The images on the right hand side show electrostatic interactions: (top) in the case of relaxed chromatin, reduced interaction between histone tail (green) and DNA (black); and (bottom) increased interactions forming compact chromatin. Green cylinders are nucleosomes and red triangles are acetyl groups.
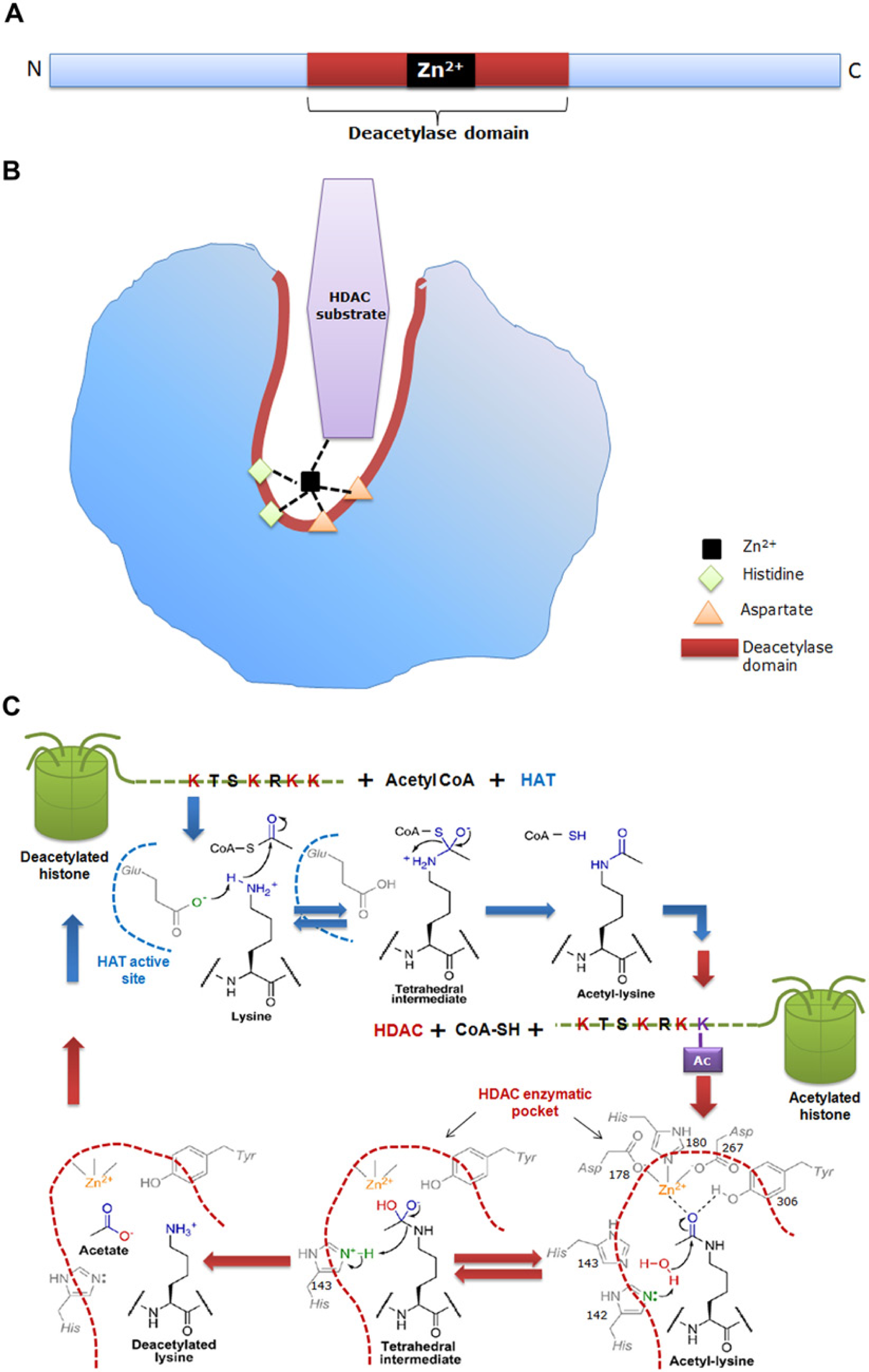
(a) Schematic representation of the primary structure of a classical histone deacetylase (HDAC) enzyme, depicting the catalytic site as red/brown box and a Zn2+ site as a black box. (b) Schematic representation of the 3-dimensional structure of the HDAC showing a narrow, tubular catalytic pocket with a Zn2+ near its base. Zn2+, with the aid of histidine-aspartate charge relay system, imparts catalytic activity to the HDAC enzyme. Black dotted lines represent charge relay between histidine, aspartate residues and HDAC substrate. (c) Detailed mechanism of the histone acetylase (HAT) and HDAC enzymatic reaction. During acetylation, the active site glutamate (Glu) of HAT acts as general base to activate the e-amine group of lysine, which helps in the nucleophilic attack of the carbonyl carbon within acetyl-CoA, thus producing a tetrahedral intermediate. Subsequently, the tetrahedral intermediate collapses to form acetylated lysine and CoA. The active site of HDAC harbors His, Asp and Tyr residues. In deacetylation, both His-142 and His-143 can act as general bases by donating a pair of electrons to the water molecule to activate it for nucleophilic attack of the acetyl carbonyl carbon. An attack by water results in a tetrahedral intermediate, which is stabilized by Zn2+ and Tyr-306. Subsequently, the tetrahedral intermediate collapses to form acetate and lysine products, with His-143 acting as a general acid to protonate the e-amine leaving group of lysine. Blue dotted line depicts the active site of HAT and brown dotted line represents the tubular pocket of active site of HDAC. (Chemical structures adopted from Smith & Denu, 2009).
In general, HATs are ascribed the function of transcription co-activators whereas HDACs act as transcriptional co-repressors. Loss of acetylating agents in conjunction with a gain in HDAC activity is one of the several epigenetic abnormalities in cancer. A wealth of published data strongly supports the repression of tumor suppressor genes as one of the prime reasons for carcinogenesis and cancer progression. Of all epigenetic modulators responsible for such repression, HDACs occupy a central position. Undoubtedly, HDACs are concerned with a multitude of cancers especially because of their tumorigenic activity. Moreover, recently, the identification of non-histone targets of HDACs make them as the most powerful tools to explore the acetylome in eukaryotes, which may be possible by studying functional genomics approaches and assessing the effects of gene over expression in the absence of HDACs (Choudhary et al. 2009). Considering the emerging interest in HDACs, it is necessary to have a detailed account of these enzymes. The present review reveals some facts behind HDACs and their roles in the disruption of the well-orchestrated homeostasis of acetylation in cancer.
1. The Biology and Chemistry of HDACs
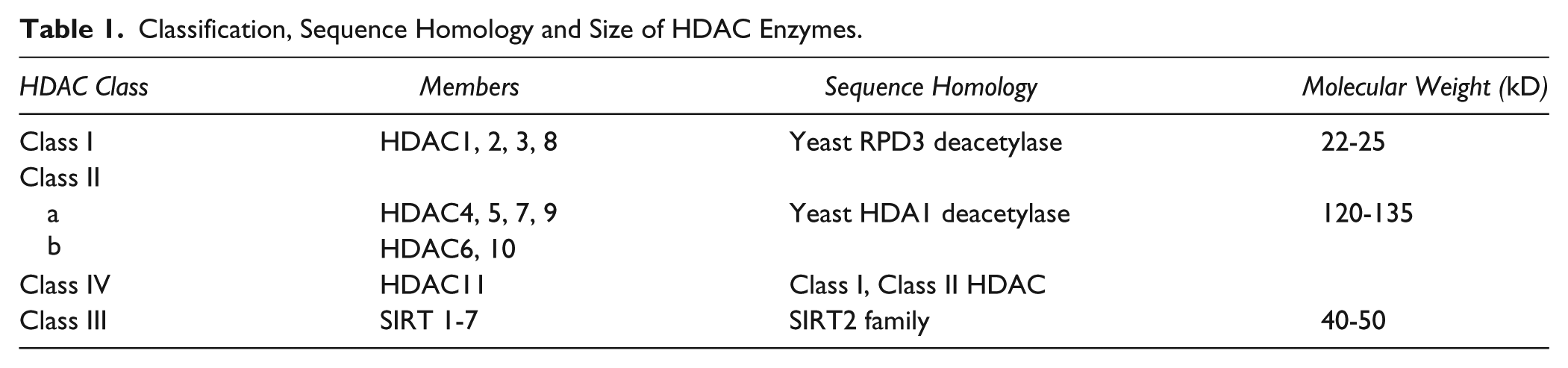
HDACs are highly conserved from yeast to human. Thus far, 18 HDACs have been documented in humans. Based on the structure, function, sub-cellular localization and pattern of expression as well as homology to yeast HDACs, the vast deacetylase family can be classified into four classes (Refer Table 1 and Figure 3 for a detailed classification) (Haberland et al. 2009). Class I, II and IV HDACs comprise classical HDACs, which are zinc-dependent amidohydrolases, sensitive to Zn2+ chelating compounds such as hydroxamic acids. Class III HDACs, in contrast, belong to the recently discovered sirtuin family, and are homologous to the yeast Sir2 (Silent information regulator 2) family of proteins. Being NAD+-dependent enzymes, sirtuins are insensitive to hydroxamic acid-derived HDAC inhibitors (HDACi) (Dokmanovic et al. 2007).
Classification, Sequence Homology and Size of HDAC Enzymes.
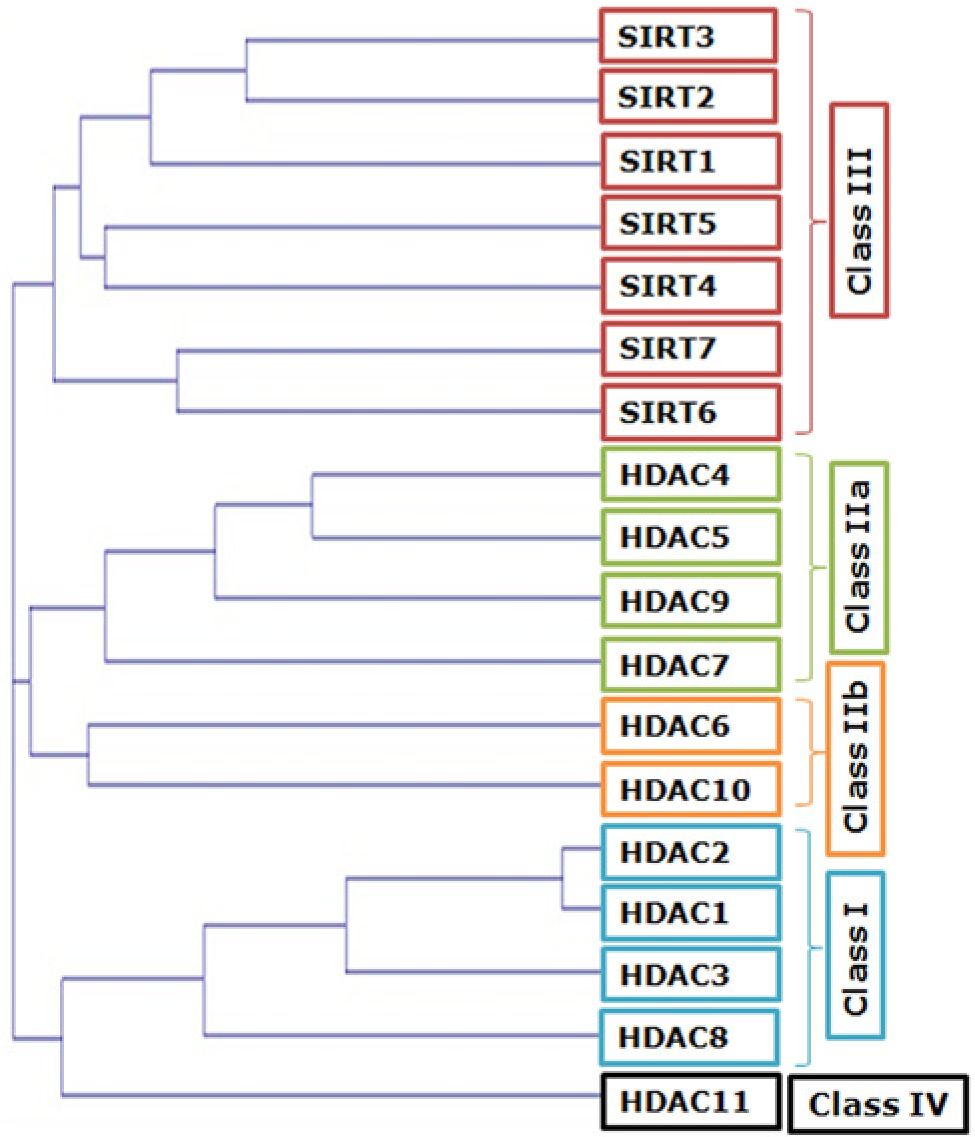
Phylogenetic tree and classification of histone deacetylase (HDAC) enzymes (Created using ClustalW software).
Each classical HDAC shares a common deacetylase domain of approximately 390 amino acids constituting the catalytic site. A narrow cylindrical structure of a length equivalent to that of a 4–6 carbon straight chain with a Zn2+near its base, forms the enzyme pocket of the catalytic domain. This catalyzes the deacetylation in assistance with two Histidine-Aspartate charge-relay systems (Lin et al. 2006; Smith and Denu, 2009) (For details see Figure 2). The ubiquitously expressed and nuclear-residing Class I HDACs have a relatively simple structure with a conserved deacetylase domain. Class II HDACs can shuttle between the nucleus and the cytoplasm, thus governing the acetylation status of non-histone substrates (Haberland et al. 2009). HDAC6, the main cytoplasmic deacetylase in mammalian cells, has two complete catalytic domains. The second catalytic domain of HDAC6 deacetylates cellular proteins, whereas that of HDAC10 is incomplete and little is known about its function. Class III HDACs harbor quite a different conserved catalytic domain of approximately 275 amino acids, containing aspartic acid and glycine residues (Frye, 1999).
Due to the lack of intrinsic DNA binding ability, HDACs are recruited to DNA via transcription factor protein complexes. Different HDACs reside in distinct protein complexes, which vary in their subunit composition. HDACs 1 and 2 dwell in complexes with Sin3, NuRD (nucleosome remodeling and deacetylation), N-CoR (nuclear receptor corepressor), mSin3A, Mi-2/NRD, PRC2 and/or CoREST. HDAC3 is present in distinct complexes, such as the N-CoR– Silencing mediator for retinoic and thyroid receptor (SMRT) complex, whereas no complex has been reported for HDAC8 (Haberland et al. 2009; Witt et al. 2009).
2. Acetylation Homeostasis and Its Disruption in Cancer
Recently, cellular-wide proteomic analyses on protein acetylation have used combinatorial approaches with high-affinity acetyl-lysine antibodies, mass spectrometry (MS) and stable-isotope amino-acid labeling (SILAC) techniques to detect almost 2000 acetylated proteins comprising almost 3,600 acetylation sites in the cell (Choudhary et al. 2009). The sum of all acetylated sites on a set of acetylated proteins in a cell is designated as the ‘acetylome’. Despite histone proteins, over 60 different transcription factors—proteins implicated in DNA repair and replication, metabolism, cytoskeletal dynamics (tubulin), apoptosis, protein folding and cellular signaling—constitute the acetylome (Yang and Gregoire, 2007).
The acetylation status of the entire acetylome are governed by a stoichiometrically balanced HAT and HDAC enzymatic activities, brought about by the alternative exchange between these two enzymes. This phenomenon, termed ‘acetylation homeostasis’ (Figure 4a), confers stability to cellular homeostasis, a condition that prevails in normal cells (Saha and Pahan, 2006). Experimental evidence has clearly shown that this well-tuned balance between HATs and HDACs is a prerequisite for true gene expression. During normal physiological conditions, the acetylation status of histones, transcription factors and other proteins of the acetylome are harmoniously regulated. Balanced protein concentration, enzymatic activity and the proper recruitment of HAT and HDAC enzymes are responsible for such homeostasis. Such equilibrium reveals the cellular homeostasis and is essential for regulated gene expression leading to normal cellular functions. However, a wealth of studies has indicated that aberrant action and the differential recruitment of HATs and HDACs disrupt acetylation homeostasis (Figure 4b). Indeed, fazed acetylation homeostasis has been linked to various neurodegenerative diseases, as well as non-cancerous and cancerous disorders. Here, we mainly discuss the oncogenic role of HDACs in various stages of different types of cancer.
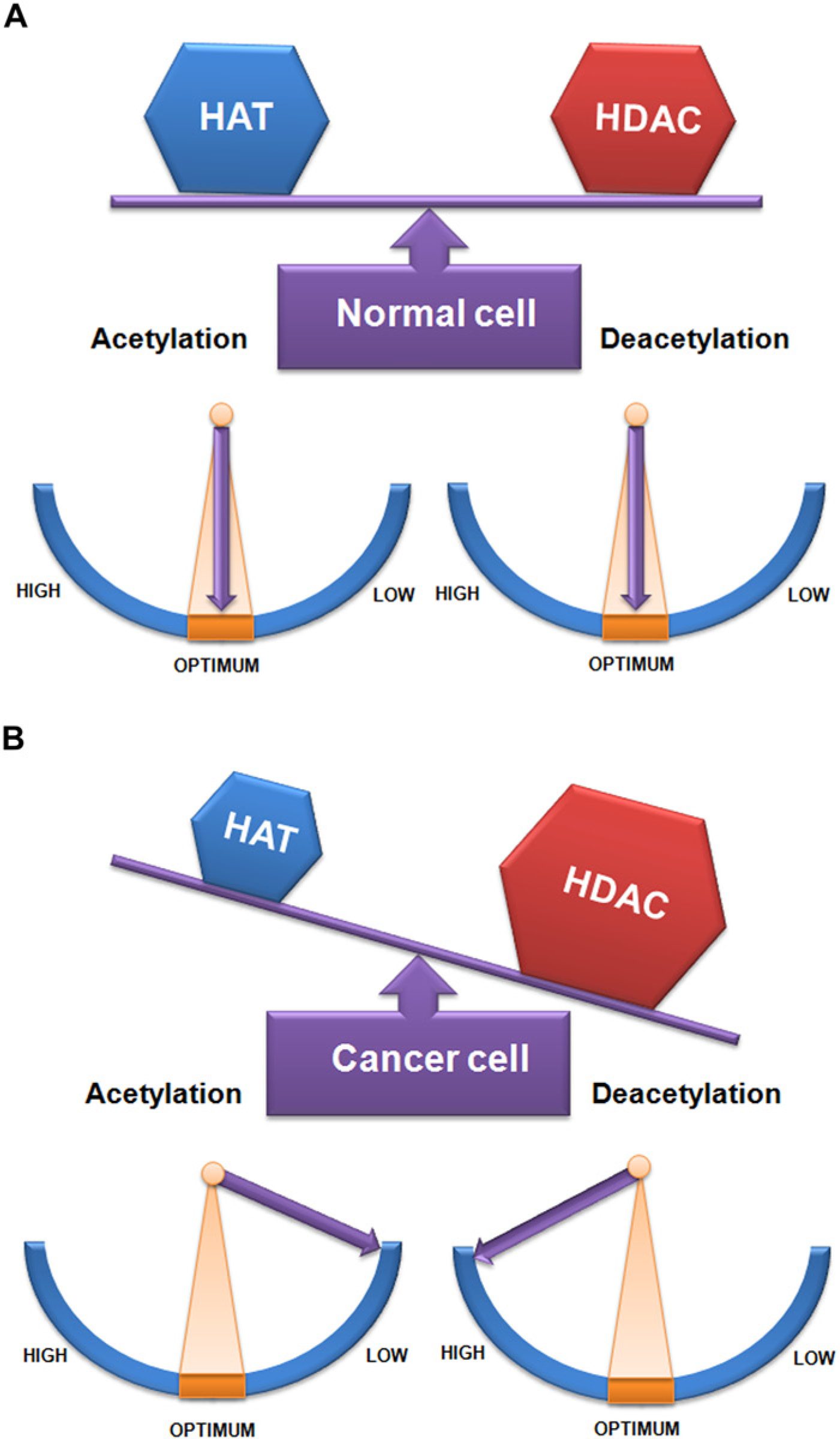
(a) The schematic depicts acetylation homeostasis in a normal cell, with a balance in the amount and activity of HAT and HDAC. (b) The schematic depicts disrupted acetylation homeostasis in cancer cells, with an imbalance in the amount and activity of HAT and HDAC shifting more towards HDAC. The size of the blue and brown hexagons represents the amount of HAT and HDAC, respectively. Enzymatic activity is optimum within the orange region indicating balanced acetylation state. High activity of HDAC and low activity of HAT indicate a cancerous state (Idea adopted from Saha & Pahan, 2006).
In tumorigenesis, the finely tuned acetylation status is greatly impaired and shifts towards deacetylation. This brings about changes in proliferation, differentiation and apoptosis of normal cells and, in simplistic terms, transforms them into malignant cells; this is also implicated in the loss of adhesion, migration, invasion and angiogenesis, thus leading to the origin and progression of cancer (Figure 5). HDACs are well known for their repressive actions on genes. The aberrant recruitment of HDAC activity has been associated with the development of multiple human tumors such as promyelocytic leukemia, non-Hodgkin lymphoma, colon cancer and gastric cancer (Chevallier et al. 2004; Huang et al. 2005a; Song et al. 2005).
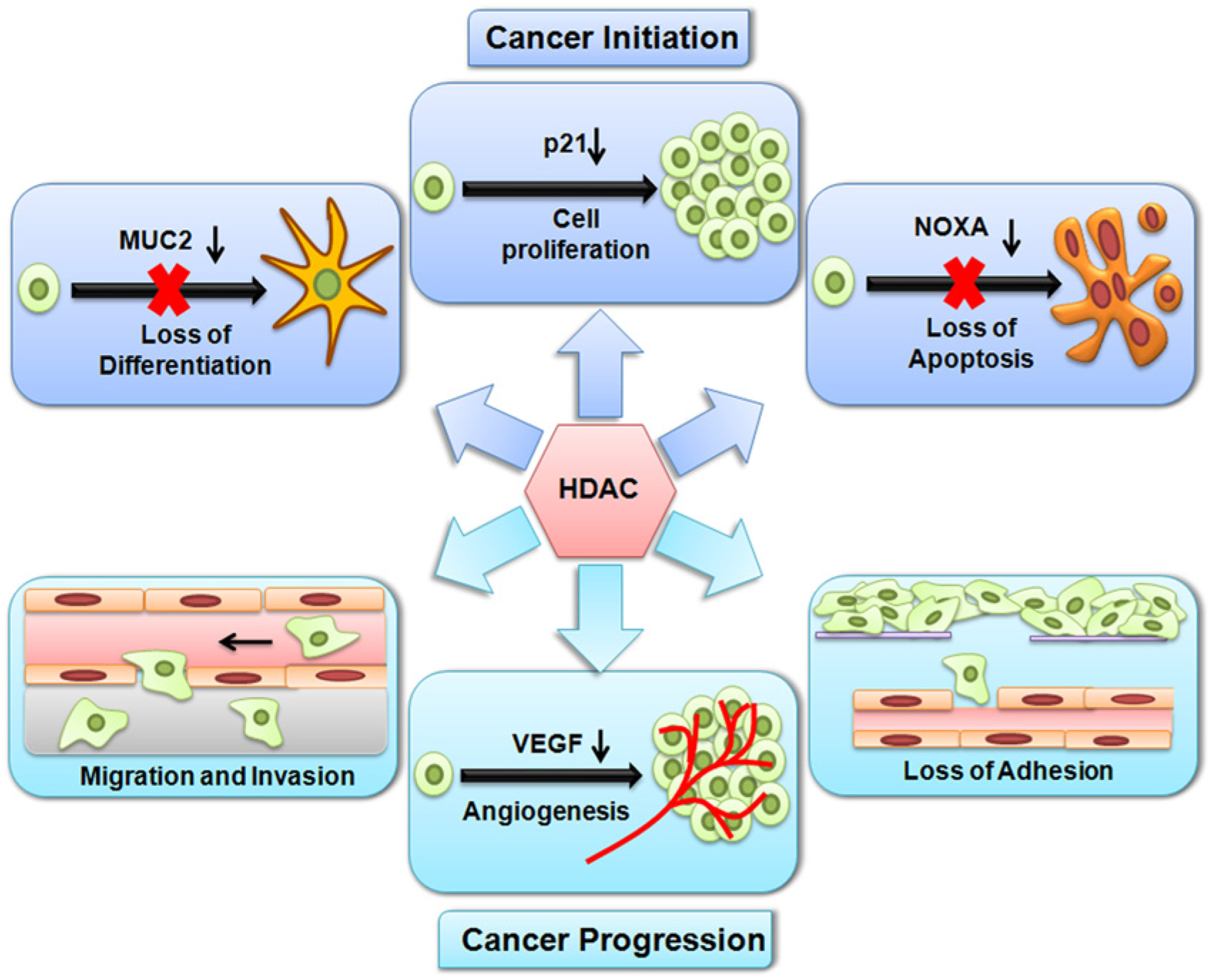
Histone deacetylation by HDACs alters the expression of genes involved in both cancer initiation and progression. During cancer initiation, HDACs repress genes, which results in uncontrolled cell proliferation, loss of differentiation and an inhibition of apoptosis. During cancer progression, HDACs repress genes, which results in a loss of adhesion, migration, invasion and angiogenesis.
3. Role of HDACs
3.1 Role of HDACs in Cancer Initiation
The histone acetylation erasers, HDACs, play a crucial role in silencing the tumor suppressor genes concerned with cancer initiation and progression. Carcinogenesis involves the transition of normal cells into malignant cells. This is characterized by uncontrolled cell proliferation, a lack of differentiation, including epithelial and hematopoietic differentiation, and the prevention of apoptosis. During the course of cancer initiation, HDACs exert their silencing effects on cell-cycle regulatory genes and genes associated with the differentiation process. Table 2 outlines the genes that are regulated by HDACs during the different stages of cancer initiation and progression.
Genes Regulated by HDACs in Different Stages of Cancers.
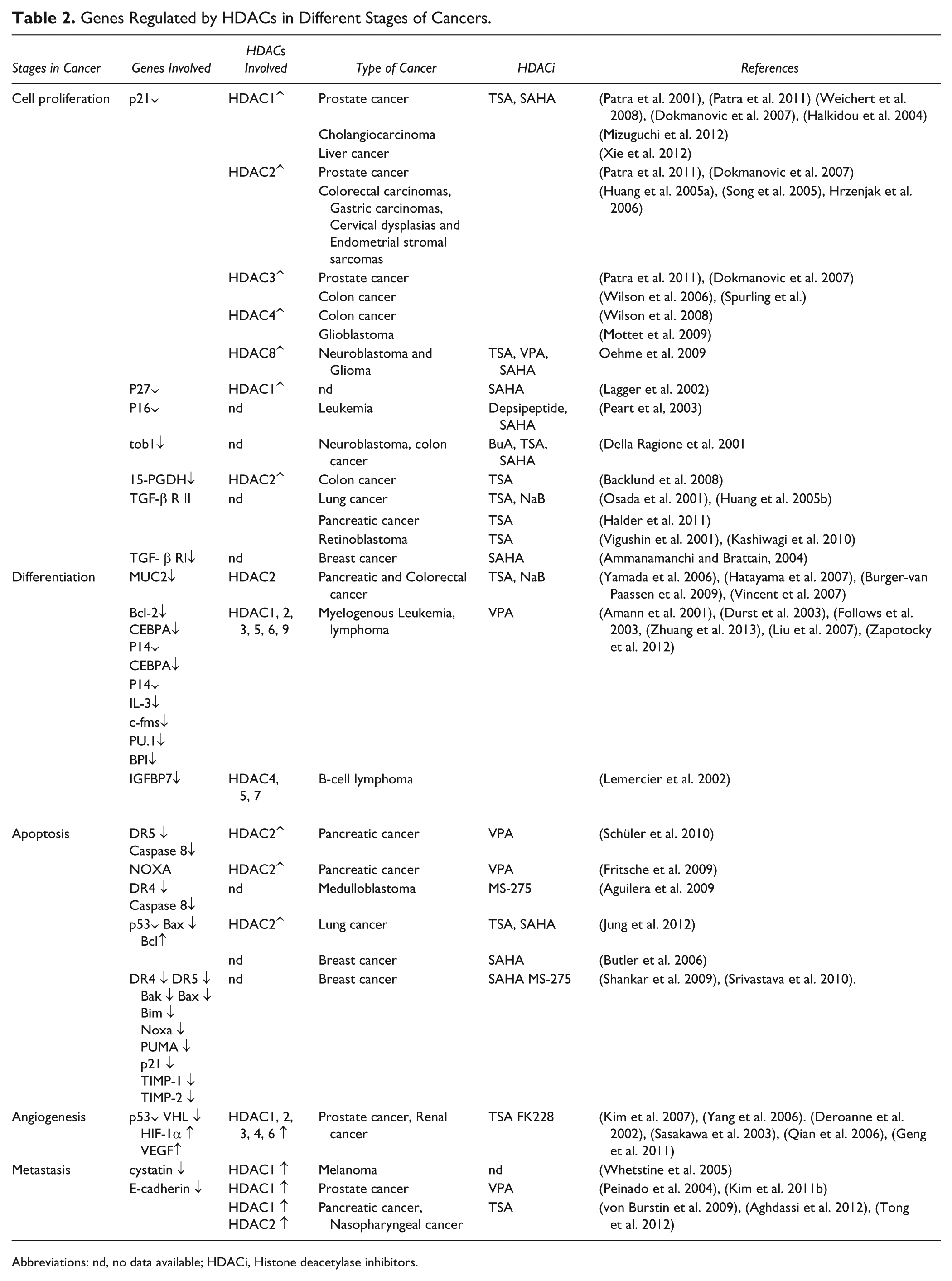
Abbreviations: nd, no data available; HDACi, Histone deacetylase inhibitors.
3.1.1 HDACs Account for Cell Proliferation
Repression of proliferation-restraining genes is one of the hallmark features of cancer cells. In this context, HDACs are essential for unhindered cell proliferation by repressing the expression of selective cyclin dependent kinase (CDK) inhibitors—p21 and p27. Several lines of evidence exist to support this notion. For example, HDAC1-null mouse embryos and ES cells exhibit an overall reduced cellular proliferation rate caused by increased levels of p21 and p27 (Lagger et al. 2002). Chromatin immunoprecipitation (ChIP) experiments demonstrate the association of HDAC1 with the p21 gene in mouse embryonic fibroblasts and further repression of p21 by HDAC1 via Sp1 binding leading to enhanced cell proliferation (Lagger et al. 2003; Zupkovitz et al. 2010). Likewise, it has been demonstrated that HDAC4 mediates repression of p21 expression via Sp1/Sp3 binding in colon cancer cells (Wilson et al. 2008) and in glioblastoma cells (Mottet et al. 2009). Moreover, it has been observed that HDAC4 silencing–mediated induction of p21 leads to cancer cell growth arrest in vitro and tumor growth inhibition in vivo in a human glioblastoma model (Mottet et al. 2009). HDAC1 (Patra et al. 2001; Patra et al. 2011; Halkidou et al. 2004), HDAC2 and HDAC3 are generally highly expressed in prostate cancer (Weichert et al. 2008) (see Figure 6).
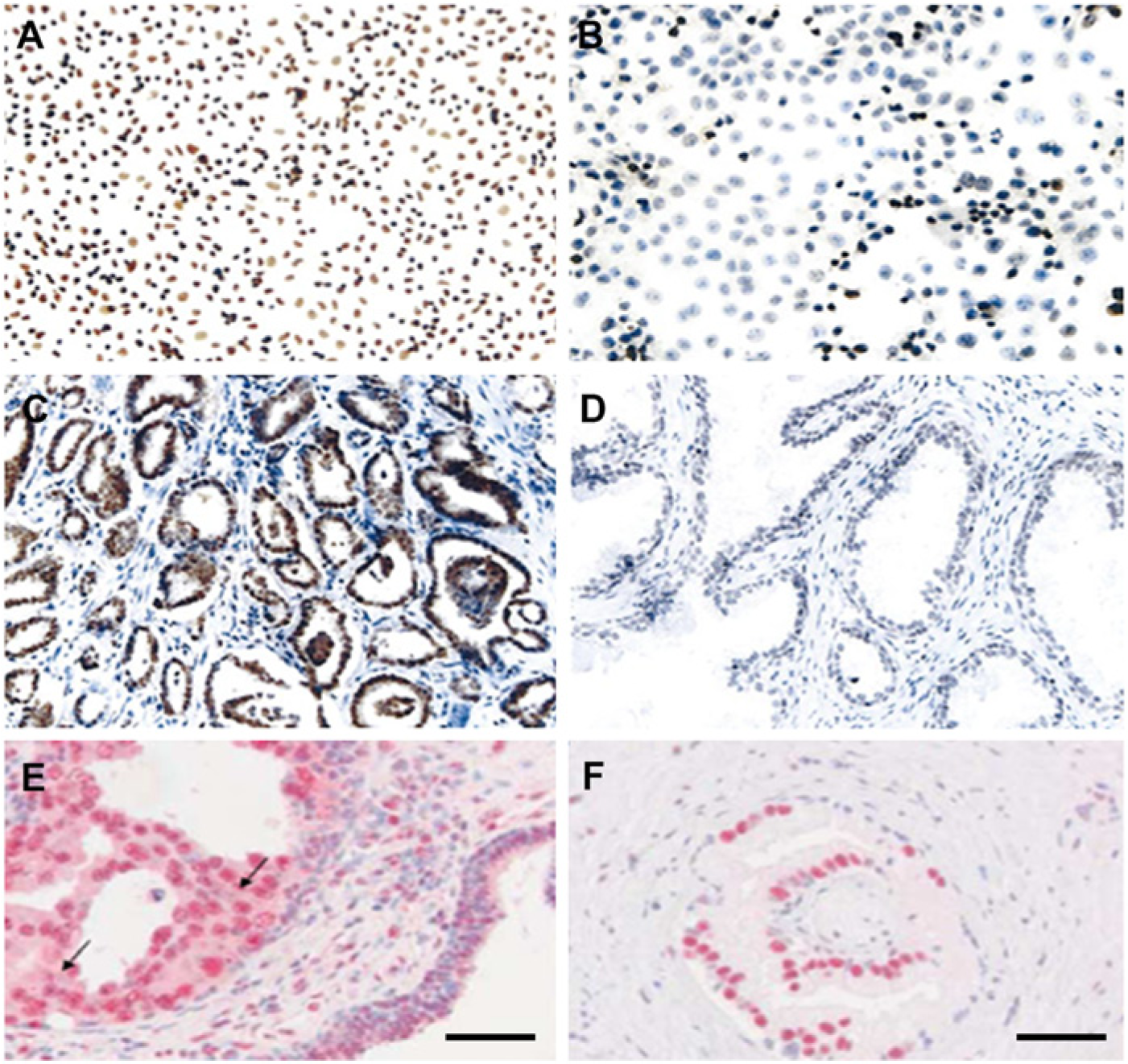
Examples of increased HDAC1 expression in human prostate cancer cell lines (A) and tissues (C) in comparison with BPH-1(human benign prostate epithelium) cells (B) and BPH-1 tissue (D) as detected by immunocytochemistry and immunohistochemistry, respectively, and described previously (Patra et al. 2001). Enhanced expression of HDAC2 (E) and HDAC3 (F) in prostate cancer tissue as detected by immunohistochemistry and described previously (Weichert et al. 2008). Bars = 50 µm.
Overexpression of HDACs is correlated with many other cancers, with subsequent decreases in p21 expression. For instance, in addition to prostate cancer, HDAC1 has been reported to be overexpressed in cholangiocarcinoma (a type of biliary cancer) (Mizuguchi et al. 2012) and liver cancer (Xie et al. 2012). Similarly, HDAC2 has been found to be overexpressed in colorectal carcinomas, gastric carcinomas, cervical dysplasias and endometrial stromal sarcomas (Huang et al. 2005a; Song et al. 2005; Hrzenjak et al. 2006). Indeed, HDAC2 is demonstrated to directly regulate the expression of p21 in a p53-independent manner. Inactivation of HDAC2 is identified as a causal factor for the induction of p21 expression in cell cycle regulation with simultaneous suppression of cyclin E2, cyclin D1, and CDK2 expression in lung cancer cells. Moreover, diminished in vitro tumorigenic properties and in vivo tumor growth of the mouse xenograft model are also noticed with sustained repression of HDAC2 in lung cancer (Jung et al. 2012). Experimental results indicate that colon cancer cells overexpress HDAC3 with a corresponding inhibition of p21 expression (Wilson et al. 2006; Spurling et al. 2008). In contrast, silencing of HDAC3 leads to increased expression of p21 (Wilson et al. 2006) with enhanced H3-K12 acetylation at the p21 promoter (Spurling et al. 2008). Overexpression of HDAC8 has been detected in multiple tumors, especially in neuroblastomas and gliomas. Knockdown of HDAC8 in neuroblastoma cells results in the inhibition of proliferation, reduced clonogenic growth and cell cycle arrest (Oehme et al. 2009). Expression of p21 coincides with hyperacetylation of histones H3 and H4 in its promoter region (Richon et al. 2000), and the reduced expression of Class II HDAC enzymes, HDAC 5 and HDAC10, is associated with poor prognosis in lung cancer patients (Osada et al. 2004).
Enhanced HDAC2 expression has been reported in the intestinal mucosa and polyps of Adenomatosis polyposis Coli (Apc/APC)-deficient mice, as well as in human colon cancer patients, which is caused by a loss of function of the Apc/APC tumor suppressor (Zhu et al. 2004). Additionally, it is observed that increased level of Hdac2 expression inversely correlates with the loss of 15-hydroxyprostaglandin dehydrogenase (15-Pgdh) expression in Apc-deficient mouse adenomas. Also, in colorectal carcinomas (CRC), elevated HDAC expression correlates well with marked diminution of 15-PGDH expression. 15-PGDH is recently identified as a tumor suppressor gene coding for an enzyme responsible for enzymatic degradation of prostaglandin E (2) (PGE(2)), which promotes cancer. Recently, it has been reported that HDAC2 interacts with the 15-PGDH promoter, thereby causing its suppression. Additionally, ChIP assays examining the 15-PGDH promoter in CRC cells shows the loss of HDAC2 binding after treatment with HDAC inhibitors (HDACi) (Backlund et al. 2008). Certain other genes are also reported to be induced by different HDACi, including tob1 and p16; these proteins play a role in cell proliferation and cell cycle arrest (Della Ragione et al. 2001; Peart et al. 2003). tob1 may function in the absence of functional p21CIP1/WAF1 to induce cell cycle arrest, as demonstrated by the arrest of p21-/- cells by butyrate; it is also upregulated by HDACi in neuroblastoma cells and in colon cancer cells (Della Ragione et al. 2001).
TGF-β (transforming growth factor-β) is a growth inhibitory signaling molecule. TGF-β receptors (R)-II and TGF-βRI are responsible for binding and signaling of TGF-β, respectively. In contrast to normal cells, cancer cells exhibits a loss of TGF-β signaling due to the low expression of TGF-βRs (TGF-βRI and/or TGF-βRII). It is demonstrated that the TGF-βRII promoter has highly condensed chromatin caused by a low level of H3 and H4 acetylation in lung cancer cells (Osada et al. 2001). This low expression of TGF-βRII in lung cancer cells underlies the activation of the mitogen-activated protein kinase /extracellular signal–regulated kinase (MAPK/ERK) pathway by either activated Ras or epidermal growth factor (EGF) signaling through histone deacetylation (Halder et al. 2011). On the contrary, treatment with HDACi namely, Trichostatin-A (TSA), induces the expression of TGF-βRII and forms open chromatin conformation in lung cancer cells (Osada et al. 2001). Moreover, it activates the TGF-βRII promoter and induces TGF-βRII mRNA expression in retinoblastoma cells (Kashiwagi et al. 2010). It is demonstrated that Sp1 and NF-YA binding sites containing the promoter is necessary for MS-275- and TSA-mediated induction of the TGF-βRII promoter in lung cancer and pancreatic cancer, respectively (Huang et al. 2005b; Halder et al. 2011). In addition, certain cancer cells are also associated with the loss of TGF-βRI expression. It is demonstrated that breast cancer cells, when treated with Suberoylanillide hydroxamic acid (SAHA), exhibit high TGF-βRI expression, with increased acetylated histones H3 and H4 at its promoter. This is likely because of impairment of HDAC activity associated with Sp1/Sp3 and involvement of HAT p300 at the promoter region (Ammanamanchi and Brattain, 2004). Also, histone H3K9 deacetylation is recognized to play a critical role in epigenetic-based repression of the TGF-β pathway genes, including TGF-βRI, TGF-βRII in breast cancer cells (Hinshelwood et al. 2007). In fact, by silencing the receptor for a growth-restraining signaling molecule, HDACs render a cancer cell insensitive to this signal and permit uncontrolled cell growth. Thus, upregulation in HDAC activity has been linked to the disruption of the normal cell cycle which leads to aberrant cell proliferation.
3.1.2 HDACs Mediate Loss of Differentiation
Cancer cells are generally characterized by a lack of differentiation: HDACs perform a vital role in this feature. The downregulation of genes associated with differentiation elicits inappropriate cell proliferation. Mucins are highly glycosylated proteins that play a pivotal role in carcinogenesis. Mucin2 (Muc2) is the most abundantly secreted gastrointestinal mucin involved in gastrointestinal cell differentiation. The loss of expression of this gene underlies pancreatic and colorectal cancer and Muc2-null mice were found to develop adenocarcinoma (Velcich et al. 2002). It has been demonstrated that H3-K9 and H3-K27 acetylation is enhanced in the promoter region of MUC2, which, in turn, stimulates the expression of this tumor-suppressor gene. Pancreatic cancer cells lacking MUC2, when treated with TSA, were shown to induce histone acetylation along with MUC2 mRNA and protein expression (Yamada et al. 2006). Similarly, treatment of colon cancer cells with sodium butyrate (NaB) has been found to stimulate MUC2 expression both at the mRNA and protein levels together with an enhanced level of acetylated H3 (Hatayama et al. 2007). This is further supported by the findings that colon cancer cells, following treatment with butyrate, exhibited increased acetylation of histone H3 and H4 at the MUC2 promoter as well as an increased level of MUC2 mRNA (Burger-van Paassen et al. 2009). In epithelial cancer cells, only HDAC2 has the potential to effect of DNA methyltransferase 1 (DNMT1) on the endogenous expression of MUC2 and MUC5B (Vincent et al. 2007). In this scenario, little information is available regarding HDACs in MUC2 gene silencing, and extensive study is required to verify the roles of the accused HDACs.
Runx1-ETO (also called AML1-ETO), the fusion transcription factor formed by t(8:21) translocation, has been reported to deregulate the expression of genes that are crucial for normal differentiation and proliferation of hematopoietic progenitors, which leads to acute myelogenous leukemia through the recruitment of HDAC-containing repressor complexes to the promoter region of AML1 target genes. The Runx1-ETO fusion protein binds HDAC1, HDAC2, HDAC3 as well as the co-repressors mSin3a, SMRT and N-CoR (Amann et al. 2001). In addition, Runx1 binds HDAC5, HDAC6 and HDAC9 with varying affinities (Durst et al. 2003). Several target genes are repressed by this chimeric protein, among which are B-cell lymphoma-2 (Bcl-2) and CCAAT/enhancer-binding protein α (CEBPA), the key regulators gene of granulocytic differentiation, and the tumor suppressor p14ARF (Linggi et al. 2002; Zhuang et al. 2013), c-fms (colony-stimulating factor 1 receptor) (Follows et al. 2003), and IL-3 (Interleukin-3) (Liu et al. 2007). Reduced level of acetylated histones H3- K9 and H3-K14 has been reported in cells expressing Runx1-ETO at the intronic regulatory element, which recruit HDAC1 and cause decreased expression of c-fms (Follows et al. 2003). ChIP studies show further evidence that Bcl-2, CEBPA and p14ARF are the direct transcriptional targets of AML1-ETO. Moreover, the global binding of AML1-ETO to genomic DNA results in the reduction of histone H3 or H4 acetylation, indicating that AML1-ETO induces heterochromatic silencing of Bcl-2, CEBPA and p14ARF (Zhuang et al. 2013). Similarly, ETO interacts with promyelocytic leukemia zinc finger (PLZF) and BCL-6 to form PLZF-ETO and BCL-ETO fusion transcriptional repressor proteins that recruit HDACs and co-repressors (Melnick et al. 2000; Chevallier et al. 2004). ETO synergistically acts with PLZF to augment transcriptional repression of genes essential for differentiation in an HDAC-dependent manner (Melnick et al. 2000). Likewise, the repressor BCL6 is often expressed inappropriately in B-cell lymphomas and recruits both Class I and Class II HDACs. Three related Class II deacetylases—HDAC4, HDAC5, and HDAC7—have been demonstrated to be associated with BCL6 both in vivo and in vitro. This is further confirmed by an electron microscopy study that showed the partial co-localization of endogenous BCL6 and Class II HDACs in the nucleus (Lemercier et al. 2002). Analogous mechanisms of transcriptional disruption have also been illustrated in t(15;17)-positive acute promyelocytic leukemia by histone deacetylation, where the PML-RAR fusion protein recruits an HDAC repressor complex to the promoter of the retinoic acid-target genes (Grignani et al. 1998). The chimeric protein generated from chromosomal translocations can inappropriately recruit HDACs to regulatory regions of genes involved in differentiation. This process effectively prevents differentiation and allows for continued proliferation of undifferentiated progenitor cells, resulting in leukemia or lymphoma. In light of above discussion, it seems that HDACs are crucial regulators of oncogenic fusion proteins, which, in turn, elicit silencing of major genes allied with the differentiation process.
On the contrary, Valproic acid (VPA) treatment has been reported to disrupt the AML1/ETO-HDAC1 physical interaction, stimulate the global dissociation of AML1-ETO/HDAC1 complex from the promoter of AML1-ETO target genes, and persuade relocation of both AML1-ETO and HDAC1 protein from nuclear to perinuclear regions. Moreover, this process diminishes HDAC activity thereby causing histone H3 and H4 hyperacetylation, and finally leading to transcriptional reactivation of target genes (Liu et al. 2007). Likewise, treatment with VPA causes human AML cell lines of different genotypes and diagnostic bone marrow samples from patients to undergo differentiation and expression of repressed AML1 target genes— PU.1, CEBPA, BPI and IGFBP7 (Zapotocky et al. 2012).
3.1.2 HDACs Arbitrate Loss of Apoptosis
HDACs play an important role in diminishing apoptosis in a variety of cancer cells. It is reported that HDAC2 reduces TRAIL (TNF-α related apoptosis inducing ligand)-induced apoptosis of pancreatic cancer cells. In contrast, HDAC2-depleted and TRAIL-treated pancreatic cancer cells showed upregulation of the TRAIL receptor 2 (DR5), accelerated processing of caspase 8, marked cleavage of Bid, and increased effector caspase activation leading to increased apoptosis. Additionally, co-treatment of VPA and TRAIL causes enhanced apoptosis of pancreatic cancer cells (Schüler et al. 2010). Depletion of HDAC2 causes upregulation of NOXA, a pro-apoptotic gene, and sensitization of pancreatic cancer cells towards etoposide-induced apoptosis. This suggests that HDAC2 accounts for epigenetic silencing of NOXA in pancreatic cancer cells (Fritsche et al. 2009). There are reports indicating that specific HDACs mediate apoptosis in distinct cancer cells. Enhanced apoptosis has been observed in osteosarcoma cells due to the knockdown of HDAC1 (Senese et al. 2007) whereas, in HeLa cells, apoptosis was caused by knockdown of HDAC2 (Huang et al. 2005a). It is reported that stable histone acetylation is required for efficient restoration of caspase-8 in small cell lung carcinomas cells (SCLCs), which may be achieved by synergistic treatment of VPA and CI-994 (HDACi). Even though HDAC2 is also suggested to be involved in caspase-8 silencing, the level of caspase-8 is not significantly increased upon silencing of HDAC2 in combination with decitabine (DNMT inhibitor). This indicates that several HDACs may be simultaneously involved in silencing caspase-8 (Kaminskyy et al. 2011). Similarly, medulloblastoma cells show resistance to TRAIL-induced apoptosis caused by the low expression of DR4 (TRAIL receptor 1). However, treatment with MS-275 increased apoptosis, which coincides with increased histone H3 and H4 acetylation at the DR4 promoter, enhanced DR4 gene and protein expression, and elevated Caspase-8 activity (Aguilera et al. 2009). HDAC2 silencing has been shown to result in activation of cellular apoptosis via p53 and Bax activation and Bcl2 suppression in lung cancer cells. In contrast, overexpression of HDAC2 suppresses p53 expression, which is a transcriptional activator of Bax, as well as induces Bcl2 expression such that the transformed cells overcome the apoptotic signal (Jung et al. 2012). Treatment with SAHA also shows promising results by sensitizing breast cancer cells, with a concomitant activation of caspase 8, caspase 3, Bid and PARP cleavage, as well as the enhanced expression of Bax and DR4 (Butler et al. 2006). Many in vivo studies have also emerged in support of the results obtained in vitro. For instance, treatment of nude mice with SAHA results in the up-regulation of DR4, DR5, Bak, Bax, Bim, Noxa, PUMA, p21, tissue inhibitor of metalloproteinase-1 (TIMP-1), and TIMP-2 in breast cancer cells (Shankar et al. 2009). The HDAC1-containing complex, PID, is involved in the regulation of p53-mediated growth arrest and apoptosis (Luo et al. 2000). Evidence has been presented to suggest that HDAC5 is required for the maintenance of pericentric heterochromatin and the prevention of apoptosis of human cancer cells. In contrast, silencing of HDAC5 causes heterochromatin de-condensation and enhanced sensitivity of the DNA to DNA-damaging agents, thus triggering cancer cell apoptosis (Peixoto et al. 2012). Here, it is clear that HDAC2 and, in certain cases HDAC1 and HDAC5, is concerned with the attenuated apoptosis underlying carcinogenesis.
3.2 Role of HDACs in Cancer Progression
Besides regulating the genes concerned with cancer formation, HDACs also restrain the genes implicated in cancer progression. A study of gastrointestinal tumors revealed that a decrease in histone acetylation is not only involved in tumorigenesis but also in tumor invasion and metastasis (Yasui et al. 2003). The MRES phenotype is a new epigenetic phenomenon characterized by the concomitant epigenetic silencing of several chromosomal regions. This phenotype is strongly associated with the carcinoma in situ (CIS) gene expression signature and a more aggressive cancer phenotype. In aggressive bladder cancer, this MRES phenotype has been recognized, and involves the concurrent silencing of seven stretches of contiguous genes. The corresponding silencing mechanism has been reported to be histone methylation and hypoacetylation (Vallot et al. 2011). Accordingly, genes regulating angiogenesis as well as those regulating metastasis (cell adhesion, cell migration and invasion) are under the extreme control of HDACs.
3.2.1 HDACs in Angiogenesis
Angiogenesis involves the development of new blood vessels from pre-existing ones. It is a critical process required for the progression of both solid tumors and hematological malignancies. Indeed, angiogenesis is enhanced in response to hypoxic conditions created in the center of a solid tumor to provide nutrients and oxygen and to dispose of CO2 and waste products. Hypoxic conditions in angiogenesis are invariably interlinked with induced expression of HDACs, including HDAC1, HDAC2 and HDAC3 (Kim et al. 2001; Kim et al. 2007). In normoxic conditions, hypoxia inducible factor-1α (HIF-1α) is acetylated and hydroxylated and undergoes von Hippel–Lindau (VHL)-dependent degradation. But, under hypoxic conditions, HDAC1 and HDAC3 interact directly with HIF-1α and lead to its deacetylation, which stabilizes HIF-1α and increases its transactivation function; this in turn enhances the expression of vascular endothelial growth factor (VEGF) (Kim et al. 2007). Overexpression of HDAC1 is correlated with silencing of p53 and VHL but induces the hypoxia-responsive genes HIF-1α and VEGF and stimulates angiogenesis (Kim et al. 2001; Yang et al. 2006). This is clearly evident by the increased levels of HDAC1 in the hypoxic regions of tumors in vivo (Kim et al. 2001; Deroanne et al. 2002). Importantly, treatment with TSA restores the expression of p53 and VHL and represses HIF-1α and VEGF, and, consequently, silences VEGF signaling (Yang et al. 2006). The HDACi, FK228, has also been demonstrated to have a suppressive effect on VEGF via histone acetylation of its promoter in PC-3 xenografts (Sasakawa et al. 2003). Similarly, Class II HDACs, including HDAC4 (Geng et al. 2011) and HDAC 6, interact directly with HIF-1α and decrease its acetylation status; thus, these two proteins are the key modifiers and positive regulators of HIF-1α protein stability (Qian et al. 2006). Taking all this into consideration, it can be comprehended that Class I and Class II HDACs (notably, HDAC1, 2, 3, 4, 6) may serve as important therapeutic targets to avert angiogenesis in cancer.
3.2.2 HDACs in Metastasis
A cell’s lack of ability to interact accurately with its external milieu and its neighboring cells is the universal feature of metastasis. The downregulation of cystatin, a peptidase inhibitor, has been shown to be associated with tumor cell invasion, and HDAC1 is recognized as the causative agent for silencing of cystatin. Consequently, the knockdown of HDAC1 reduces cellular invasion by overexpressing cystatin (Whetstine et al. 2005).
E-cadherin (CDH1) is a cell-cell adhesion protein molecule, the loss of which frequently leads to epithelial-to-mesenchymal transition (or, EMT) in cancer. A wealth of studies has shown that HDACs are the causative agents that repress CDH1. However, HDACs do not directly interact with CDH1; rather they interact indirectly through different transcriptional repressor such as, snail, slug, ZEB, among others. It has been experimentally confirmed by ChIP assays that snail directly interacts with the CDH1 promoter and recruits HDAC1, HDAC2 and the co-repressor mSin3A. These HDACs in turn inhibit CDH1 expression that correlates with hypoacetylated histones H3 and H4 at its promoter (Peinado et al. 2004). It has been demonstrated that CDH1 expression often is lost in prostate cancer cells in response to the high expression and function of HDACs, especially HDAC1, a key repressive enzyme for this silencing (Kim et al. 2011b). Experimental evidence suggests that both HDAC1 and HDAC2 are involved in silencing CDH1 during the metastatic process of pancreatic cancer cells. The mechanism underlies the recruitment of transcriptional repressors, Snail and ZEB to the CDH1 promoter which, in turn recruit HDAC1 and HDAC2. Then HDAC1 and HDAC2 function to suppress the CDH1 expression. Conversely, treatment of pancreatic cells with HDACi restores the CDH1 with diminished EMT (von Burstin et al. 2009; Aghdassi et al. 2012). Additionally, HDAC1 and HDAC2 are also vital components of the co-repressor complex— EZH2/HDAC1/2/Snail—which is implicated in the silencing of CDH1 causing EMT in nasopharyngeal cancer (Tong et al. 2012). It has been demonstrated that HDAC3 and PPARγ together bind to the CDH1 promoter, and HDAC3 causes repression of target genes. Moreover, combined treatment of a PPARγ agonist and an HDACi make PPARγ and HDAC3 unable to bind to the promoter, resulting in hyperacetylated histone H4 and re-expression of CDH1 (Annicotte et al. 2006). Similarly, in esophageal cancer, the combined treatment of HDAC and proteasome inhibitors (vorinostat and bortezomib, respectively) upregulates CDH1 and reduces EMT (Taylor et al. 2010).
ICAM1, an intercellular adhesion molecule, is generally downregulated in tumor-derived endothelial cells. It is reported that the ICAM1 promoter contains hypoacetylated histone H3 and hypomethylated histone H3-K4. However, treatment with HDACi and methyltransferase inhibitors reverses these alterations with re-expression of ICAM1. This enhances the ability of endothelial cells to adhere to lymphocytes, allowing for better tumor infiltration by the lymphocytes (Hellebrekers et al. 2006). In this case, although there is evidence for the involvement of deacetylation, which causes ICAM1 gene silencing, there is still a lack of information regarding the involved HDACs.
3.3 Disruption of Acetylation Homeostasis and the Role of HDACs beyond Histones in Cancer
In addition to histones, a plethora of non-histone proteins, including transcription factors and other proteins of the acetylome mentioned previously, are also reversibly regulated by HATs and HDACs. The acetylation and deacetylation of non-histone proteins affect their own stability and DNA binding ability. The balanced acetylation state of non-histone proteins is thus an essential criterion for homeostatic gene regulation program under physiological conditions. However, in pathological conditions, especially in cancer, acetylation marks on these non-histone proteins is altered. In this section, we elaborate on the function of HDACs beyond histones, giving emphasis to transcription factors and other cellular proteins.
3.3.1 Disconcerted Acetylation of Transcription Factors by HDACs Invites Cancer
The acetylation of lysine residues is well known as a key post-translational modification that governs the DNA interaction ability and transactivation potential of transcription factors. A wide range of transcription factors, including, p53, Runx3, GATA, Sp1, CEBPA, NF-κB, and STATs, have been identified as the non-histone targets of the acetylation/deacetylation system. The tumor suppressor transcription factor p53 is a critical regulator of cell proliferation because of its governing action over the apoptotic and cell cycle regulatory genes during a variety of stress responses (Luo et al. 2004). Following DNA damage, p53 is acetylated, which increases its DNA-binding ability, nuclear localization, co-activator recruitment functions and, consequently, increases its ability to activate the transcription of its target genes (Prives and Manley, 2001; Luo et al. 2004), thereby checking unrestricted cell proliferation. On the other hand, deacetylation of p53, brought about by HDACs including HDAC1 (Luo et al. 2000), HDAC2 and HDAC3 (Juan et al. 2000), reduces the transcriptional activity of p53 by inhibiting the p53–DNA interaction. This is further supported by a low expression of BAX, a natural p53 responsive system in the presence of HDACs (Juanet al. 2000). Similarly, the overexpression of HDAC2 is found to repress the ability of p53 to activate the anti-proliferative gene, p21, in colon and breast cancer cells, thus leading to enhanced cell proliferation. Conversely, knockdown of HDAC2 has been shown to enhance p53–DNA binding activity, resulting in reduced cell proliferation (Harms and Chen, 2007). In the case of the Ewing Family Tumors (EFTs), the EWS-Fli1 chimeric protein is reported to deacetylate p53 and attenuate its transcriptional function and protein stability, and enhance mdm2-mediated p53 degradation via its N-terminal region HDAC1; this, in turn, leads to the repression of its target genes p21 and Puma. However, TSA treatment promotes p53-p300 interactions and recruitment of p53 Lys-382 to the promoter regions of its target genes, resulting in their upregulation (Li et al. 2012). At the molecular level, HDAC6 interacts with p53 and lessens its transcriptional activity by promoting its degradation (Ding et al. 2013). In conclusion, Class I HDACs, being predominantly nuclear, critically regulate the DNA binding and transactivation potential of p53, whereas Class IIb HDACs lead to its degradation, thereby terminating its functions and ultimately fueling cell proliferation.
In addition to p53, the runt domain transcription factor (Runx) is an important class of transcriptional regulators playing a prominent role in neoplasia. Runx3 is reported to be a putative tumor suppressor in gastric and many other solid tumors (Ito et al. 2008; Lin et al. 2012). Three lysine residues at Runx3 are recognized as acetylation sites. As both acetylation and ubiquitination occur at lysine residues, increased acetylation prevents the ubiquitin ligase Smurf-mediated degradation of Runx3 and increases its stability (Jin et al. 2004). Akt1, a cell survival gene (Lin et al. 2012), and claudin-1, a tight junction protein that promotes cell-cell contact, are identified as the target genes of Runx3 (Chang et al. 2010). Runx3 has both activating and repressing actions in accordance with the target genes, acting as a transcriptional repressor for Akt1 but functioning as an activator for claudin-1. Thus, it can be said that acetylated Runx3 leads to regulated cell growth and proliferation, with no EMT. However, in response to deacetylation by HDACs, including HDAC1, 2, 4 and 5, Runx3 undergoes degradation (Jin et al. 2004). This subsequently precludes the well-orchestrated cell division and leads to EMT, which are hallmarks of cancer.
Similarly, BCL-6 is a transcriptional repressor that is constitutively expressed in B-cell lymphomas where it suppresses the expression of the genes involved in the control of lymphocyte activation, differentiation and apoptosis. BCL-6 contains a binding domain for the co-repressor complex, SMRT/HDAC, which causes repression of its target genes. Recent studies suggest that acetylated BCL-6 fails to recruit HDACs, thereby favoring transcription of DNA damage response and cell cycle checkpoint activating genes, such as ATR, CHK1, TP53, and CDKN1A (Compton and Hiebert, 2010), thus reducing cell transformation (Bereshchenko et al. 2002). However, recruitment of HDAC3, 4, 5 and 7 by BCL-6 (Lemercier et al. 2002; Compton and Hiebert, 2010) represses the target genes and allows rapid B cell proliferation. Conversely, inhibition of HDACs results in the accumulation of the inactive acetylated BCL-6, which can promote cell-cycle arrest and apoptosis of B-cell lymphoma cells (Bereshchenko et al. 2002).
In case of B-cell follicular lymphomas, over-expression of HDAC2 is positively correlated with the expression of the antiapoptotic Bcl-2 with resistance to apoptosis. The mechanism underlies the abnormal hypoacetylation of transcription factors Sp1 and CEBPA by HDAC2, which allows for promoter binding and activation of Bcl-2. As such, HDACi promotes lymphoma cell death by inhibiting HDAC2, which in turn enhances the acetylation of Sp1 and CEBPA, reduces their binding to Bcl-2 promoter, and consequently leads to the significant decrease in Bcl-2 expression (Duan et al. 2005).
It has been shown that acetylation of GATA1 transcription factor by CBP/p300 within the zinc finger is required for chromatin binding and to activate the transcription of genes required for hematopoietic differentiation (Lamonica et al. 2006). However, deacetylation of GATA1 by HDAC5 is reported to silence GATA-mediated transcription (Watamoto et al. 2003) and give rise to highly proliferative, undifferentiated precursor cells, resulting in hematopoietic malignancy.
NF-κB, another transcription factor, plays a crucial role in oncogenesis. The activity of NF-κB is governed by acetylation and deacetylation at multiple levels. It has been found that acetylation of p65, a subunit of NF-κB, enhances the DNA binding and transcriptional activation ability of NF-kB. This prevents its association with IκB and also prevents its nuclear export (Chen et al. 2002), thus stimulating target gene expression. But as the target genes of NF-κB are anti-apoptotic in nature, so acetylation in this case leads to decreased apoptosis and promotes carcinogenesis. However, deacetylation by HDAC3 enhances NF-κB and IκB binding and facilitates its nuclear export (Chen et al. 2001). Moreover, SIRT1 also deacetylates p65 at K310, resulting in decreased expression of NF-κB responsive genes, which includes anti-apoptotic genes and Bcl-xL (Chen et al. 2000). It has been reported that, in osteosarcoma cells, HDAC1–3 binds to p65 and leads to a decrease in histone H3 acetylation at the Bcl-xL promoter, with subsequent repression of antiapoptotic gene Bcl-xL (Campbell et al. 2004). So, in this case, HDACs play a positive role, and thus silencing by HDACs would be beneficial for the downregulation of the antiapoptotic genes, thereby enhancing apoptosis of cancer cells.
STATs are cytokine-induced transcription factors known to play essential roles in cancer development. Acetylation has been reported in STAT1, 2, 3, 5a, 5b and 6, whereas HDAC1, 2, 3, 4 and SIRT 1 have been recognized to modulate the acetylation status of STAT1 and 3 (Wieczorek et al. 2012). Dimerization of STAT3 and STAT5 (Ma et al. 2010) depends on the acetylation of specific lysine residues, which enhances both DNA binding and activates transcription. Deacetylation by HDAC3 acts positively and inhibits transcription of STAT3 target genes, including growth-promoting genes such as cyclin D1 and antiapoptotic genes such as Bcl-xL (Wang et al. 2005; Yuan et al. 2005). However, deacetylation of STAT6 by HDAC1 and 2 contributes actively to the oncogenesis of lymphomas and leukemia in humans and in rodent models. Studies argue that treatment with HDACi can correct aberrant STAT6 functions in Hodgkin lymphoma. Still, the correlation of these alterations and STAT6 acetylation is not properly understood (Wieczorek et al. 2012). In certain tumors, acetylated STAT1 preferentially binds p65 and enhances its nuclear export. This prevents the expression of antiapoptotic NF-κB target genes. Therefore in this context, inhibition of HDAC activity can certainly be useful as a therapeutic intervention against cancer.
The acetylation/deacetylation status of the transcription factor HIF1α plays an indispensable role in carcinogenesis by regulating angiogenesis and cellular metabolism. It has been reported that the acetylation and deacetylation at different lysine residues within HIF1α can lead to different biological consequences. Acetylation at the N-terminus (Lys-10, Lys-11, Lys-12, Lys-19, and Lys-21) and at the oxygen-dependent degradation domain (Lys-532) can promote HIF1α protein degradation and inhibition of downstream HIF-1 activity (Geng et al. 2011). Deacetylation of HIF1α at the N-terminus by HDAC4 increases its stability, transcriptional activity and thus the expression of a subset of Hif-1α target genes, including VEGF-α, lactate dehydrogenase A (LDHA), and Glut1 (Geng et al. 2011); this, in turn, directs the cells towards transformation. Other HIF-1α targets generally upregulated in cancer cells include glycolytic genes such as 6-phosphofructo-2-kinase (pfkfb3) (Obach et al. 2004), phosphoglycerate kinase 1 (PGK 1) and pyruvate kinase M2 (PKM2) (Kress et al. 1998), and glucose transporters GLUT1 and GLUT3 (Liu et al. 2009). In this context, it is probable that HDACs play a role in the metabolic changes seen in cancer cells and the deacetylation machinery help meet the demand of extra energy requirement of cancer cells by enhancing the expression of the aforementioned HIF-1α target genes to favor growth and proliferation. Here, HDACs play their usual function by acting as oncoproteins; however, it is not clear how these usually “repressing” enzymes result in activation of the target genes in question.
In this scenario, it is evident that, in the majority of cases, HDACs play a negative role by acting as oncogenic proteins, only acting positively as tumor suppressor proteins in a few cases. Recently, HDACi are emerging as promising anti-cancer agents and extensive research for better HDACi is still ongoing. Therefore, the design of inhibitors should be such that only the ‘offending’ HDACs are targeted without targeting all of the HDACs.
3.3.2 Modulation of the Acetylation State of Cytoplasmic Proteins by HDACs Favors Cancer
Altered patterns of acetylation and deacetylation in cytoplasmic proteins that are part of acetylome are frequently seen in various cancer types. Ku70, a cellular protein, plays a role in suppressing Bax-mediated apoptosis by interacting with Bax. In response to the acetylation of Ku70 at K539 and K542 by CBP and PCAF, Bax is translocated to the mitochondria to initiate apoptosis. However, it has been found that SIRT1 (Cohen et al. 2004) and HDAC6 (Subramanian et al. 2011; Kerr et al. 2012) deacetylate Ku70, which serves to the keep Ku70–Bax complexes in the cytosol. This prevents apoptosis and leads to uncontrolled cell proliferation. As such, treatment with HDACi promotes Bax-dependent apoptosis in several cancer cell types, including neuroblastoma (Subramanian et al. 2005) and prostate cancer (Chen et al. 2007).
Heat shock protein-90 (HSP90) is another protein of the acetylome, the acetylation of which at K294 generally inhibits its ability to form complexes with oncogenic client proteins such as BCR-ABL or Runx1-ETO (Scroggins et al. 2007). This prevents activation or enhances degradation of client proteins by ubiquitination. HDAC6, on the other hand deacetylates HSP90, allowing for the maturation or stabilization of client oncoproteins leading to cancer. As such, treatment with HDACi can promote the degradation of client proteins and restore the acetylation state (Bali et al. 2005; Kovacs et al. 2005). For example, HDACi 17-AAG inhibits HDAC6 and abrogates hsp90 chaperone function in human leukemia cells (Rao et al. 2008).
HDAC6 is a tubulin deacetylase having an essential role in cell motility during metastasis. Generally, stable microtubules are hyperacetylated in the normal cell. When this acetylation status is disturbed by the altered activity of HDAC6, it forms dynamic microtubules. These hypoacetylated dynamic microtubules promote cell movement (Matsuyama et al. 2002). The Class III HDAC SIRT2 has also been found to deacetylate α-tubulin at K40 (North et al. 2003). HDAC6 is also reported to enhance the migration of lymphocyte and breast cancer cells by tubulin deacetylation. Treatment with tubacin, an HDAC6 inhibitor, has been reported to inhibit such migration (Saji et al. 2005). Thus, HDAC inhibition is an important means of preventing cell migration and, subsequently, metastasis.
The above discussion clearly reveals the involvement of constituent proteins of the acetylome such as a cadre of transcription factors and other cellular proteins in cancer. However, it is quite difficult to target these non-enzymatic proteins because of an absence of an active site. But with the recognition of HDACs as the regulating agents of such proteins, it is now possible to target these proteins in the fight against cancer. The localization and nuclear-cytoplasmic translocation of HDACs greatly affect their functions. Mostly, the nuclear residing Class I HDACs, such as HDAC1, 2 and 3, are associated with modifying the acetylation status of transcription factors. In comparison, Class II HDACs have the ability of nuclear-cytoplasmic translocation and thus HDAC4, 5, 7 are linked with the acetylation status of transcription factors and HDAC6 with that of cytoplasmic proteins.
4. Sirtuins: A Double-Edged Sword in Cancer
Sirtuins are one of the important classes of NAD-dependent HDACs, which have been the focus of intense scrutiny because of their involvement in cancer. The sirtuins for which a role in cancer has been proposed are SIRT1, SIRT2, SIRT3, SIRT6 and SIRT7. Experimental studies with genetic mouse models reports SIRT1, SIRT2, and SIRT3 as tumor suppressors (Wang et al. 2008; Kim et al. 2010; Kim et al. 2011a). SIRT1, SIRT2, SIRT3 and SIRT6 have dual roles: tumor suppressor and oncogenic activities. The tumor suppressor activity of SIRT1 has been ascribed because of its capacity to deacetylate and consequently inhibit the β-catenin and RelA/p65 subunit of NF-κB, thereby attenuating cell proliferation. Its oncogenic activity mainly involves deacetylation and inactivation of p53 (Luo et al. 2001; Vaziri et al. 2001) and proapoptotic FOXO (Brunet et al. 2004); deacetylation of Ku-70, with subsequent sequestration of Bax away from mitochondria (Cohen et al. 2004); and inhibition of senescence and of apoptosis in c-Myc- and in PML-driven cancers (Langley et al. 2002; Menssen et al. 2012). Similarly, in its tumor suppressor role, mice with a deficiency of SIRT2 have been reported to develop gender-specific tumorigenesis, where females develop mammary tumors and males develop hepatocellular carcinoma. Also, human breast cancers and hepatocellular carcinomas were reported to exhibit reduced SIRT2 levels as compared with normal tissues. In contrast, the tumor promoting role is clearly evident from the antileukemia effects of SIRT2 inhibition (with AC93253) accompanied by Akt acetylation and its subsequent inhibition (Dan et al. 2012). The tumor opposing activity of SIRT3 has also been demonstrated, with an absence of SIRT3 triggering cell transformation in response to oncogenic Ras or Myc and to leading to HIF-1α stabilization with consequent induction of the Warburg effect. The role of SIRT3 as a tumor suppressor does not seem to extend to all types of cancer. In particular, oral squamous carcinoma cells are shown to overexpress SIRT3 and its downregulation causes antiproliferation and sensitization of carcinoma cells to radiation and cisplatin (Alhazzazi et al. 2011). Finally, SIRT6 anticancer activity could be ascribed to its capacity to negatively regulate NF-κB and HIF-1α (Kawahara et al. 2009; Zhong et al. 2010); HIF-1α is a transcription factor that promotes the expression of glycolytic and proangiogenic genes. Conversely, the oncogenic role of SIRT6 has been demonstrated because of its role in TNF synthesis, which causes some of the systemic manifestations of cancer via a mechanism that involves the augmented translation of Tnf mRNA (Van Gool et al. 2009; Truyers et al. 2010). Finally, a recent study suggests that SIRT7 may promote tumorigenesis by deacetylating H3K18 and thus repressing the function of tumor suppressor genes. H3K18 deacetylation by SIRT7 is reported to maintain anchorage-independent cell growth and loss of contact inhibition, which are essential characteristics of cancer cells. In fact, depletion of SIRT7 reduces the tumorigenicity of human cancer xenografts in mice (Barber et al. 2012).
The illustration is rather convoluted: the same sirtuins may actually favor certain aspects of neoplastic growth and, in some cases, their inhibition may be preferable to their activation. Nonetheless, it is ultimately becoming apparent that sirtuins play important roles in cancer pathophysiology.
5. Reinstatement of Acetylation Homeostasis in Cancer: HDAC Inhibitors
Given their global effect on histone acetylation, HDACs are the key players in the deranged acetylation homeostasis that prevails in cancer cells by regulating transcription either at the histone or non-histone level. Moreover, acetylation-deacetylation reactions are a reversible epigenetic phenomenon. These facts establish HDAC inhibition as one of the major mechanisms implicated in anti-cancer therapy and bring the HDACi to the fore of anticancer drugs. In light of this, it is important to know how HDACi function to re-establish the acetylation threshold. The basic mechanism underlies the binding of HDACi to the Zn2+ ion which resides at the active site of HDACs. As mentioned previously, the Zn2 +ion is essential for the catalytic function of classical HDACs. Hence, the binding of HDACi interferes with the activity of HDACs, thereby inhibiting their enzymatic function (Finnin et al. 1999). But still, the job of the HDACi is not finished yet, as it has also to allow the HATs to re-acetylate the histones at the appropriate gene promoter (Vaissiere et al. 2008). Thus, HDACi enable the altered cell to re-instate the disrupted acetylation balance by promoting acetylation and demoting deacetylation.
Generally, the structurally diverse HDACi can be categorized into four groups: short-chain fatty acids (aliphatic acids), hydroxamic acids, benzamide derivatives, and cyclic peptides (Minucci and Pelicci, 2006; Dokmanovic et al. 2007). Indeed, TSA, SAHA, VPA and NaB have been reported to exhibit encouraging results in mammalian cell cultures as well as in mouse models (Marks, 2007; Backlund et al. 2008; Oehme et al. 2009). Intriguingly, the first natural HDACi, TSA, shows an encouraging effect against HDAC1, 2, 3, 4, 6, 7 and 9 at the nanomolar level but it is less effective against HDAC8 (micromolar level). The high toxicity of TSA, however, precludes its therapeutic use (Vanhaecke et al. 2004). On the other hand, a synthetic derivative of hydroxamate, SAHA, is a pan HDACi that is effective against all classical HDACs. It is a Food and Drug administration (FDA)-approved HDACi for the treatment of cutaneous T-cell lymphoma (CTCL) (Marks, 2007). Besides, there are isoform-selective HDACi that also exhibit encouraging results. Remarkably, MS-275 is found to preferentially inhibit HDAC1 as compared with HDAC3 and has little or no effect against HDAC6 and 8 (Hu et al. 2003). A myriad of research is ongoing to develop isoform-specific HDACi. Nevertheless, it is not apparent whether selective inhibition will be beneficial over a pan inhibitor for HDACs for the treatment cancer. Selective HDAC inhibition, inhibiting specific HDACs and affecting the acetylation status of a relatively small number of HDAC substrates, may offer better efficiency and a broader therapeutic window with reduced adverse effects such as, thrombopenia, neutropenia, diarrhea, nausea, vomiting and fatigue that is associated with pan HDAC inhibition (Su et al. 2008). But, the ability to target individual HDACs in patients remains challenging. The highly conserved active site of Class I and II HDACs make it difficult to selectively inhibit these enzymes with small molecules. Recently, tumor-suppressive miRNAs such as, miR-449a (Noonan et al. 2009), miR-449a/b (Jeon et al. 2012) and miR-874 (Nohata et al. 2013), have been regarded as promising alternative methods for such selective inhibition and their evolution is presently underway. Additionally, dietary HDACi are generally weak ligands, inhibiting HDAC activity at higher concentrations than pharmacologic inhibitors, which are effective in the nanomolar to low micromolar range. However, due to their low cytotoxic nature, intense research is attempting to make them drugable candidates. Certain phytochemicals, such as SFN, AM, EGCG, curcumin and quercetin, are found to exhibit promising results in regaining acetylation balance via increased global H3/H4 histone acetylation owing to their ability to inhibit HDAC activity and induce histone hyperacetylation (Rajendran et al. 2011).
The effective inhibition by HDACi relies on the selection of optimal drug, the choice of the right dose and a schedule that may vary according to the different clinical settings. A better comprehension of the mechanism of action in general, and specifically for each particular clinical setting, will allow for the development of more selective HDACi.
6. Methods and Pitfalls in Detecting HDAC Activity
The substantial role of HDACs in transcriptional repression is thus recognized as an emerging key target for anticancer drugs. Moreover, the activity of HDACs in this process is invariably associated with the diagnosis and prognosis of a broad range of cancers. In view of the growing interest in these enzymes, it is very important to have assays that can efficiently detect their activity. This is essential for the isolation and characterization of enzymes and to screen possible candidate drugs. A broad survey reports the existence of a number of assay methods to determine the HDAC activity. The traditional methods are very complicated and laborious, generally employing radio-labeled histones from chicken reticulocytes or cell culture (Kolle et al. 1998). The main drawbacks include batch-to-batch variation in substrate properties, anemia in pretreated chickens and, moreover, their sacrifice. In order to overcome the above limitations, an alternative strategy of recombinant histone technology has been implemented. Here, the histones are replaced by chemically tritiated ([3H])-acetylated, 8- or 24-amino acid residues containing oligopeptides derived from histone peptide sequences (Taunton et al. 1996) and deacetylase activity is monitored by the extraction of liberated [3H]-acetic acid by scintillation counting. However, the process has its own disadvantage, as it is time-consuming and requires sophisticated instruments with a low sample throughput. Although a high-throughput version, comprising a radiolabeled and biotinylated octapeptide with scintillation proximity, is available (Nare et al. 1999), the radio-active oligopeptides create a health risk. Therefore, in order to overcome the necessity for radioactive assays, recently studies have used non-isotopic fluorescent labeled acetylated lysine substrate such as tripeptides-AMC (Aminomethylcoumarin) derivatives (Wegener et al. 2003) and lysine-AMC derivatives, MAL (N-(4-methyl-7-coumarinyl)-N-α-(tert.-butyloxycarbonyl)-N-Ω-acetyllysinamide) (Hoffmann et al. 1999; 2000). In the case of latter being used as HDAC substrate, it has been found that the deacetylated metabolite (ML) has the same fluorescence properties as the parent substrate (MAL). Thus, this method suffers from extraction of the remaining unmodified MAL, which is then monitored by either HPLC (Hoffmann et al. 1999; 2000) or a plate reader (Heltweg and Jung, 2002). However, the extraction procedure and the use of internal standards are found to be the main pitfalls in this protocol. Thereafter, histone deacetylase assay-homogenous (HDASH) procedure was developed as a homogeneous non-isotopic assay for the determination of HDAC activity, which involves quenching of fluorescent deacetylated metabolites by naphthalene dicarboxaldehyde (NDA); this technique thus allows the monitoring of the remaining unmodified MAL by a plate reader method (Heltweg and Jung, 2003). Although, this assay is extremely useful for the activity assay of Class I and II HDACs, it is not suitable for measuring the activity of sirtuins as it involves NDA detection, which does not work well with sirtuins. The most recent advancement, Z-(Ac) Lys-AMC (ZMAL), incorporates an optimized analog of MAL, which has been used as substrate for determining the activity of human deacetylases including Class I, II and sirtuins. The HPLC method can be employed for the simultaneous detection of both substrate and resulting fluorescent metabolites. However, for routine inhibitor screening, the homogeneous assay is recommended. This assay involves no NDA detection as the deacetylated metabolites (ZML) can be easily tryptically digested and the remaining ZMAL can be monitored by a plate reader (Heltweg et al. 2005). The main advantages of this assay include speed, its high-throughput suitability, the low consumption of enzymes, and last, but not the least, the use of an inexpensive substrate. Because of the aforementioned advantages, this assay is most preferably and extensively used for detecting HDAC activity.
7. Conclusions and Perspectives
Lysine acetylation/deacetylation is a dynamic, reversible posttranslational modification, with a well-defined role in regulating histones as well as non-histone proteins belonging to the acetylome. The epigenetic modulators, HDACs, are major repressive enzymes that erase acetylation marks from the proteins of the acetylome. Thus, HDACs are concerned with the maintenance of proper acetylation homeostasis, which is indeed a primary requirement for cell survival. It is currently increasingly becoming apparent that the acetylation status is largely disturbed during cancer. This is mainly owing to the altered recruitment and hyperactivity of HDACs, which leads to repression of the genes concerned with cell proliferation, differentiation, apoptosis, metastasis, and angiogenesis, among other processes. Because of their broad involvement in carcinogenesis and cancer progression, HDACs are recognized as appealing targets for the development of anti-cancer agents. Although, the HDACs concerned with many tumor suppressor genes have been identified, there are still certain repressed genes, such as MUC2 and ICAM1, which are known to be controlled by the acetylation/deacetylation system for which the responsible HDAC(s) has not been identified. Issues like this should be addressed to clearly understand the underlying mechanism of cancer.
In view of this, HDACi provide an attractive avenue to restore the acetylation pattern and thus contribute to the re-expression of the genes in question. Considering the significance of specific HDAC inhibition over pan inhibition, extensive research should be directed toward the design of a specific HDACi that may also include phytochemicals and tumor suppressive miRNAs. This approach opens up a new therapeutic opportunity by which improved, tailored epidrugs can be identified to target cancer.
Footnotes
Acknowledgements
S. Parbin is thankful to DST, Govt. of India for INSPIRE fellowship awarded to her. S. Kar, A. Shilpi, D. Sengupta, M. Deb and S. K. Rath are thankful to NIT-Rourkela for granting them fellowships under the Institute Research Scheme. We apologize to those, whose works and related publications we have not been able to discuss and cite due to space limitations.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was supported in part by the departmental operating grants of LS, NIT-Rkl.