
Abstract
Objective
Epidemiologic studies have linked smoking to various malignancies, including bladder cancer, but its underlying biological functions remain elusive. Currently, we aimed to identify the smoking-related epigenetic modifications and disclose their impacts on prognosis and therapies in bladder cancer.
Methods
DNA methylation, transcriptome, and clinical profiles were acquired from The Cancer Genome Atlas (TCGA) using “TCGAbiolinks” Differential expression analyses were performed with “limma” and visualized by the “pheatmap” package. Smoking-related interactions were displayed using Cytoscape. Least absolute shrinkage and selection operator (LASSO) algorithm was for generation of a smoking-related prognostic model. Kaplan–Meier analysis with log-rank test was for survival analysis, followed by a prognostic nomogram. The Gene Set Enrichment Analysis (GSEA) was used for functional analysis. The “oncoPredict” package was applied for drug sensitivity analysis.
Results
We recruited all types of bladder cancers and found that smoking was involved in poor prognosis, with the hazard ratio (HR) of 1.600 (95%CI: 1.028–2.491). A total of 1078 smoking-related DNA methylations (526 hypermethylation and 552 hypomethylation) were identified and 9 methylation-driven genes differentially expressed in bladder cancer. Also, 506
Conclusions
Totally, we initially identified the smoking-related epigenetic modifications in bladder cancer and constructed a corresponding prognostic model, which was also linked to disparate sensitivities to chemotherapeutics. Our findings would provide novel insights into the carcinogenesis, prognosis, and therapies in bladder cancer.
Keywords
Introduction
Epidemiology data show that bladder cancer is still involved in high morbidity and mortality, with an estimated 573,000 new cases and 213,000 deaths in 2020 worldwide. Almost 25% of cases are diagnosed with muscle-invasive bladder cancer (MIBC), a biologically aggressive pathological type with propensity for early distant metastases. Statistically, MIBC carries a poor prognosis, with a 5-year survival rate of 40%–65%. 1 Bladder cancer is more prevalent in males than in females, representing approximately 9.5 per 100,000 of incidence among males. 2 Until recently, accumulating evidence has disclosed multiple etiologies for bladder cancer, that is, age, smoking, schistosomiasis infection, chemical exposure, poor diet and lifestyle, air pollution, and genetic susceptibility.3,4 Numerous explanations for gender discerption in incidence of bladder cancer have been uncovered, especially for disparate exposures and sex hormone.3,5,6 The incipient symptoms of bladder cancer generally manifest as hematuria, dysuria, higher frequency and urgency of urination. Of these, painless hematuria does not seriously devastate the health of affected patients but its underestimation and ignorance often leads to delayed diagnosis and treatment. Currently, the cisplatin-based treatment is the standard-of-care first-line chemotherapy regimen for affected patients with advanced stage or distant metastases. However, the prognosis of most patients remains bleak, primarily ascribing to the disparate drug sensitivity. 7 Therefore, the identification of potential prognostic and therapeutic biomarkers for bladder cancer is essential to make progress in its precision diagnosis or treatment.
Epigenetic modifications refer to heritable alterations under the premise of non-gene sequence changes, that is, DNA methylation, histone modification, non-coding RNA (ncRNA), and chromatin remodeling. 8 Among them, the well-known one is DNA methylation, which initially accelerates the progress in human epigenetic diagnosis and therapies. 9 In mammals, DNA methylation commonly occurs in cytosines prior to guanines, destinated as dinucleotide CpGs. 10 The CpG-rich regions in the genome are known as CpG islands which are frequently detected in promoter CpGs regions. Substantial evidence has witnessed that DNA methylation functionally drives the expression of certain genes, thereby affecting the hallmarks of malignancies, that is, cell cycle, apoptosis, cell interactions, and drug sensitivity.11–14 Over the past decades, a multitude of ncRNA species have been observed in carcinogenesis of human cancers, including long ncRNAs (lncRNAs), microRNAs (miRNAs), small interfering RNAs (siRNAs), and circular RNAs (circRNAs).15–17 ncRNAs commonly exert their functions as sponging miRNAs, interacting with RNA-binding proteins, regulating the transcription, and translating proteins. Of note, a majority of studies have focused on their competitive endogenous RNA (ceRNAs) regulation, a hypothesis put forward by Salmena et al. in 2011. 18 As recently documented, we found that ncRNAs could play critical roles in the proliferation, metastasis, and invasion of bladder cancer. 19 Although great advances have been made in epigenetic modifications, it should be noted that previous literature studies have been limited to simple species of modifications, rather than comprehensive exploration.
It is well-recognized that health of an individual is a result of crosstalk between various environmental and genetic factors. The former, especially for behavior, nutrition, and environmental toxicant, has been reported to induce specific epigenetic modifications, thereby leading to variations in gene expression. For instance, several methylated CpGs were reportedly associated with long-term exposure of ambient fine particulate matter (PM2.5). 20 The miR-4466 from nicotine-activated neutrophils could promote tumor cell stemness and metabolism in lung cancer metastasis. 21 Dong et al. found that lncRNA uca1 might affect the arsenic-induced cell cycle through facilitating the ubiquitination of EZH2 to evaluate NFATc2 expression. 22 The smoking-induced demethylation of AhRR was associated with bladder cancer. 23 Considering the profound roles of smoking behavior in bladder cancer, it is of great significance to define the smoking-related epigenetic modifications and evaluate the corresponding clinical implications.
In this study, we queried the publicly available The Cancer Genome Atlas (TCGA) database to retrieve the level-3 data on DNA methylation, transcriptome data, and corresponding clinical information from patients with bladder cancer. Subsequently, we investigated the prognostic values of smoking behavior in bladder cancer and determined its epigenetic modifications, as well as its prognostic, pathogenic, and therapeutic implications.
Materials and methods
TCGA data retrieval and processing
The level-3 expression profiles and their corresponding clinical features were downloaded from TCGA database (TCGA) using “TCGAbiolinks”. 24 The DNA methylation expression was generated by HM450k platform (21 normal tissues and 419 bladder cancer). The methylation level is measured as a beta value, which is defined as the ratio of the methylated signal to the sum of methylated and unmethylated signal (ranging from 0 to 1). The TPM-normalized RNAseq data (19 normal tissues and 412 bladder cancer) and raw count of miRNA (19 normal tissues and 418 bladder cancer) were generated by IlluminaHiSeq RNASeq platform. The RNA-expression levels were log2-transformed to dilute extremely skewed gene expression distributions before analysis. Meanwhile, the corresponding clinical information included age, gender, days to last follow-up, survival status, grade, clinical stage, and smoking behavior. Here, non-smokers included all patients who had never smoked or reformed smoking for longer than 15 years. To define long-term exposure to smoking, we defined smoking behavior as current smoking ≥20 pack-years. We recruited all types of bladder cancers; of which, the majority were diagnosed with muscle-invasive bladder cancer (MIBC) and only 3 cases were with non-muscle invasive bladder cancer (NMIBC). All datasets involved in our work are open access, so there was no requirement for any ethical committee.
Screening for smoking-related epigenetic modifications
The “limma” package was applied for the identification of the tumor- and smoking-related differential expression. The tumor-related DNA methylations were identified with a filter as log2|Fold change| >0.5 and FDR <0.05, and smoking-related ones were filtered with log2|Fold change| >0 and p-value <0.05. The tumor-related lncRNAs and mRNAs referred to those with the log2|Fold change| >1 and FDR <0.05, and smoking-related ncRNAs were destinated as log2|Fold change| >0 and p-value <0.05. Ultimately, these differentially expressed genes were displayed by the “pheatmap” package.
Generation of the smoking-related ceRNA network
Subsequently, we generated the smoking-related endogenous RNAs (ceRNA) regulatory network through lncRNA–miRNA and miRNA–mRNA pairs. First of all, we predicted the potential miRNAs for smoking-related lncRNAs (miRcode) and then intersected with those miRNAs expressed in bladder tissues, followed by predicted mRNAs for these overlaps (miRDB, miRTarBase, and Targetscan). Afterward, the predicted mRNAs overlapped with tumor-related mRNAs to constitute the smoking-related lncRNA–miRNA–mRNA network. Similarly, we predicted the targeted mRNAs for smoking-related miRNAs and then intersected with tumor-related mRNAs for the smoking-related miRNA–mRNA network. Both the smoking-related lncRNA and miRNA regulatory network were shown by Cytoscape software. 25
Construction of the smoking-related prognostic model and risk-score calculations
To further explore the prognostic values of smoking-related genes in bladder cancer, we generated a prognostic model and then calculated the risk score for each individual for further analysis. The univariate Cox regression with a filter as p < 0.05 was performed, followed by the least absolute shrinkage and selection operator (LASSO) regression for gene selection and risk-coefficient determination. The LASSO regression analysis was performed using the “glmnet” package, with the maxit defined as 10,000. Furthermore, a smoking-related risk score was calculated for each individual, and then all cases were divided into high-risk and low-risk groups on basis of the median risk score for further exploration.
Gene set enrichment analysis and drug sensitivity exploration
The gene set enrichment analysis (GSEA), a computational method based on gene sets, was used to investigate the potential biological pathways in smoking-related epigenetic modifications in bladder cancer. 26 Here, the “c2. cp.kegg.symbols.gmt” set was regarded as a reference. The filter was defined as q-value <0.05, with the parameters as “minGSSize = 15” and “maxGSSize = 500.” Drug sensitivity exploration was carried out using the “oncoPredict” package, and the tumor therapy genome data were downloaded from GDSC. 27
Statistical analysis
The Cox regression analysis was performed with the “survival” package, and then presented using the “forestplot” package. The Kaplan–Meier analysis with a log-rank test was for survival analysis. Wilcoxon signed ranking test was used to compare the differences in drug sensitivity between high-risk and low-risk groups. All analyses were conducted with R software (version 4.0.3), and the statistical significance was defined as p-value <0.05.
Results
Smoking behavior is correlated with poor prognosis in bladder cancer
To assess the impacts of smoking behavior on prognosis in bladder cancer, we performed a Cox regression analysis. Totally, there were 219 (219/225) non-smokers and 59 (59/60) smokers with full information on age, gender, grade, clinical stage, survival status, and following time. At diagnosis, the mean age of all participants was 68.97 ± 10.57 years, and the male participants accounted for 71.22%, almost 2.5 times of females. The median follow-up time was 516 days and ultimately 129 deaths were observed. In the univariate Cox regression analysis, the hazard ratio (HR) of stages III–IV was 2.683 (95%CI: 1.663–4.329) as compared to stages I–II, and the HR of smoking was 1.322 (95%CI: 0.868–2.013) compared with no-smoking (Figure 1(a)). In the multivariate Cox regression analysis, both stages III–IV and smoking behavior were significantly linked to the poor prognosis in bladder cancer, with the HR of 2.543 (95%CI: 1.560–4.146) and 1.600 (95%CI: 1.028–2.491), respectively (Figure 1(b)). Hazard ratio (HR) with 95% confidence interval (CI) for smoking-related death in bladder cancer. (a) Univariate Cox regression analysis; (b) Multivariate Cox regression analysis.
Smoking-related epigenetic modifications are frequently observed in bladder cancer
To identify the smoking-related epigenetic modifications in bladder cancer, we performed differential expression analyses between no-smoking and smoking groups. First of all, we profiled the tumor-related mRNAs in bladder cancer, including 6168 up-regulated mRNAs and 4120 down-regulated mRNAs (Figure 2(a)). For smoking-related DNA methylation, the differential expression analysis was conducted on 225 non-smokers and 60 smokers with DNA methylation data. A total of 1078 smoking-related DNA methylations were identified (526 hypermethylation and 552 hypomethylation), and there were 9 methylation-driven genes (CSAG1, HOXA11-AS, TMEM171, SCARF1, SPON1, HHEX, RGN, NUPR1, and PPP1R14D) that differentially expressed between bladder cancer and normal tissues (Figure 2(b)). For smoking-related ncRNAs, the differential expression analysis was performed on 222 non-smokers and 58 smokers with transcriptome data. A total of 506 smoking-related lncRNAs were determined, consisting of 448 upregulated and 58 downregulated lncRNAs (Figure 2(c)). The top five upregulated lncRNAs were ranked as MIR31HG, LINC01116, LINC00648, LINC01456, and AC011632, and the top five downregulated ones were MIR200CHG, PSLNR, SNHG9, AL137785, and LINC00665. A total of 102 smoking-related miRNAs were uncovered among 223 non-smokers and 60 smokers, involving 74 upregulated and 28 downregulated miRNAs (Figure 2(d)). Among them, the top five upregulated miRNAs were hsa-miR-31-5p, hsa-miR-31-3p, hsa-miR-548f-3p, hsa-miR-581, and hsa-miR-3662, and the top five downregulated ones were hsa-miR-184, hsa-miR-1270, hsa-miR-141-5p, hsa-miR-200a-5p, and hsa-miR-410-3p. Differential expression analyses in bladder cancer. (a) Heatmap of the tumor-related mRNAs; (b) Heatmap of the smoking-related DNA methylations; (c) Heatmap of the smoking-related lncRNAs; (d) Heatmap of the smoking-related miRNAs; (e) The smoking-related lncRNAs regulatory network; (f) The smoking-related miRNAs regulatory network.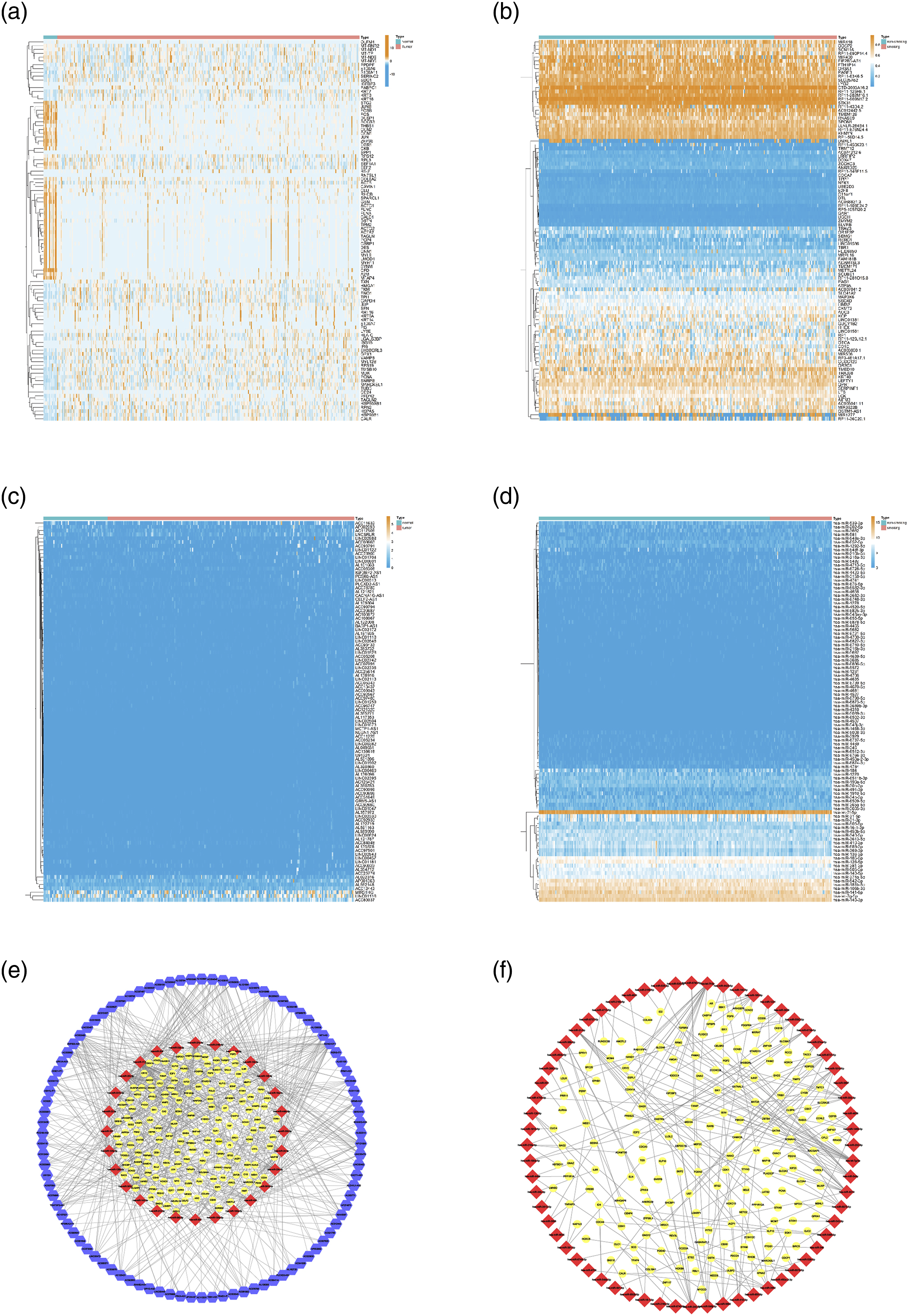
Construction of the smoking-related ceRNA network and functional analysis
With regard to profound functions of ceRNA regulatory network in human cancers, we further predicted the smoking-related ceRNA network in cases of bladder cancer. For smoking-related lncRNA network, 206 miRNAs were predicted to interact with smoking-related lncRNAs; of which, 43 miRNAs were expressed in bladder cancer. Afterward, we predicted the targeted mRNAs for these 43 miRNAs, and 1792 predicted ones were simultaneously predicted by miRDB, miRTarBase, and Targetscan. Among them, 167 mRNAs were differentially expressed between bladder cancer and normal tissues. Finally, the smoking-related lncRNAs–ceRNA network was produced through 493 lncRNA–miRNA and 247 miRNA–mRNA pairs (Figure 2(e)). For the smoking-related miRNA network, we firstly predicted 1256 targeted mRNAs for these miRNAs, and 179 were differentially expressed between bladder cancer and normal tissues. Ultimately, the smoking-related miRNA network consisted of 235 miRNA–mRNA pairs (Figure 2(f)).
Smoking-related signatures are potential prognostic indicators
Furthermore, we generated a prognostic model based on mRNAs that were regulated by smoking-related epigenetic modifications. We firstly intersected the mRNA regulated by smoking-related epigenetic modifications with those mRNAs differentially expressed in bladder cancer. In all, 292 mRNAs modulated by smoking-induced DNA methylation, lncRNA and miRNA, were differentially expressed in bladder cancer. A total of 84 mRNAs were filtered by the Cox regression analysis and then 28 prognostic signatures were identified by LASSO regression (CBX7, MAP1B, MXRA7, RAC3, ARHGAP6, HMGA1, ZNF106, PDGFRA, TMTC1, ELN, ARHGEF26, RPS6KA1, ERC1, GNG5, MAP3K8, EPHB1, ID2, MYC, ARHGEF6, SCD, LDLR, TXNIP, TPD52L1, AHNAK2, GOLGA8A, FBN2, GJA1, and CSAG1) (Figures 3(a) and (b)). For further analysis, we calculated the smoking-related risk score for each individual, and all patients were then divided into the high-risk and low-risk groups with the threshold value of the median risk-score. Of these, high-risk group was defined as the risk score greater than the median value. We found that a majority of patients with high-risk scores tended to have shorter survival time (Figure 3(c)). The expression parterns of prognostic signatures between high-risk and low-risk groups were shown in Figure 3(d). Among them, the RAC3 and MXRA7 levels were significantly upregulated in the high-risk group, while the RPS6KA1 and CBX7 levels were obviously upregulated in the low-risk group. Generation of the smoking-related prognostic model and risk-score calculations. (a, b) The LASSO Cox regression analysis; (c) The survival time distribution of patients with different risk scores; (d) Heatmap of smoking-related signatures among patients with high-risk and low-risk groups.
In the univariate Cox regression analysis, the HR of stages III–IV was 2.606 (95%CI: 1.613–4.209) as compared to stages I–II and that of risk score was 1.149 (95%CI: 1.113–1.185) when compared to corresponding references (Figure 4(a)). In the multivariate Cox regression analysis, the stages III–IV and high-risk score were significantly associated with the poor prognosis in bladder cancer, with an HR of 2.301 (95%CI: 1.404–3.771) and 1.143 (95%CI: 1.105–1.183), separately (Figure 4(b)). Then, a prognostic nomogram was established among 274 patients with complete clinical features and expression data, to predict the 1-, 3-, and 5-year overall survival rates (Figures 4(c) and (d)). Hazard ratio (HR) with 95% confidence interval (CI) for smoking-related risk score in bladder cancer. (a) Univariate Cox regression analysis; (b) Multivariate Cox regression analysis; (c) Calibration curves of risk signature to predict the 1-, 3-, and 5-year overall survival rates; (d) A prognostic nomogram to predict the 1-, 3-, and 5-year overall survival rates.
Drug sensitivity is differentially predicted between high-risk and low-risk groups
For functional analysis, the GSEA analysis was performed to explore the potential pathways underlying different risk-score groups. In total, there were 31 pathways enriched in the high-risk group and 10 pathways in the low-risk group. Among them, several cancer-related pathways were enriched in the high-risk group, including focal adhesion, WNT signaling pathway, pathways in cancer, MAPK signaling pathway, and JAK STAT signaling pathway (Figure 5(a)). Meanwhile, the drug metabolism and other enzymes pathway were enriched in the low-risk group (Figure 5(b)). Gene set enrichment analysis (GSEA) and drug sensitivity exploration. (a, b) The GSEA analyses of smoking-related prognostic signatures; (c) Comparisons of drug sensitivity between high-risk and low-risk groups.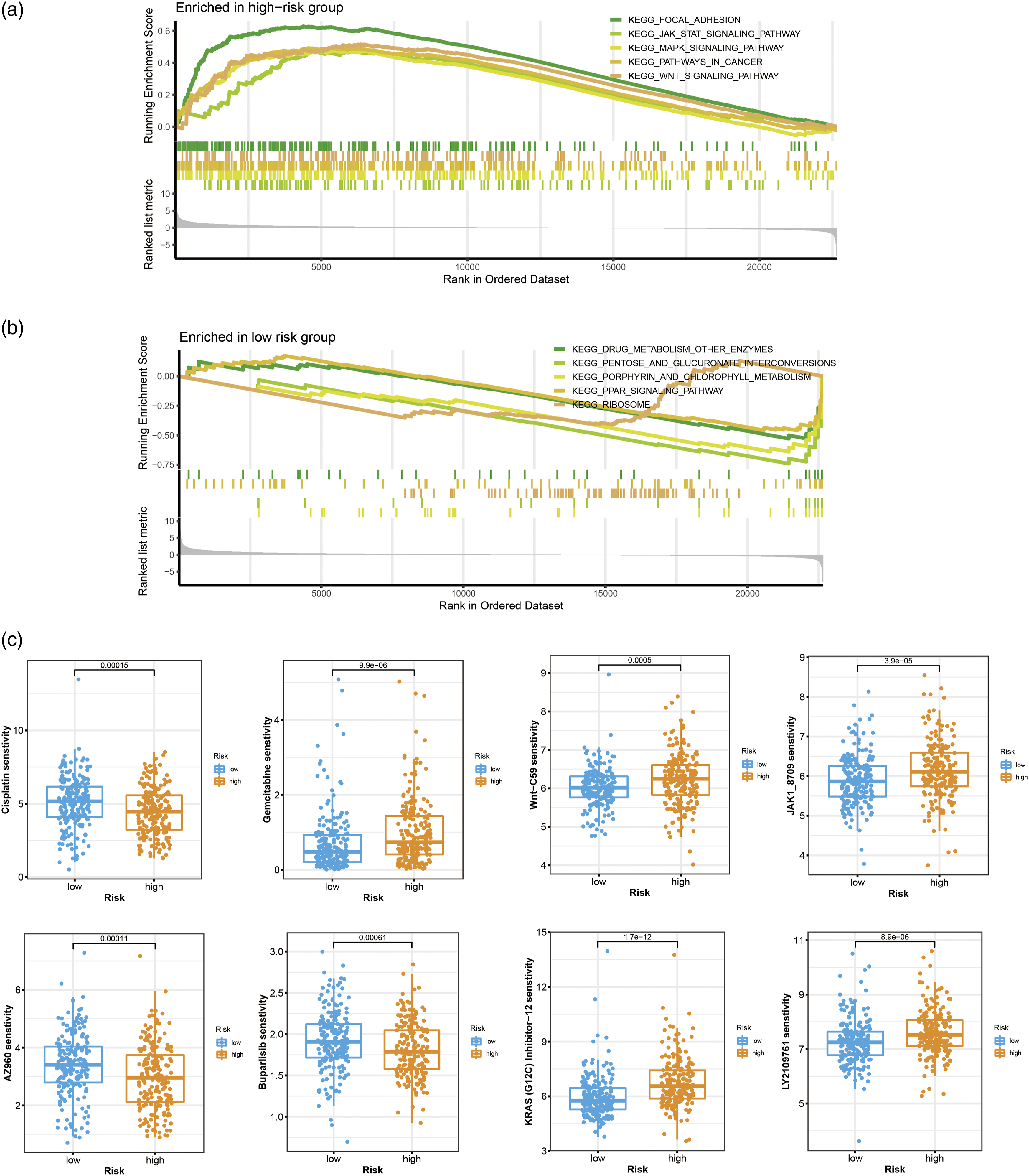
To delineate the effects of smoking-related epigenetic modifications on the treatment of bladder cancer, we also compared differences in the drug sensitivity between high-risk and low-risk groups. We found that patients with different risk scores displayed different susceptibilities to multiple antineoplastic drugs. In particular, patients with low-risk scores were more sensitive to Cisplatin, whereas they exhibited lower sensitivity to Gemcitabine (Figure 5(c)). In terms of several well-recognized cancer-related pathways, cases with high-risk scores presented better sensitivity to Wnt-C59, JAK1_8709, LY2109761, and KRAS (G12 C) Inhibitor-12, while they were less sensitive to AZ960 and Buparlisib (Figure 5(c)).
Discussion
Advances in deep sequencing and bioinformatics have extensively expedited the identification of multiple epigenetic modifications in human cancers, especially for DNA methylation, ncRNAs, histone modification, etc. Numerous evidence has suggested that these epigenetic modifications were dynamic, guaranteeing their sensitivity to environmental factors, that is, disinfection by-products, metals exposure, and smoking.28–30 Considering the potential carcinogenesis of smoking behavior in bladder cancer, it is essential for further exploration of its-induced epigenetic modifications, as well as their corresponding clinical implications. In this study, we screened the smoking-related DNA methylations, lncRNAs, and miRNAs in bladder cancer based on the TCGA database and investigated the underlying prognostic, therapeutic, and carcinogenic effects. We found that smoking behavior was associated with poor prognosis in bladder cancer. A total of 1078 DNA methylations, 506 lncRNAs, and 102 miRNAs were determined as smoking-related epigenetic modifications. We also calculated smoking-related risk scores and then observed that cases of high-risk scores had poor prognosis. Several cancer-related pathways were enriched in the high-risk group, and disparate sensitivities to chemotherapeutics were witnessed between high-risk and low-risk groups.
The geographic and temporal variations in the incidence of bladder cancer extensively implied its risk factors. Among them, smoking was reported as a well-established risk factor for bladder cancer, even explaining a comparable risk of bladder cancer in gender, with population attributable risks (PARs) as 50% in males and 52% in females. 31 Among Koreans, smoking accounted for approximately 20% new cases and 32% deaths of bladder cancer. A population-based, prospective health, and lifestyle study on Australians revealed that the current smokers had an increased risk of all types of cancers (HR: 1.43, 95%CI: 1.34–1.51) and bladder cancer (HR: 3.08, 95%CI: 2.00–4.73) compared with non-smokers. 32 Especially, Li et al. reported that former smokers (quit smoking) had a lower risk of bladder cancer than current smokers (HR:0.61, 95% CI: 0.40–0.94) among postmenopausal women. 33 In our study, our results indicated that smoking behavior was associated with the poor prognosis (HR: 1.600, 95%CI: 1.028–2.491). A meta-analysis indicated that patients with bladder cancer who had a history of smoking may present worse recurrence-free and progression-free survival compared to non-smokers. 34 Encouragingly, the attempts of smoking cessation that are motivated by bladder cancer diagnosis present higher cessation success. 35 In all, our current findings strongly underpin the urgent need to invest more in smoking cessation strategies. It is also convincible to investigate the underlying biological functions of smoking in bladder cancer.
Initially, disease divergences between identical twins led to the realization that DNA sequence is not the single determinant of clinical phenotypes. With the rapid development of deep sequence, the term “epigenetic modifications” introduces the understanding of the “genotype” and “phenotype.” Of which, epigenetic modifications, referring to the dynamic modifications that occur on DNA and its subsequent modifications associated with regulatory proteins, exert crucial functions. Accumulating evidence demonstrated that the epigenetic disruption is an important hallmark of human cancers.15,36–38 Intriguingly, these modifications are dynamic and easily influenced by external stimuli, thereby explaining the environmental exposures on the carcinogenesis and progression of cancers. Liu et al. profiled 194 lncRNAs that were differentially expressed in oncogenic HPV infection along with cervical carcinogenesis, and uncovered that oncogenic HPV could promote the level of lnc-FANCI-2 through E7 and YY1. 39 A set of methylated differentially CpGs and regions correlated with HPV infection were identified in head and neck squamous cell carcinomas.40–42 Nicotine could attenuate DNA methylation by regulating the expression of DNMTs in human endometrial stromal cells. 43 However, previous studies commonly explored solo types of epigenetic modifications. In our study, we systematically investigated the smoking-related DNA methylation, lncRNA, and miRNA in bladder cancer, and identified 1078 DNA methylations, 506 lncRNAs, and 102 miRNAs. Of which, the LINC01106, an upregulated lncRNA in smoking, could regulate ELK3 and HOXD8 to promote the progression of bladder cancer. 44 We were interested in further investigation on the potential roles of these smoking-related modifications.
Recently, epigenetic modifications have been heralded as a novel type of promising diagnostic, prognostic, and therapeutic targets in cancers. According to Balázs et al., aberrant DNA methylation might affect the prognosis of patients with breast cancer. 45 The DNA methylation-based signatures could evaluate the immune response and prognosis in cases of colorectal cancer. 46 In this study, we further explored the prognostic and therapeutic implications of smoking-related epigenetic modifications in bladder cancer by using their most downstream regulatory mRNAs. We generated a smoking-related risk score for each patient and then observed that high-risk cases had poor prognosis. Increasing evidence has disclosed multiple cancer-related pathways involved in epigenetic modifications. For instance, the lncRNA JPX might participate in the carcinogenesis and metastasis of lung cancer via miR-33a-5p/Twist1 axis, thereby activating the WNT/β-catenin signaling pathway. 47 The miR-433 was reported to suppress the proliferation of breast cancer via the MAPK signaling pathway. 48 In our study, we found that several cancer-related pathways were significantly enriched in the high-risk group, containing the focal adhesion, WNT signaling pathway, pathways in cancer, MAPK signaling pathway, and JAK STAT signaling pathway. Reversely, the drug metabolism enzyme pathway was enriched in the low-risk group. Our findings indicated that smoking behavior might participate in the progression of bladder cancer via inducing these epigenetic modifications. Meta-analysis suggested that smoking behavior was involved in the lower neoadjuvant chemotherapy response rate. 49 In our research, we found that patients with different risk scores also had disparate sensitivities to chemotherapeutics, providing novel insights into the selection of therapeutic regimens.
However, several limitations involved in this study should be emphasized. First of all, this study was performed based on TCGA database, and large-scale and multicenter validations are necessary. Second, the chemotherapeutic implications of smoking-related modifications were derived from bioinformatics analysis, and it is necessary to conduct more drug-sensitivity validations in vivo or vitro.
Conclusions
Collectively, we comprehensively profiled smoking-related epigenetic modifications and generated a corresponding prognostic model. Then, we calculated a smoking-related risk score, which was extensively involved in different prognoses and disparate sensitivities to chemotherapeutics. Our findings would provide novel insights into the carcinogenesis, prognosis, and therapies in bladder cancer.
Footnotes
Author’s contributions
Ya Ling performed all data analyses and drafted this manuscript. Jindong Li provided the statistical support. Lijuan Zhou designed this research and reviewed the manuscript. All authors have reviewed and approved this manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethical approval
All datasets involved in our work are open access, so there was no requirement for any ethical approval.
Informed consent
All datasets involved in our work are open access, so there was no requirement for any informed consent. All datasets (Level-3) involved in current work were downloaded from the publicly available TCGA database, so there was no additional patient consent.