
Abstract
The nucleolus is a nuclear compartment that plays an important role in ribosome biogenesis. Some structural features and epigenetic patterns are shared between nucleolar and non-nucleolar compartments. For example, the location of transcriptionally active mRNA on extended chromatin loop species is similar to that observed for transcriptionally active ribosomal DNA (rDNA) genes on so-called Christmas tree branches. Similarly, nucleolus organizer region–bearing chromosomes located a distance from the nucleolus extend chromatin fibers into the nucleolar compartment. Specific epigenetic events, such as histone acetylation and methylation and DNA methylation, also regulate transcription of both rRNA- and mRNA-encoding loci. Here, we review the epigenetic mechanisms and structural features that regulate transcription of ribosomal and mRNA genes. We focus on similarities in epigenetic and structural regulation of chromatin in nucleoli and the surrounding non-nucleolar region and discuss the role of proteins, such as heterochromatin protein 1, fibrillarin, nucleolin, and upstream binding factor, in rRNA synthesis and processing.
E
Eukaryotic genomes contain hundreds of head-to-tail tandem repeats called nucleolus organizer regions (NORs). In human diploid cells, which contain ×400 rRNA genes, these regions map to five pairs of acrocentric chromosomes: 13–15, 21, and 22 (Henderson et al. 1972). In the mouse genome, NORs are found on chromosomes 12 and 15–19 (Dev et al. 1977; McStay and Grummt 2008). The position of NORs on the short arms of acrocentric chromosomes isolates them from genes transcribed by RNA polymerase II (Pol II) and Pol III. Nucleolar rRNA genes are transcribed by Pol I, and their expression is regulated by chromatin remodeling factors and some specific epigenetic events (McStay and Grummt 2008). However, the transcription and silencing of genes encoding rRNAs is governed by histone modification, as occurs with non-nucleolar genes.
rRNA genes provide an interesting model for studying how epigenetic processes and chromatin arrangements can regulate gene expression and silencing (Santoro et al. 2002; Santoro and Grummt 2005). Genes encoding mRNA and rRNA can be found in two different chromatin configurations: an open chromatin structure corresponding to a transcriptionally active state and a more “condensed” configuration representing a silent state. rRNA genes are localized to the nucleolus, a large “transcription factory” (Cook 1999) that is often in the central region of the nucleus. Interestingly, trancriptionally active mRNA genes that occupy Pol II-positive sites are also centrally located and share “transcription factories” (Osborne et al. 2004; Harničarová et al. 2006; Takizawa et al. 2008).
Recently published experiments have shown that specific epigenetic factors, including histone acetylation and methylation, affect the transcription of rRNA genes (Santoro et al. 2002; Santoro and Grummt 2005). This suggests that rDNA transcription is a result of cooperation between DNA methylation, histone modification, and chromatin remodeling factors (McStay and Grummt 2008). This review will summarize current knowledge about the epigenetic regulation of rDNA genes in comparison with the epigenetic and structural regulation of mRNA species.
General Structure of Nucleoli
Mammalian nuclei mainly contain one nucleolus, with tumor cells tending to contain multiple nucleoli. The nucleolus is a non-membrane-bound nuclear compartment and is a relatively dynamic structure, as are some components of the surrounding non-nucleolar chromatin (Phair and Misteli 2000; Misteli 2001; Chen et al. 2005; Olson and Dundr 2005; Shav-Tal et al. 2005). Nucleolar morphology has been correlated with various steps in ribosome biogenesis, and the primary function of the nucleolus is synthesis of ribosomal subunits. NORs, which contain rDNA repeats oriented in a telomere-to-centromere direction, are responsible for formation of nucleoli (Worton et al. 1988; Gonzalez and Sylvester 1997). NORs consist of canonical and non-canonical repeats (Caburet et al. 2005), some of which are palindromic. In human chromosomes, rDNA repeats map to short arms of acrocentric chromosomes. However, not all NOR-containing chromosomes associate with nucleoli, and chromosomes without NORs can also surround nucleoli. In some cases, NOR chromosomes that are not associated with nucleoli extend their rDNA-chromatin fibers to the nucleolus, connecting the extranucleolar chromosomal territory with nucleoli (Kalmárová et al. 2008). Some transcriptionally active non-ribosomal genes are located on large chromatin loops (Volpi et al. 2000; Wiblin et al. 2005), and some mRNA genes associate with the nucleolus (Shimizu et al. 2007; Bártová et al. 2008a; Royo et al. 2009). Thus, in addition to the transcriptional activity of NOR-bearing chromosomes, the transcription and splicing of non-ribosomal genes can be closely linked to the nucleolar compartment.
Many experiments have shown that nucleolar integrity is preferentially maintained by transcription of rRNA genes. The nucleolus contains several important compartments: different domains are responsible for the production of preribosomal particles, the transcription of rRNA genes, the processing of primary transcripts into mature 18S, 5.8S, and 28S rRNA, the addition of proteins to nascent preribosomes, and the incorporation of 5S rRNA, which is synthesized in the nucleoplasm by RNA Pol III (Figure 1A). Nucleoli contain many subdomains, such as the fibrillar center (FC), the dense fibrillar component (DFC), and the granular component (GC), that are visible by transmission electron microscopy after staining. Transcription of ribosomal genes, which is mediated by RNA Pol I, likely occurs at the border between the FC and DFC (González-Melendi et al. 2001; Raška 2003). However, some experiments indicate that rDNA transcription occurs in the FC (e.g. Granboulan and Granboulan 1965; Scheer and Benavente 1990; Raška et al. 1995; Cmarko et al. 2000; Thiry et al. 2000). During the next step of rRNA processing, the pre-rRNA accumulates in the DFC, where posttranscriptional events take place. Maturation of pre-rRNA in the DFC leads to formation of 18S, 5.8S, and 28S rRNA (Figure 1A). Granules in the GC are considered preribosomal particles in various stages of maturation (Shaw and Jordan 1995; Raška et al. 2006). Synthesis of ribosomal subunits takes place in the GC, and nearly mature ribosomal subunits leave the nucleolus and can be observed in the cytoplasm (Granboulan and Granboulan 1965; Olson and Dundr 2005). Nucleolar export likely occurs by simple diffusion, as has been documented for 60S subunits (Politz et al. 2003). Actin and myosin I have been detected within nucleoli, suggesting that motor proteins may be involved in the movement of preribosomal ribonucleoprotein (RNP) complexes through the nucleolus. However, there is no direct evidence that actin and myosin are responsible for the movement of nucleolar pre-rRNP complexes (Philimonenko et al. 2004; Miralles and Visa 2006).
The nucleolus contains many other key proteins. For example, fibrillarin is a small nuclear RNP component and plays an important role in processing of pre-rRNA. Moreover, fibrillarin associates with the U3, U8, and U13 small nuclear RNAs and is located in the DFC (Nicol et al. 2000). Similarly, nucleolin is an abundant protein in the nucleolus that interacts with different proteins and RNA sequences. Nucleolin is involved in regulation of chromatin structure, rDNA transcription, rRNA maturation, ribosome assembly, and nucleocytoplasmic transport (Ginisty et al. 1999). Nucleolin is responsible for chromatin condensation and decondensation and has enhanced affinity to histone 1 (H1) (Erard et al. 1988) in linker regions. These experiments clearly document a close relationship between histone signature and nucleolar function.
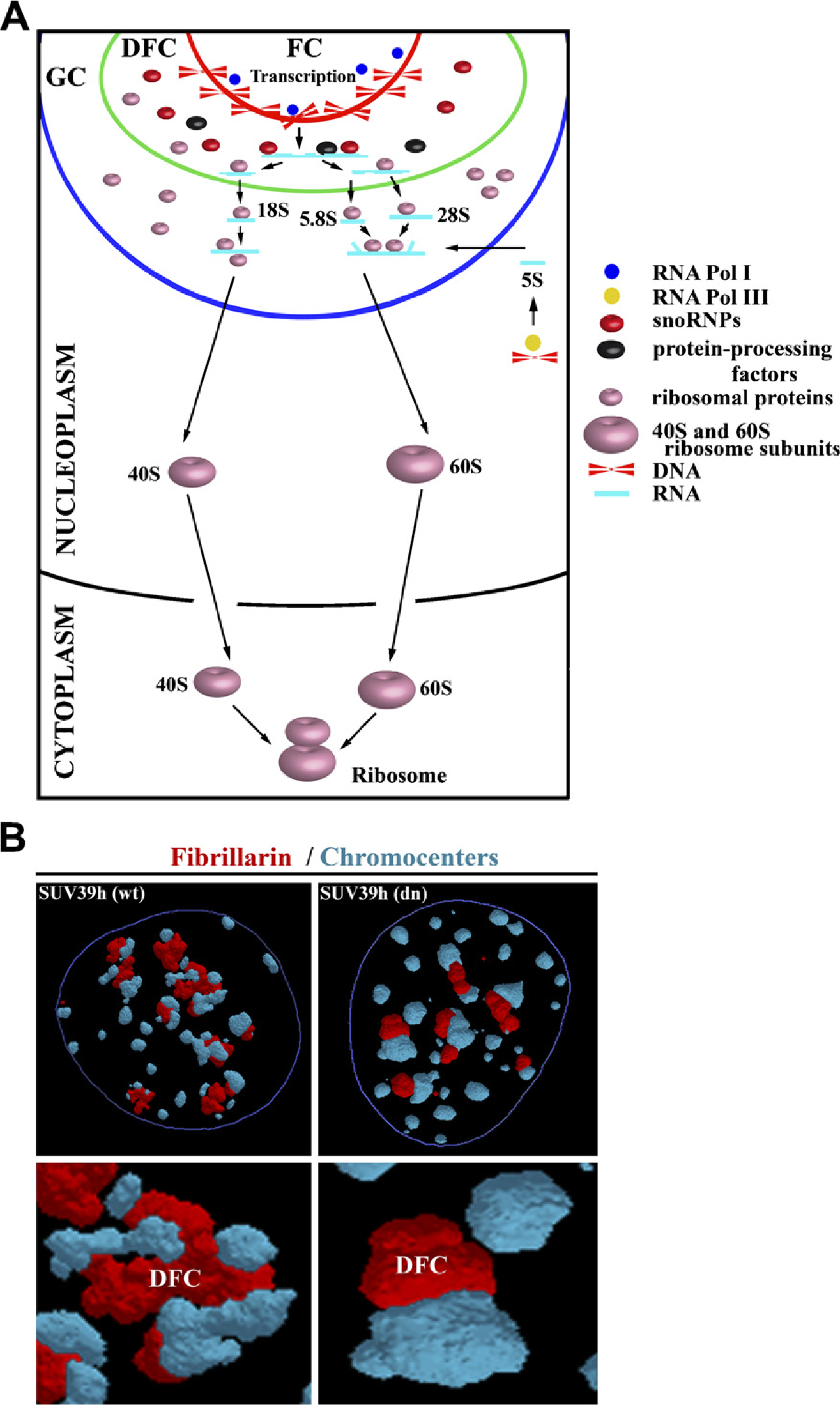
Structure and organization of nucleoli and nucleolar organizer regions (NORs).
Higher-order Chromatin Structure of mRNA and rRNA Genes
Functional significance of nuclear arrangement of mRNA genes is still matter of discussion. Fluorescence in situ hybridization (FISH) was a breakthrough technique in the chromatin architecture field, since it enabled visualization of the spatial nuclear arrangement of chromosome territories (CTs) and their subdomains (Cremer et al. 1988; Pinkel et al. 1988). Similarly, immunofluorescence combined with high-resolution confocal microscopy permits examination of the functional and structural characteristics of chromatin-related proteins and bodies, including Cajal, promyelocytic leukemia bodies, nuclear speckles containing splicing factors, and others (Zirbel et al. 1993; Clemson et al. 1996; Bridger et al. 1998). There are several general principles governing the nuclear and territorial organization of mRNA genes (Scheuermann et al. 2004). For example, transcriptionally active genes are positioned on the periphery of their CTs, while an anonymous DNA fragment was observed in more interior parts of CTs (Kurz et al. 1996). Moreover, in cells with decondensed chromatin, such as pluripotent embryonic stem cells (ESCs), active genes are positioned on chromatin loops extended away from their CTs (Volpi et al. 2000; Mahy et al. 2002, Williams et al. 2002; Wiblin et al. 2005, Bártová et al. 2008c). Some of these genes share the same “transcription factory” or site of nascent RNA synthesis, even though they are from different territories (Osborne et al. 2004). Extreme chromatin loops are especially prevalent for genes in clusters. For example, the Oct3/4 genes map to the major histocompatibility gene cluster, and when they are active, they are positioned on chromatin loops in pluripotent human ESCs (hESCs) (Wiblin et al. 2005; Bártová et al. 2008c). However, the c-myc gene, mapped in close proximity to the telomere of human chromosome 8, is associated with the periphery of the chromosome territory in pluripotent and differentiated hESCs (Bártová et al. 2008c). Similarly, the Nanog gene, which is responsible for hESC pluripotency, is not observed in loops when transcriptionally active; rather, it is positioned in the very interior of the nucleus. Thus, individual genes show distinct structural characteristics that are likely to be influenced by gene density in the surrounding chromatin (Küpper et al. 2007). Generally, GC-rich domains have often been observed closer to the nuclear center, while gene-poor regions occupied the nuclear periphery (Saccone et al. 2002; Goetze et al. 2007). For instance, gene-rich chromosome 19 is positioned closer to the nuclear interior than gene-poor human chromosome 18. The nuclear position of the sequences of these two chromosomes was found to be conserved in several cell types and during evolution (Tanabe et al. 2002; Neusser et al. 2007). On the other hand, inverted nuclear architecture was observed in rod photoreceptor cells in animals with nocturnal vision. The rods of nocturnal retinas have a unique inverted pattern, such that heterochromatin is located in the nuclear center, while transcription of euchromatin and posttranscription processes are associated with the nuclear periphery (Solovei et al. 2009). This inverted pattern of nuclear architecture is not found in other eukaryotic cells and can be explained by adaptation of animals to nocturnal vision.
The arrangement of ×400 copies of the 43-kb human rRNA genes is in some aspects very similar to the nuclear architecture of mRNA genes. Nucleoli are positioned in the interior of the nucleus; thus, highly transcriptionally active rDNA, which accounts for ×50% of nascent RNA synthesis (Bernstein and Allis 2005; Russell and Zomerdijk 2005), occupies the nuclear center. Moreover, ribosomal transcripts are arranged in a formation known as a “Christmas tree” (Miller and Beatty 1969; Mosgoeller et al. 2001), which resembles the positioning of mRNA genes on the loops that extend away from the body of related chromosome territory. In “Christmas tree”-shaped structures, which are visible in electron micrographs, transcriptionally active rRNA genes that carry nascent pre-rRNA are located on the imaginary branches. Arrangement of rRNA genes into “Christmas tree” formation has been observed in many human (Koberna et al. 2002) and plant (Shaw et al. 2002) cells. This structure was first detected by the use of bromouridine labeling, which enabled the visualization of compact “Christmas tree” structures in the plant Pisum sativum (González-Melendi et al. 2001). However, there are contradictory data showing exact location of rRNA transcription sites within individual compartments of nuclei (FC, DFC, and GC). Some authors mapped rRNA primary transcripts to the interior of the FC, while other experiments showed that transcription of rRNA genes occurs at the border of the FC and DFC (Granboulan and Granboulan 1965; Raška et al. 1995; Cmarko et al. 2000; Thiry et al. 2000). These discrepancies could be due to the different experimental strategies and approaches used to visualize nucleolar structure. Moreover, cell type specificity may also be a factor.
Efficient transcription of rDNA by Pol I requires the formation of a preinitiation complex (PIC), which includes upstream binding factor (UBF) and selectivity factor 1 (SF1), on the promoter (Prieto and McStay 2005). Surprisingly, recent live imaging experiments showed that only 7–10% of Pol I in the nucleolus participates in transcription. Thus, many Pol I molecules are not engaged in rRNA processes (Dundr et al. 2002a,b). UBF mediates recruitment of Pol I by its direct interaction with this enzyme and/or indirectly via still unknown factors. UBF and other PIC components at the rDNA promoter are responsible for topological changes in the rDNA (Russell and Zomerdijk 2005). Moreover, increased rDNA transcription is related to increased levels of promoter occupancy by the SL1 factor, which directs formation of the PIC complex. Synthesis of rRNA is also influenced by other factors and events, especially by changes in the cell cycle and cell differentiation. For example, the p53 protein, which is responsible for cell cycle regulation, prevents interaction between UBF and SL1, thus repressing Pol I–mediated transcription (Grummt 2003). Similarly, retinoblastoma (Rb) protein disrupts interactions between UBF and SL1, resulting in decreased Pol I–mediated transcription (Hannan et al. 2000). UBF expression was also changed during differentiation, accompanied by changes in the level of the c-myc proto-oncogene (Poortinga et al. 2004). These data indicate that products of proto-oncogenes, such as c-myc, and/or the TP53 and Rb1 tumor suppressor genes affect rRNA synthesis. Thus, the level and processing of rRNA are likely influenced by mutations in these genes which lead to malignant cell transformation and uncontrolled tumor growth. Therefore, coordinated regulation of the Pol I–mediated transcription machinery, which involves many epigenetic modifications such as pseudouridylation and rRNA methylation, is important for optimal ribosome biogenesis, protein synthesis, cell renewal, growth, differentiation, and cell death.
Many Histone Modifications Are Observed in Both Nucleolar and Non-nucleolar Regions
Eukaryotic chromatin is formed by a complex of core histones (H2A, H2B, H3, and H4) surrounded by 146-bp DNA. These structures are called nucleosomes and are connected to one another by linker DNA that acts as a binding site for H1 (Rice and Allis 2001). Nucleosomes are organized into more coiled structures, leading to the formation of CTs, which are the highest order chromatin structures. CTs are specifically organized within interphase nuclei (Cremer and Cremer 2001; Lanctot et al. 2007), and mutual contact and/or intermingling between chromosomes territories is currently a topic of interest (Cremer and Cremer 2001; Branco and Pombo 2006; Goetze et al. 2007). According to the model of chromosome nuclear arrangement, with gene-dense territories positioned more interiorly than gene-poor ones (Scheuermann et al. 2004), it is highly probable that histone epigenetic modifications can also dictate nuclear radial arrangement of chromosome subdomains and/or whole territories (Cremer et al. 2004; Zinner et al. 2006; Strašák et al. 2009).
The core N-terminal histone tails are subject to covalent modifications, such as acetylation, methylation, phosphorylation, sumoylation, and/or ubiquitination. The presence of these epigenetic marks is mediated by specific enzymes, including histone acetyltransferases, histone deacetylases (HDACs) that regulate acetylation and/or histone methyltransferases (HMTs), and histone demethylases (Kouzarides 2007; Zhang et al. 2007). These enzymes are responsible for transcriptional activation and/or repression (Hassig et al. 1998; Rice and Allis 2001; Zhang and Reinberg 2001). Although the existence of histone methylation has been known since 1964 (Murray 1964), only recently has the importance of the histone code in the regulation of nuclear functions been appreciated; indeed, imbalances in this process can lead to human diseases, including cancer (Dialynas et al. 2008; Krejčí et al. 2009).
Histones can be modified at many amino acid positions. There are more then 60 different histone residues at which epigenetic modifications have been identified (Kouzarides 2007). Histone acetylation is one of the best-characterized epigenetic marks and is generally associated with activation of gene expression, while deacetylation causes gene silencing and heterochromatinization (Rice and Allis 2001; Schübeler et al. 2001). However, many other modifications have been detected on histones. Recent studies have shown that methylation at lysines or arginines is an important means of regulating transcription. Moreover, there are three different forms of methylation: mono- and di-methylation at lysines and arginines and tri-methylation at lysines (Kouzarides 2007). Specific histone modifications can be studied by immunohistochemistry, Western blotting, chromatin immunoprecipitation combined with PCR and/or microarray technologies [e.g., chromatin immunoprecipitation (ChIP-on-chip)], and the most sophisticated method, mass spectrometry (Kouzarides 2007).
Global analysis of all histone modifications has shown that some forms of histone methylation, such as H3K4, H3K36, and H3K79, are responsible for transcriptional activation, while H3K9, H3K27, and H4K20 methylation occupies heterochromatin regions and/or transcriptionally silent loci. Moreover, orchestrated activation of Polycomb-group (PcG) protein complexes (PRC1 and PRC2), which act in concert with H3K27me3, leads to inactivation of one female × chromosome as facultative heterochromatin. Some proteins from PcG, especially BMI-1, were also observed within nucleoli (Cmarko D, unpublished data). Interaction of H3K9 methylation with heterochromatin protein 1 (HP1) guarantees the stability of pericentromeric heterochromatin (Lehnertz et al. 2003), which is arranged into chromocenters that surround nucleoli. However, most histone modifications are dynamic, and basic enzymes that mediate and remove specific histone marks were described previously (Kouzarides 2007). For example, acetyltransferases are divided into three families, GNAT, MYST, and CBP/p300, and deacetylation is mediated by class I–III histone deacetylases. SUV39h HMT is responsible for H3K9me3, and G9a HMT is responsible for H3K9me1 and H3K9me2 (Kouzarides 2007). Moreover, recently discovered histone demethylases (Shi et al. 2004; Kouzarides 2007), such as LSD1 (Shi et al. 2004), are responsible for H3K4 demethylation, while methylation at H3K36 can be reversed by the demethylase JHDM1 (Tsukada et al. 2006; Whetstine et al. 2006). Similarly, murine JMJD2 proteins antagonize H3K9me3 at pericentromeric heterochromatin (Fodor et al. 2006). Thus, JMJD2b could be an important factor that regulates the morphology and composition of nucleoli, which are surrounded by H3K9me3-dense chromocenters.
Histone modifications could also significantly influence higher-order chromatin structure. The histone signature likely affects interactions between histones and DNA, and different modifications can act simultaneously. For example, ubiquitination of H2B is required for H3K4me3 (Kouzarides 2007). LSD1, which is normally responsible for H3K4 demethylation, forms a complex with androgen receptor and causes H3K9 demethylation, thus inducing transcriptional activation (Metzger et al. 2005). These data show that histone epigenetic profiles help to establish euchromatin and heterochromatin domains and thus nuclear architecture. For instance, in early G1 phase, histone-lysine–methylated sites were scattered throughout the nucleus, whereas during S phase, methylated lysines were located mostly at the nuclear periphery and around nucleoli (Cremer et al. 2004). Similarly, Zinner et al. (2006) and Skalníková et al. (2007) revealed that methylation patterns in interphase nuclei show distinct nuclear layers with a certain degree of overlap, which depends on the type of epigenetic modification. Testing the association of the heterochromatin markers H3K9me3 with H4K27me3 and modifications characterized for active chromatin, including H4K20me1 and H3K4me3, Zinner et al. (2006) showed that specific histone modifications appear in distinct nuclear layers. This observation supports the theory of compartmentalization of interphase chromatin (Cremer and Cremer 2001; Bártová and Kozubek 2006; Lanctôt et al. 2007; Bártová et al. 2008b).
Epigenetics and Histone Modification in Nucleoli
Similar to the observtions in non-nucleolar compartments, transcription of rDNA within the nucleolus (Figure 2A) is regulated by distinct epigenetic marks responsible for both transcriptional silencing and activity (Figures 2B and 2C). The histone signature of acetylation, methylation, and ubiquitination determines the accessibility of chromatin structures for rDNA gene transcription (McStay and Grummt 2008; McKeown and Shaw 2009; Salminen and Kaarniranta 2009). The equilibrium between the epigenetic state and the activity of chromatin remodeling factors is responsible for the physiological processes that occur within nucleoli. Conversely, non-physiological changes in the nucleolar histone signature can lead to pathological changes with diagnostic significance, especially in oncology (Maggi and Weber 2005). The integrity of the nucleolus is likely maintained by the epigenetic state of rRNA genes, particularly by DNA methyltransferases and histone-modifying enzymes, which act simultaneously with chromatin remodeling complexes involved in rRNA synthesis. These orchestrated processes are responsible for optimal biosynthesis of ribosomes and subsequently for translation. Inhibition of rRNA synthesis leads to rearrangement of the nucleolus, which is accompanied by increased levels of p53 protein, which acts in the nucleolus as a critical stress sensor (Parlato et al. 2008). Moreover, the RNA polymerase inhibitor actinomycin D causes disassembly of nucleoli and loss of mRNA-FISH signals associated with the nucleolar periphery (Royo et al. 2009). Reorganization of nucleoli was also observed after abrogation of DNA methyltransferase 1 (Dnmt1) (Espada et al. 2007) and loss of SUV39h in cells (Figures 2D and 2E). In addition, the functional assembly of the nucleolus is thought to be self-organizing and dynamic (Misteli 2001). The morphology of the nucleolus is directly linked to its functional status and can react to external stimuli (Raška et al. 2004; Louvet et al. 2005; Shav-Tal et al. 2005).
Specific nucleolar functions are also ascribed to the c-Myc protein that accumulates within nucleoli. The c-Myc and Max proteins interact in nucleoli and associate with ribosomal DNA. In addition, c-Myc can activate Pol I transcription, suggesting that c-Myc could play a key role in regulating ribosome biogenesis and subsequently cell growth (Arabi et al. 2005). c-Myc also associates with nucleolar proteins such as Pol I and pontin, a putative mammalian DNA-helicase involved in the c-Myc–dependent regulation of rRNA synthesis (Cvačková et al. 2008). In addition, c-myc transcripts occupy nucleoli in various cell types (Bond and Wold 1993; Shimizu et al. 2007; Bártová et al. 2008a). In all cases tested, c-myc transcripts were often positioned at the periphery of and/or inside nucleoli, which correlates with the observation that transcription and probably splicing of mRNA genes proceeds in the central part of interphase nuclei (Zink et al. 2004; Harničarová et al. 2006).
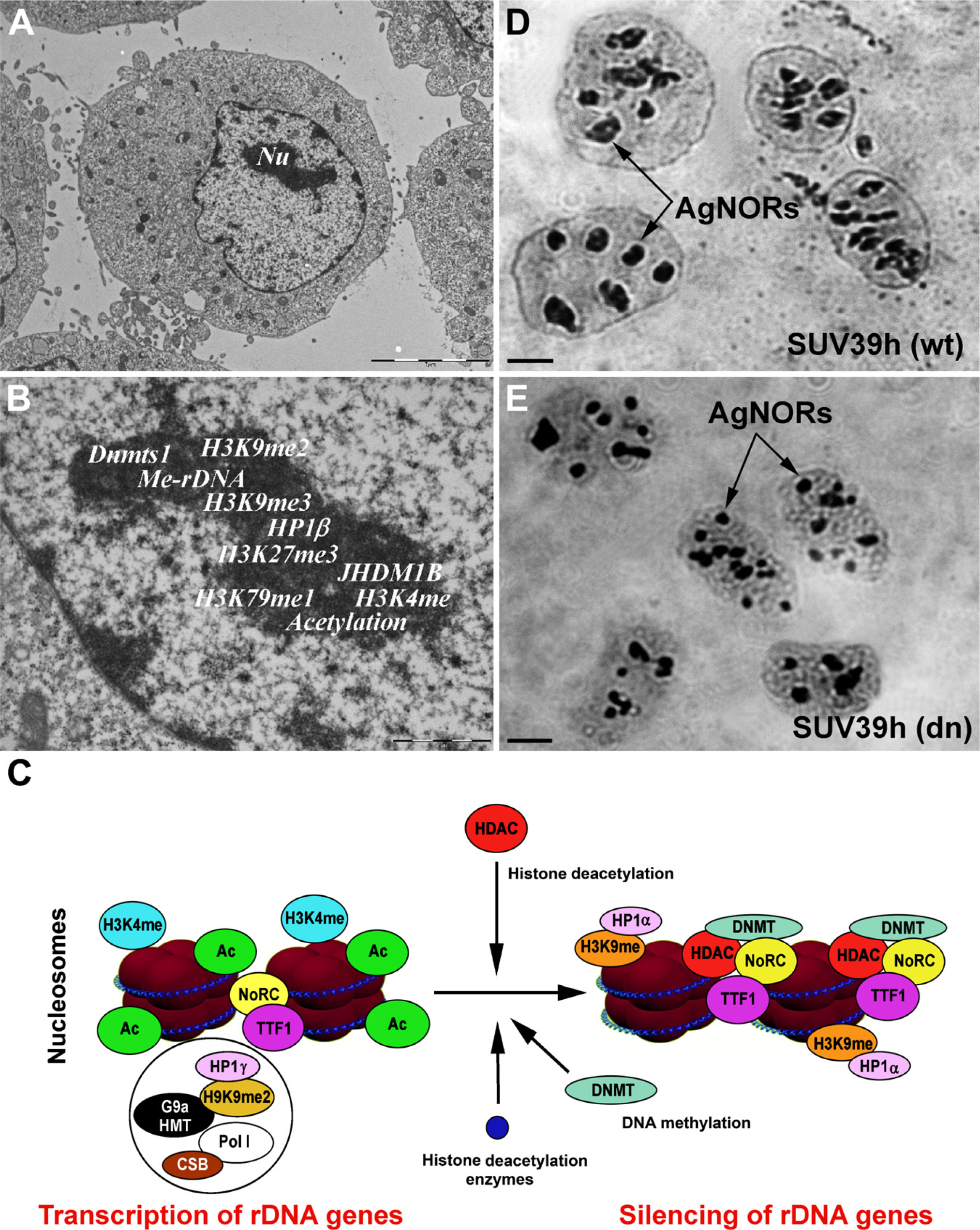
Illustration of epigenetic modifications at rDNA.
The nucleolus (Figure 2A) is characterized by specific epigenetic patterns (Figure 2B). Human cells lacking Dnmt1, but not Dnmt3b, show a loss in DNA methylation and an increase in the acetylation level of H4K16 in rRNA genes (Espada et al. 2007). Moreover, the recruitment of SirT1, a NAD+-dependent histone deacetylase with a preference for H4K16, to rRNA genes is abrogated in Dnmt1-deficient cells. These events are accompanied by structural disorganization of nucleolus and dispersal of nucleolar proteins, such as fibrillarin (Figure 1B) and pKi-67 [an inter-phase pattern was published by Harničarová et al. (2006); see also Krejčí et al. (2008)], within the nucleus of the cells carrying mutations. Thus, epigenetic events and factors such as Dnmt1 are likely responsible for the maintenance of stable nucleolar structure and function.
Recent work has also demonstrated that the histone demethylase JHDM1B is located in the nucleolus, which suggests a possible functional role of this enzyme in this compartment. JHDM1B appears to induce H3K4 demethylation and cause transcriptional repression of rDNA genes. However, it is not clear how JHDM1B is targeted to rDNA or how it relates to silencing by the nucleolar remodeling complex (NoRC), which is responsible for nucleosome sliding in an ATP- and H4 tail-dependent manner (Strohner et al. 2001; Frescas et al. 2007; McStay and Grummt 2008). The NoRC is composed of two subunits, the ATPase SNF2h and the 205-kDa TTF-I-interacting protein 5 protein (TIP5) (McStay and Grummt 2008). This complex can interact with Dnmt1, Dnmt3, and the Sin3 correpressor complex containing histone deacetylases HDAC1 and HDAC2 but not SIRT1 (Santoro et al. 2002; Zhou et al. 2002; McStay and Gruμmt 2008). This complex is subsequently responsible for acetylation and methylation of H3K9, H4K20, and H3K27. This epigenetic mark triggers heterochromatinization, which stabilizes the structure of nucleoli (Salminen and Kaarniranta 2009). Thus, NoRC is an important epigenetic repressor that silences rDNA repeats via DNA methylation and histone deacetylation (Figure 2C). Moreover, overexpression of TIP5, which is required for localization of NoRC to nucleoli in human and mouse cells, results in rDNA silencing (Santoro et al. 2002). Interestingly, TIP5-mediated transcriptional repression of rDNA genes is lost after treatment of cells by Dnmt and HDAC inhibitors, such as 5-azacytidine and trichostatin A (TSA). This suggests that NoRC may repress rDNA transcription in parallel with inducing DNA methylation and histone deacetylation. DNA methylation induced by the NoRC complex is important for recruitment of UBF to the promoter of rDNA genes and inhibits the activation of Pol I and thus the transcription of rDNA genes.
The balance between activity and silencing of rDNA genes is likely regulated not only by TIP5 and TTF-1, but also by Cockayne syndrome B protein (CSB), which is also involved in transcription of ribosomal genes. CSB, together with SNF-like ATPase and G9a HMT, are responsible for H3K9 dimethylation, which plays an important role in rDNA gene activation (Yuan et al. 2007) (Figure 2C). Thus, H3K9 methylation, which binds heterochromatin protein 1γ (HP1γ), is a surprisingly important epigenetic signature for transcriptionally active rDNA repeats (Yuan et al. 2007) (Figure 2C). Simultaneous activation and silencing of a given gene is not observed only in rDNA genes; monoallelic expression occurs with several mRNA species, including the c-myc oncogene and the CCND1 gene (Levsky et al. 2002). The transcriptionally active allele is positioned more interiorly in the nucleus than the non-active allele (Harničarová et al. 2006; Takizawa et al. 2008). This suggests the simultaneous existence of distinct epigenetic profiles in active and silent genomic regions.
An example of selected histone modifications and how HDAC and Dnmts inhibitors change their nucleolar compartment localization.
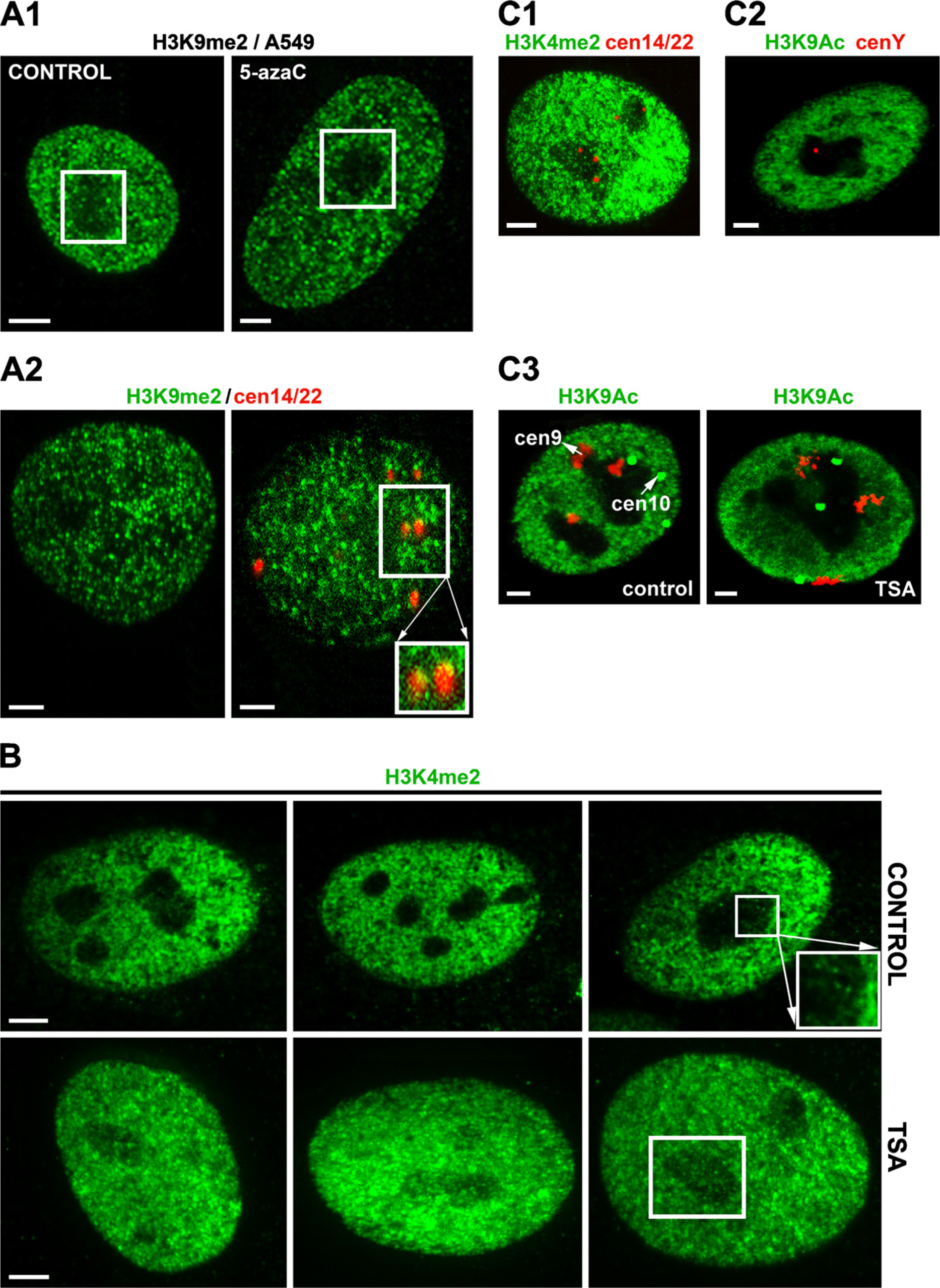
Different epigenetic patterns were also observed after the cell treatment by inhibitors of epigenetic processes such as 5-azacytidine and TSA. We found decreased levels of H3K9me2 in nucleoli after 5-azacytidine treatment (Figure 3A1) and increased levels of H3K4me2 in nucleoli after TSA cell stimulation (Figure 3B). The centromeres of acrocentric chromosomes are known to border the nucleoli. We found dense H3K9me2 at the centromeres of two human acrocentric chromosomes, 14 and 22 (Figure 3A2), while H3K4me2 and H3K9 acetylation was reduced in these centromeric regions (Figure 3C). Similarly, H3K9 acetylation was absent from the centromeres of human submetacentric chromosomes 9 and 10, which frequently occupied the border of nucleoli (Figure 3C3). Surprisingly, even centromere of the human Y chromosome was associated with the nucleolus (Figure 3C2). Thus, close relationships exist between chromatin structure, histone modifications, and arrangement of nucleoli, suggesting that the nucleolus contains many epigenetic factors that are responsible for both rDNA gene silencing and transcriptional activity.
Heterochromatin carries epigenetic marks, such as H3K9 methylation, HP1 protein subtypes (HP1α, HP1β, HP1γ), and rDNA methylation that can be also observed within nucleoli (Pikaard 1999; Kovařík et al. 2000, McStay and Grummt 2008). On the other hand, rDNA genes are also characterized by epigenetic marks specific to the more “open” chromatin configuration of transcriptionally active loci. For instance, histone acetylation has been observed in active rDNA genes (Santoro et al. 2002). However, when NoRC-mediated H4 deacetylation occurs, H3K9 dimethylation and de novo DNA methylation is increased. This leads to modifications of heterochromatin at the rRNA gene promoter. TSA treatment, inducing histone hyperacetylation, does not influence NoRC-mediated H3K9 dimethylation (Santoro et al. 2002), which is in agreement with previous findings that histone acetylation and methylation act independently (Maison et al. 2002; Yan et al. 2003). Moreover, the association of NoRC with chromatin requires H4K16 acetylation, which is required for subsequent deacetylation of H4K5, H4K8, and H4K12, indicating that H4K16 acetylation plays a dominant role in NoRC-mediated heterochromatinization (Zhou and Grummt 2005).
H3K9 methylation is known to be a mark of heterochromatin and transcriptionally silent loci. Interestingly, inhibition of H3K9 methylation by mutation of the Su(var)3–9 gene destabilizes rDNA, leading to the formation of extrachromosomal rDNA circles and nucleolar rearrangement (Peng and Karpen 2007). This suggests that disrupting the molecular mechanisms that establish the epigenetic state of rRNA genes destabilizes the interactions between DNA methyltransferases, histone-modifying enzymes, and chromatin remodeling complexes that regulate transcription of rDNA genes. Moreover, methylation at rDNA promoters is a dynamic and reversible process (Schmitz et al. 2009), as is the arrangement of nucleoli under external stimuli.
Conclusions
The nucleolus is the largest transcription factory and is a key nuclear compartment for biosynthesis of ribosomal subunits. The dynamic nature of nucleoli has been recently demonstrated, with nucleolar composition changing in response to external stimuli. It is highly probable that the stability of nucleoli is guaranteed by the stability of nucleolar proteins, such as modified histones. The histone signature, together with chromatin remodeling factors, plays an important role in the regulation of rRNA gene transcription, thus influencing the stability and function of nucleoli. Disorders in this process lead to pathophysiological states, including cancer.
Footnotes
Acknowledgements
This work was supported by the research projects LC535, LC06027, ME919 (Ministry of Education, Youth and Sports of the Czech Republic), AVOZ50040702, 1QS500040508, and AVOZ50040507 (Academy of Sciences of the Czech Republic, v.v.i).