
Abstract
Psoriasis is the most common autoimmune disease in man and is characterized by focal to coalescing raised cutaneous plaques with consistent scaling and variable erythema. The specific pathogenesis of psoriasis is not completely understood, but the underlying mechanisms involve a complex interplay between epidermal keratinocytes, T lymphocytes as well as other leukocytes (including dendritic cells and other antigen presenting cells [APCs]), and vascular endothelium. Mirroring the complexity of mechanisms that underlie psoriasis, there are a relatively large number of models of psoriasis. Each model is based on a slightly different pathogenic mechanism, and each has its similarities to psoriasis as well as its limitations. In general, psoriasis models can be very broadly divided on the basis of the pathogenic mechanisms that interplay to cause psoriasis, with the addition of several relatively poorly defined spontaneous murine mutant models. Other than the spontaneous mutant models, murine models of psoriasis can be divided into those that are genetically engineered (transgenic and knockout—with manipulation of either the epidermis, leukocytes, or the endothelium), and those that are induced (either by immune transfer or by xenotransplantation of skin from psoriatic patients). In addition to the murine models, in vitro human epidermal models have recently become more widely utilized. While no one single model of psoriasis is ideal, many have proven to be extremely valuable in investigating and better understanding the molecular mechanisms that underlie the complex interplay between epidermal keratinocytes, the innate and adaptive immune system, and the vascular endothelium in psoriasis.
Introduction
Psoriasis is a common inflammatory condition of human skin characterized by focal to coalescing raised cutaneous plaques with consistent scaling and variable erythema. 26 Typical histologic features of psoriasis include epidermal hyperplasia (acanthosis) with elongated rete ridges, a less discrete epidermal granular layer (hypogranulosis), parakeratosis, and leukocytic infiltration of the dermis and epidermis. 26, 29, 34 The cellular composition of the psoriatic leukocytic infiltrate is variable but very consistently contains both CD4+ and CD8+ T lymphocytes, with CD4+ T lymphocytes predominating in the dermis, and CD8+ T lymphocytes, particularly those expressing the αEβ7 integrin, preferentially infiltrating the epidermis. 26, 38 Psoriasis is now recognized as the most common autoimmune disease in man, with a prevalence of 2–4% worldwide. 26, 29 While the specific etiology of psoriasis is unknown, a genetic basis has been suspected for some time, and a number of different psoriasis susceptibility gene clusters, designated PSORS1, PSORS2 . . . PSORS6, have been identified, underscoring the heterogeneous nature of psoriasis. 26, 34 Similarly, the specific pathogenesis of psoriasis is still not fully understood, but there has been much recent progress in understanding many of its complex underlying mechanisms, which involve an interplay between epidermal keratinocytes, leukocytes (including dendritic cells and other antigen presenting cells [APCs]), and vascular endothelium (Fig. 1 illustrates some of the key features underlying the pathogenesis of psoriasis). 26, 29 Originally, psoriasis was considered to be primarily a disorder of disregulated epidermal proliferation and differentiation, and indeed, agents such as retinoids and vitamin D analogs that treat epidermal differentiation defects have shown some efficacy in the treatment of psoriasis. 2, 24, 32 With the discovery that immunosuppressive agents such as cyclosporin A and corticosteroids are often effective in treating psoriasis, the pendulum has swung toward psoriasis being considered primarily a disorder of the immune system, with T cells and specifically CD4+ T cells felt to play a central role. 35, 51 Initially, psoriasis was believed to primarily be a Th1 T helper cell–mediated process, driven by IFNγ and related cytokines. 26, 29 More recently it has been shown that IL-23, a cytokine involved in the development of the newly defined Th17 T helper cell subset, 36 plays a major role in psoriasis. 5, 22, 27, 33 Even more recent data suggest that psoriasis is caused by an interaction between epidermal keratinocytes and the immune system, 32, 39, 41 and that one possible candidate linking the immune system and epidermal keratinocytes is IL-22, a T-cell-derived cytokine that is produced by Th17 polarized T cells that are stimulated by IL-23, 56 but that acts on epidermal keratinocytes to induce acanthosis and differentiation toward a psoriatic phenotype. 7, 33, 40 Regardless of the specific underlying pathogenesis, psoriasis is characterized by disregulated epidermal acanthosis, dermal and epidermal leukocytic infiltration, and dilation of dermal blood vessels—lesions that are maintained by the complex interplay between T cells and their cytokines, other leukocytes, vascular endothelium, and epidermal keratinocytes. 26, 29 As noted above, epidermal keratinocytes as well as vascular endothelial cells are active participants in the psoriatic inflammatory process via secretion of cytokines and growth factors, and the upregulation of signaling and adhesion molecules on their surfaces. 26, 29
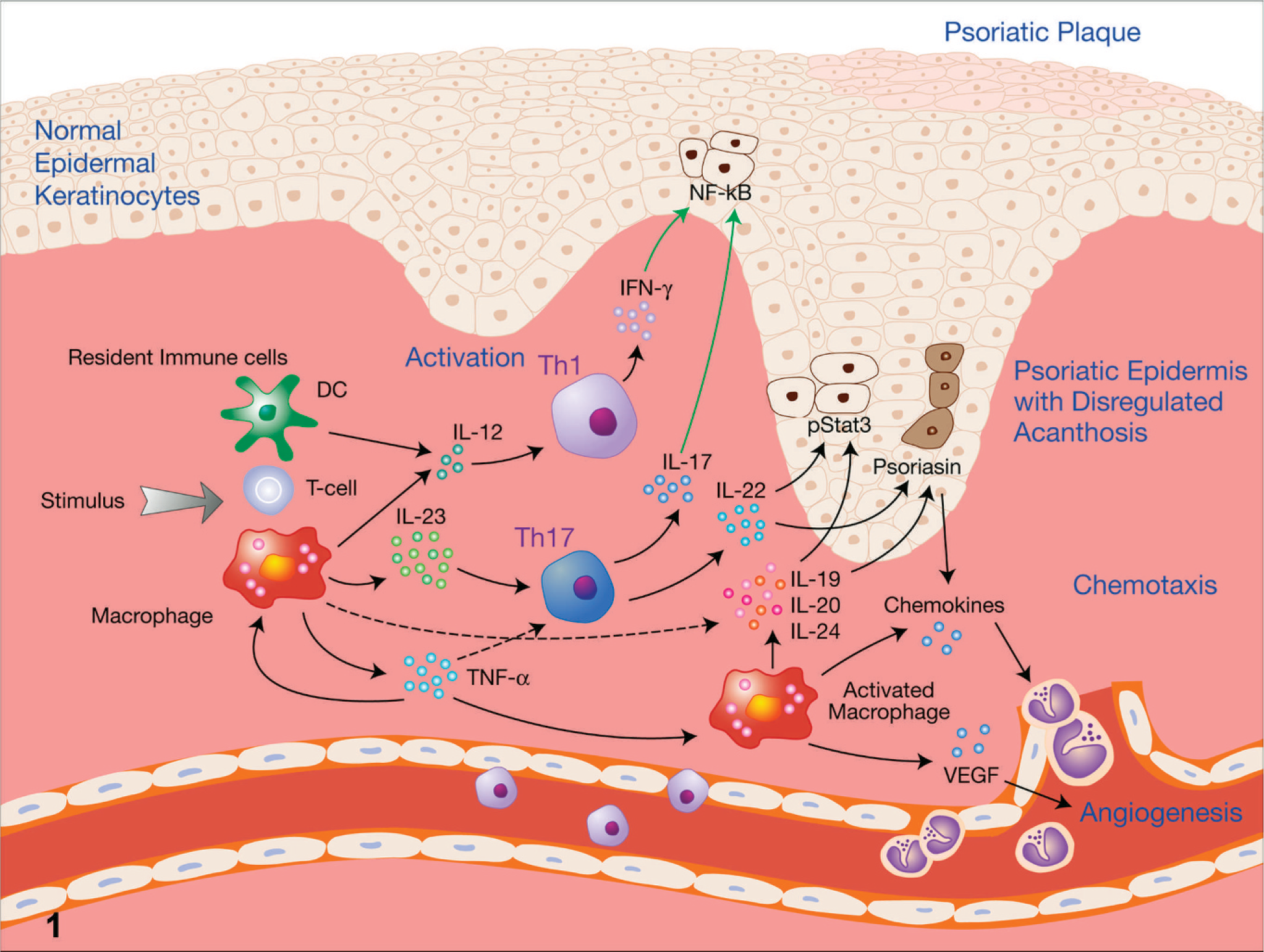
Schematic diagram illustrating some of the key features that make up our current understanding of the pathogenesis of psoriasis. Psoriasis in genetically susceptible individuals is believed to be triggered by specific stimuli, such as trauma or bacterial infections, which in turn induce resident dendritic cells (DC), macrophages, and T cells to produce cytokines that initiate a cascade of events leading to the hallmarks of psoriasis: immune cell activation, disregulated epidermal acanthosis, and angiogenesis. IL-12 produced by DC and macrophages induces a Th1 immune response, characterized by production of IFNγ, while IL-23 from DC and macrophages induces a Th17 immune response, characterized by production of IL-17 and IL-22. These cytokines, in turn, induce the changes characteristic of psoriatic epidermal keratinocytes, including upregulation of NF-κB, induction of nuclear pStat3, and induction of psoriasin. Macrophages activated by TNF-α produce additional cytokines that induce psoriatic epidermal changes, as well as VEGF, which stimulates angiogenesis. Activated macrophages and psoriatic keratinocytes also produce chemokines that recruit leukocytes from the vasculature to infiltrate the developing psoriatic plaque. DC = dendritic cell; IFNγ = interferon gamma; IL = interleukin; NF-κB = nuclear factor-kappa B; pStat3 = phospho-signal transducer and activator of transcription 3; Th = helper T cell; TNF-α = tumor necrosis factor-α; VEGF = vascular endothelial growth factor..
Mirroring the complexity of mechanisms that underlie psoriasis, there are a relatively large number of models of psoriasis. While most of these are murine models, there are also several non-murine and a few in vitro human epidermal models (Table 1). Each model is based on a slightly different pathogenic mechanism, and each has its strong points/similarities to psoriasis, as well as its limitations, not the least of which are the fundamental morphologic differences between human psoriatic skin and murine psoriasis models (excepting xenotransplantation models). 29, 34, 42 Morphologic differences in murine models versus psoriatic skin include lack of parakeratotic scaling and epidermal hypogranulosis, and marked follicular hyperplasia rather than true exaggeration of epidermal rete ridges (Figs. 2, 3). In general, murine models of psoriasis can be broadly divided on the basis of the pathogenic mechanisms that interplay to cause psoriasis, with the addition of a few relatively poorly defined spontaneous mutant models. Aside from these spontaneous models, murine models of psoriasis can be divided into those that are genetically engineered (transgenic and knockout—with manipulation of the epidermis, leukocytes, or the endothelium), and those that are induced (either by immune cell transfer or by xenotransplantation). In general, to be considered as a useful model of psoriasis, the model has to either share some histopathologic features with psoriasis, have a pathogenesis and/or disease mechanism that is similar to psoriasis, and/or respond similarly to therapeutic agents that psoriasis has been demonstrated to respond to. The ideal psoriasis model would have all 3 features. However, as mentioned earlier, psoriasis is a heterogeneous disease with a complex pathogenesis, Therefore, identifying a single model of psoriasis that completely mirrors all aspects of psoriasis is very likely not possible. A discussion of the most widely accepted models of psoriasis follows in the subsequent sections.
Animal and in vitro models of psoriasis.
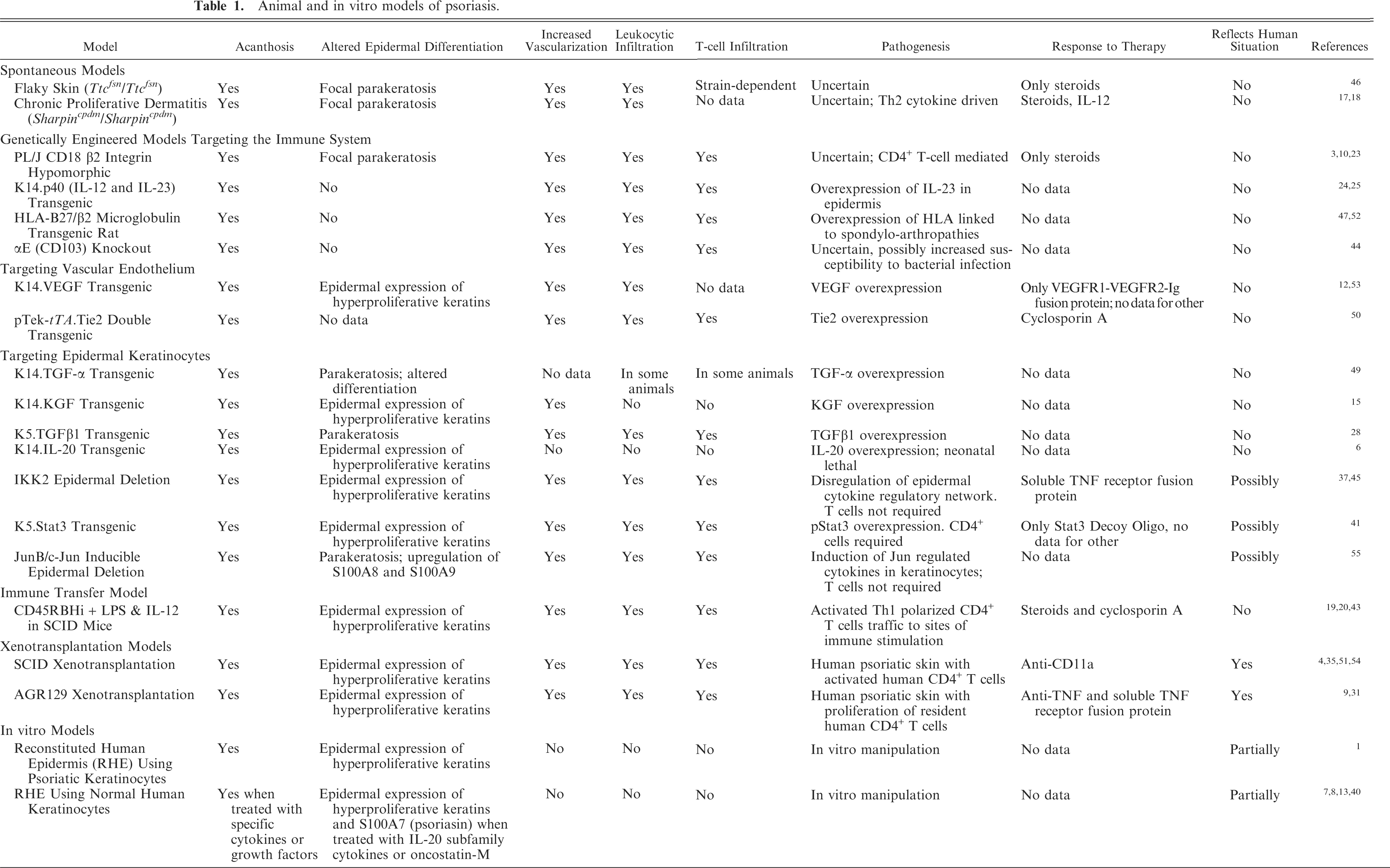
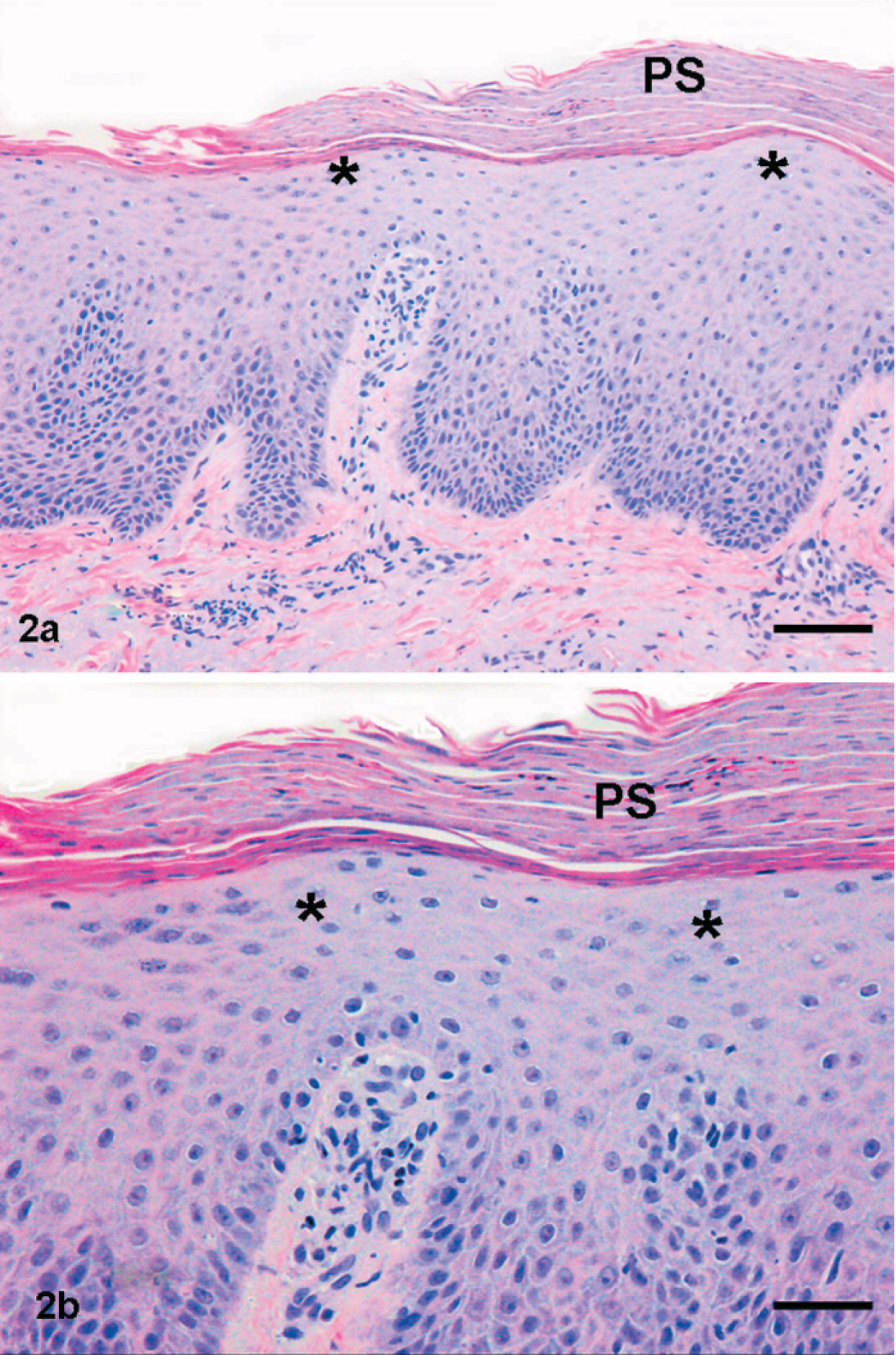
Skin; human psoriatic skin illustrating exaggerated epidermal rete ridges exhibiting epidermal hypogranulosis (asterisks) with extensive parakeratotic scaling (PS). HE. Bar = 100 µm in a and 50 µm in b.
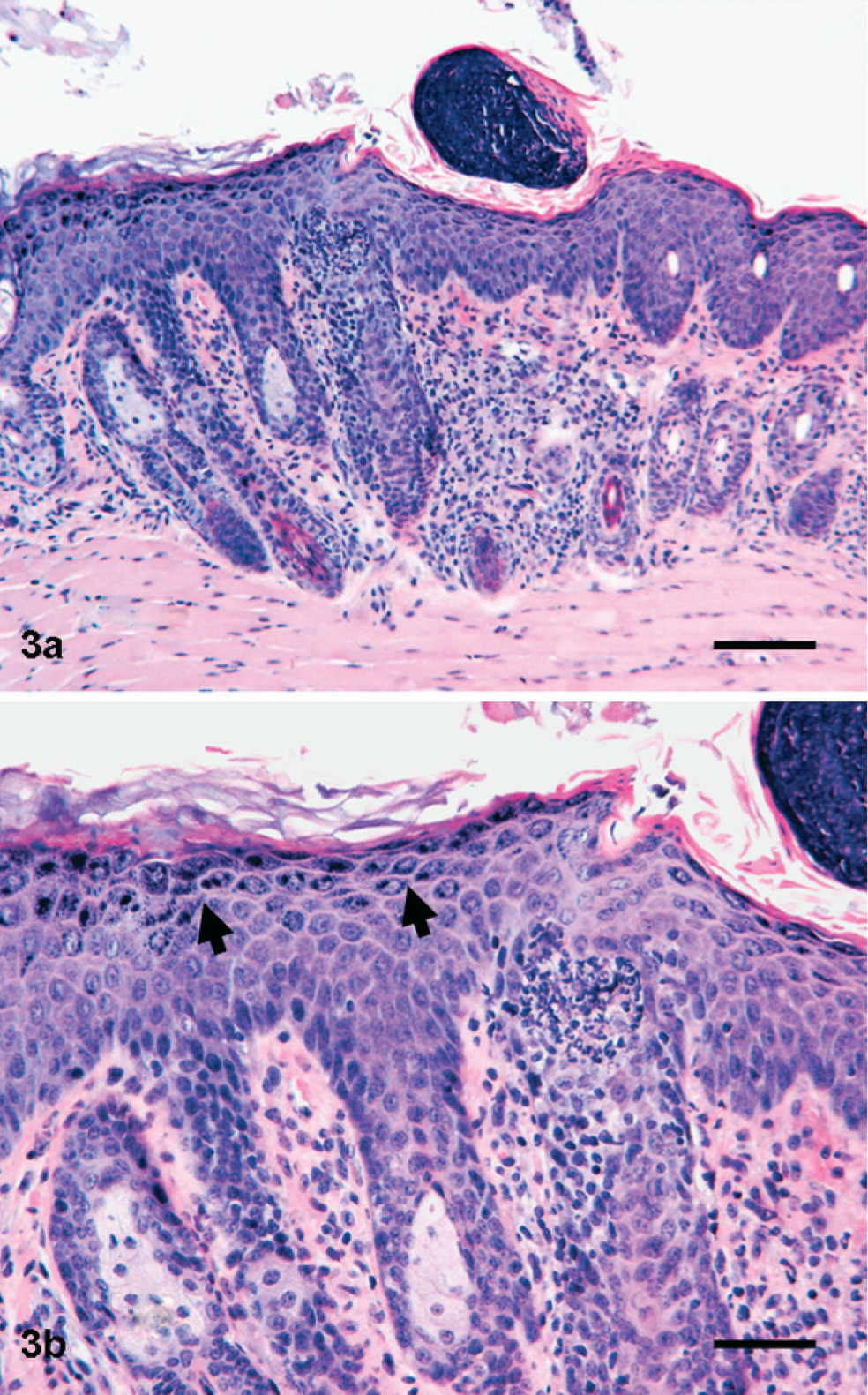
Skin; CD45RBHi CD25− T-cell immune transfer mouse model. In contrast to the human psoriatic skin illustrated in Fig. 1, the epidermis in this mouse model has follicular hyperplasia rather than exaggerated epidermal rete ridges, exhibits epidermal hypergranulosis (arrows) rather than hypogranulosis, and lacks the parakeratotic scaling that is one of the hallmarks of a psoriatic plaque. HE. Bar = 100 µm in a and 50 µm in b.
Spontaneous Models
There are a number of different spontaneously occurring mouse models of psoriasis, none of which are very well understood mechanistically. 42 Flaky skin mice (Ttcfsn/Ttcfsn ) are probably the best described, and have a spontaneous mutation that induces proliferation and hyperkeratosis of stratified squamous epithelia, including the nonglandular forestomach. 46 While these mice have interesting squamous proliferative and inflammatory lesions that do respond to corticosteroids, the pathogenesis of these lesions is unknown, and they do not express all of the features of psoriasis, such as lack of an epidermal T-cell infiltrate and the lack of hyperproliferative keratin expression. 47 In addition, these mice have a limited useful lifespan because of nonregenerative anemia and the massive papillomatous hyperplasia of their forestomachs. 47 Another spontaneous mouse model that has been relatively well described is the chronic proliferative dermatitis mouse (Sharpincpdm/Sharpincpdm ), a mouse that develops marked eosinophilic inflammation in a number of tissues, including the skin, which leads to marked acanthosis. 17, 18, 45 Unlike psoriasis, however, the inflammation in this model is driven by Th2 cytokines such as IL-4, IL-5, and IL-13, and lesions respond to IL-12 treatments. 18 For these reasons, this model is not felt to be very representative of psoriasis, which is predominantly a Th1- and Th17-driven disease, and which has been shown to respond to an anti-IL-12 (and anti-IL-23) p40 MAb. 22, 49
Genetically Engineered Models
Genetically engineered mice and rats are the largest category of psoriasis models and include both transgenics and knockouts. In this review, I have divided these into 3 broad categories: those that target epidermal keratinocytes, those that target leukocytes, and those that target vascular endothelium. The CD18 hypomorphic mouse is a genetically engineered model that targets leukocytes. This mouse does not completely lack the CD18 β2 integrin, but rather has decreased expression of this common β2 chain of the leukointegrin adhesion molecule complex. When on a PL/J strain background, these CD18 hypomorphic mice, PL.129S7-Itgb2tm1Bay , develop a psoriasiform inflammatory skin condition with a predominantly lymphocytic infiltration. 3, 10, 23 While lesions in these mice are responsive to dexamethasone, 10 their pathogenesis has not been well characterized. In addition, unlike psoriasis, these mice exhibit nonpsoriasiform epidermal hyperplasia with lack of hyperproliferative keratin expression. Therefore, this model has not been widely used for efficacy testing of potential therapeutic agents.
Another genetically engineered mouse model that develops cutaneous inflammation with some features similar to psoriasis is the p40 keratin 14 (K14) transgenic mouse, Tg(KRT14-Il12b)1Tsk, in which the p40 subunit of both IL-12 and IL-23 is overexpressed via the K14 promoter in basal epidermal keratinocytes. 24, 25 These mice develop cutaneous inflammation that is felt to be more similar to eczema and atopic dermatitis, lacking a significant cutaneous CD8+ T-cell infiltration, and so are not considered to be a particularly useful model for psoriasis. Similarly, the αE integrin (CD103) knockout mouse, Itgaetm1Cmp , also develops a cutaneous inflammatory disease. The αE integrin complexes with the β7 chain and is thought to play a role in cutaneous and mucosal immunity through interaction with its counter receptor, E cadherin. 44 In addition, αEβ7 is preferentially expressed by the CD8+ T cells that infiltrate the psoriatic epidermis. 38 However, the cutaneous inflammatory condition that αE knockout mice develop is not felt to mimic psoriasis closely enough, having significant ulceration and very few cutaneous CD8+ T cells, for these mice to be a useful psoriasis model. Still, these mice have a phenotype that does shed some light on the potential functions of the αEβ7 integrin complex in cutaneous inflammatory disease. 44
Another genetically engineered animal model that targets the immune system more broadly is the HLA-B27/human β2 microglobulin transgenic rat, Tg(HLA-B∗2705, B2M)33-3Trg. 48, 53 This rat overexpresses a form of human leukocyte antigen (HLA)-B27 that has been linked to spondyloarthropathies. 21 HLA-B27 transgenic rats develop epidermal acanthosis with epidermal infiltration of both CD4+ and CD8+ T cells, as well as immune-mediated arthritis and inflammatory bowel disease. 48, 53 The occurrence of psoriatic skin lesions is less consistent than is the occurrence of the other immune-mediated disorders, 48, 53 and there have been no published reports of therapeutic efficacy testing for the psoriatic lesions. Therefore, while this model has attained some utility as a model of HLA-B27 associated autoimmune disease, it has not gained widespread use as a model of psoriasis.
Genetically engineered mouse models of psoriasis that target vascular endothelium include VEGF K14 transgenic mice and Tie2 transgenic mice. Mice overexpressing VEGF epidermally via a K14 promoter, Tg(KRT14-Vegfa)1Gdy, develop a very vasculocentric cutaneous inflammatory disease with hyperplastic and inflamed dermal vasculature, and psoriasiform epidermal acanthosis. 12, 54 Although these mice have many vascular, epidermal and inflammatory features that resemble psoriasis, the lesions appear to be largely vascular-based, and there is marked dermal infiltration by mast cells. In addition, other than responding to treatment with a soluble VEGFR1-VEGFR2-Ig fusion protein, data demonstrating response to therapeutic candidates in this model are lacking. 12, 54 Therefore, there are a number of caveats that limit the usefulness of the VEGF K14 transgenic mouse model. Tie2 transgenic mice, Tg(Tek-tTA)1Dmt × Tg(TetOS-Tek)1Dmt, were constructed using a driver transgene, pTek-tTA, which localizes Tie2, the receptor for angiopoietin-1 and angiopoietin-2, to vascular endothelium as well as to keratinocytes in the epidermis and hair follicles. 51 Tie2 transgenic mice also develop a very vasculocentric cutaneous inflammatory disease with vascular hyperplasia and epidermal acanthosis with inflammation. While many of the same caveats that hold for the VEGF K14 transgenic mice also pertain for the Tie2 transgenic mice (vasculocentric inflammation with many mast cells and incomplete characterization of epidermal changes), cutaneous lesions in the Tie2 transgenic mice have at least been shown to be responsive to a therapeutic that has shown efficacy in psoriasis, namely cyclosporin A. 50
There have been several genetically engineered mice, all transgenics, that have targeted epithelial growth factors such as TGF-α, KGF, and TGF-β via K14 or K5 promoters (Tg(TGFa)1Efu, Tg(FGF7)2Efu, and Tg(KRT5-TGF-B1)F2020Xjw, respectively) to the basal epidermis, with resulting phenotypes that somewhat mimic the epidermal acanthosis of psoriasis because of the disruption of normal epidermal growth and differentiation, while generally lacking the inflammatory component of psoriasis. 15, 28, 50 While these growth factor transgenics are not good models of psoriasis, they have proven to be useful in the study of epidermal growth and differentiation. 15, 28, 50 A relatively recently discovered cytokine in the IL-10 family, IL-20, also induces a similar psoriasiform phenotype in K14 promoter epidermal targeted transgenic mice, Tg(KRT14-Il20)1Yac, but again these mice lack cutaneous inflammation, and in addition exhibit neonatal lethality, essentially rendering them useless as a model of psoriasis. 6 Recently however, several genetically engineered mouse models that target epidermal keratinocytes have put the epidermal keratinocyte back into a central role in the pathogenesis of psoriasis. These models appear to fulfill most of the criteria necessary for a model of psoriasis to be representative and useful. The first of these models targeting epidermal keratinocytes is the epidermal-specific IKK2 knockout mouse, Tg(KRT14-cre)1Cgn × Ikbkbtm1Mpa , in which deletion of the IKK2 catalytic subunit of the IκB kinase complex (necessary for NF-κB activation by inflammatory stimuli) caused mice to develop psoriasiform cutaneous inflammation that shared many features of psoriasis, including dependence on intact TNF signaling, but which was T-cell independent. 37, 46 A second model targeting epidermal keratinocytes is the JunB/c-Jun epidermal inducible double knockout mouse, Tg(Krt1-5cre/ERT)1lpc × Junbtm3Wag × Juntm4Wag . 56 JunB is a component of the AP-1 transcription factor and has been localized to the PSORS6 psoriasis susceptibility locus, while c-Jun is felt to be an antagonist to JunB. Inducible deletion of both JunB and c-Jun leads to psoriasiform cutaneous inflammation, as well as to arthritis in some mice. The cutaneous inflammation is not dependent on T cells, nor is it dependent on TNF signaling, as both Rag2 knockout mice and TNFR1 knockout mice still developed cutaneous lesions. 56 Interestingly, however, the TNFR1 knockout mice did not develop arthritis. 55 A final recent transgenic model targeting epidermal keratinocytes is the K5-Stat3 transgenic mouse, Tg(KRT5-stat3∗A661C∗N663C)1Jdg, in which Stat3, a transcription factor implicated as playing a major role in signal transduction in psoriatic keratinocytes, is overexpressed in epidermal keratinocytes. 41 In this transgenic mouse model, mice develop psoriasiform epidermal acanthosis that is most pronounced in areas of friction, such as the tail head, and that is accentuated by tape stripping and wounding, all features that are similar to psoriasis. 41 In addition, these mice have a cutaneous lymphocytic infiltrate that is predominantly CD4+ in the dermis, and CD8+ in the epidermis, another feature that is similar to psoriasis. 41 Finally, both Stat3 transgenic skin and the injection of activated lymphocytes, specifically CD4+ T cells, are necessary to generate a psoriatic phenotype in transplanted SCID mice, 41 a finding that firmly establishes a link between keratinocytes and CD4+ T lymphocytes in the pathogenesis of psoriasis. 39 Since phosphorylated Stat3 overexpression has also been detected in the nuclei of psoriatic keratinocytes, the link between the K5 Stat3 transgenic mouse and psoriasis is even further strengthened. 39, 41 Cutaneous lesions in these transgenic mice respond to Stat3 decoy oligonucleotides, suggesting that therapies aimed at this signaling pathway may be of potential clinical benefit. 41 A number of IL-10 family members, including the previously mentioned IL-20, as well as IL-6 family members, such as oncostatin M, induce Stat3 phosphorylation, and are being investigated as potential therapeutic targets by a number of investigators. 7, 8, 33, 40
Immune Transfer and Transplantation Models
This category of models includes the CD45RBHi CD25− T-cell immune transfer model and the human psoriatic skin xenotransplantation model, both using SCID mice as the transplant recipients. Both of these models have been relatively extensively validated in efficacy studies, and thus are frequently favored by investigators hoping to test a novel potential therapeutic entity. 43, 55
In the CD45RBHi T-cell immune transfer model, MHC minor mismatched CD4+ CD45RBHi CD25− naïve T cells (lacking in regulatory T cells) are injected into ICR-Prkdcscid/Prkdcscid SCID mice, and target areas of antigenic stimulation, such as the lower intestinal tract, skin, and lung. When these CD25− naïve T cells are stimulated with either LPS or IL-12 (SDI mouse, BioSeek, Burlingame, CA), the predominant disease is chronic persistent cutaneous inflammation that has many features resembling psoriasis 19, 20, 43 (Fig. 3). The underlying pathogenesis of the cutaneous inflammation is Th1-driven, similar to psoriasis (although psoriasis is now felt to have a major Th17-driven component), but unlike psoriasis, IFNγ does not appear to play a major role in the development of lesions in this model. 19, 20 In addition, since all of the transferred lymphocytes are CD4+, no CD8+ lymphocytes are present in the inflammatory infiltrate, which is unlike psoriasis. To address this limitation, we and other investigators have added back different lymphocyte subsets to the cells being immune transferred with relatively good results, such that CD8+ T cells can be found in the epidermal infiltrate with the maintenance of the severity of the cutaneous inflammation (Khattri and Danilenko, unpublished data). As mentioned above, cutaneous lesions in this model have been shown to respond to a number of therapeutic agents, including cyclosporin A, corticosteroids, and anti-IL-12. 19, 20, 43
The final set of animal models of psoriasis are probably the most faithful of all to the human condition, since these models use human psoriatic skin transplanted onto immunodeficient mice. However, these models are also the most difficult to utilize, as they rely on having a steady supply of human psoriatic skin available. Hence, only a few laboratories are able to run these models with any consistency. 4, 9, 31, 35, 52, 55 In the prototype xenotransplantation model, the epidermis and dermis from a patient with psoriasis is transplanted onto the flank of a CB.17-Prkdcscid/Prkdcscid SCID mouse. As was the case with the Stat3 transgenic mice, psoriatic lesions are only induced when activated T cells, and specifically CD4+ T cells but not CD8+ T cells, are injected into the transplanted SCID mice. 35, 52 The transplanted epidermis then undergoes psoriasiform acanthosis with the induction of proliferation markers such as keratin 16. In addition, transplanted skin becomes infiltrated by lymphocytes, with primarily CD4+ T cells in the dermis and primarily CD8+ T cells in the epidermis. 35, 52 Intraepidermal microabscesses (Munro's microabscesses) are also sometimes present. 52 An anti-CD11a MAb (efalizumab), which has demonstrated efficacy in human psoriasis, 14, 34 has also been shown to have efficacy in this mouse model. 54 A variant of this model using symptomless psoriatic skin transplanted onto AGR129 mice, 129-Ifnar1tm1Agt × Ifngr1tm1Agt × Rag2tm1Fwa, which lack both interferon type 1 and type 2 receptors as well as RAG2, has also recently been described. 9, 31 In this model, transplanted symptomless psoriatic skin spontaneously becomes psoriatic without the addition of activated T cells because of the proliferation of resident human T cells within the xenograft. 9, 31 An anti-TNF MAb (infliximab) and a soluble TNF receptor fusion protein (etanercept), both of which have been shown to have efficacy in human psoriasis, 11, 30, 34 also demonstrate efficacy in inhibiting the development of psoriatic lesions in this model. 9
In Vitro Models
As an alternative to the in vivo models, several laboratories, including our own, have begun to use reconstituted human epidermal culture model systems whereby epidermal keratinocytes are grown to the air-liquid interface and differentiate and stratify to mimic the morphology of normal stratified squamous epidermis. Epidermal keratinocytes in this model can be obtained from individuals with psoriasis 1 or from normal individuals and then treated with a variety of cytokines and/or growth factors (e.g., IL-20, IL-22, oncostatin-M) such that the resulting reconstituted human epidermal model develops phenotypic characteristics that can mimic psoriatic epidermis 7, 8, 13, 40 (Fig. 4). In both the psoriatic keratinocyte-derived model and the normal human keratinocyte-derived model treated with either IL-20, IL-22, or oncostatin-M, keratinocytes exhibit many of the same phenotypic features evident in psoriasis, such as upregulation of chemokines IL-8 and GRO-α, and induction of hyperproliferative keratin 16. 1, 7, 8, 13, 40 Many additional features in common with psoriatic epidermis are seen in the normal keratinocyte reconstituted epidermis model treated with cytokines such as IL-20, IL-22, or oncostatin-M, including upregulation of S100 family members such as S100A7/psoriasin, and activation of pStat3 7, 8, 40 (Figs. 3–5), one of the major signal transducers in psoriatic epidermis, as described previously. 41 While these in vitro model systems have obvious limitations in that they lack leukocytes and blood vessels, they have still proven useful in studying many aspects of the psoriatic epidermis, including keratinocyte differentiation and response to stimuli.
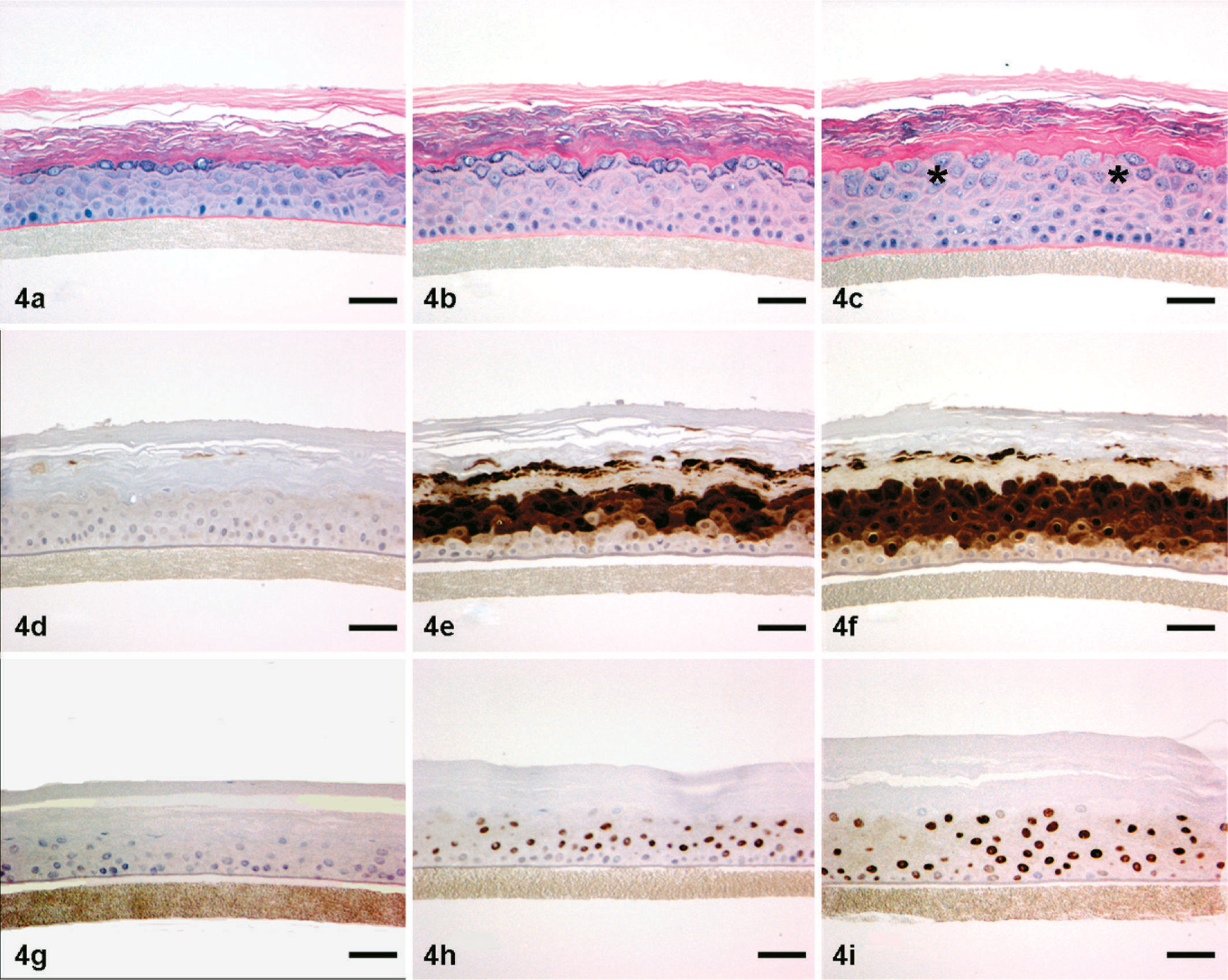
Skin; in vitro reconstituted human epidermis. Epidermal hyperplasia, induction of epidermal S100A7 (psoriasin) expression, and induction of pStat3 in epidermal keratinocyte nuclei following 4 days in vitro treatment with IL-20 (b,e,h) and IL-22 (c,f,i) compared with media control (a,d,g). IL-22-treated epidermis also exhibits hypogranulosis (asterisks in c), a feature frequently evident in psoriatic epidermis. HE (a,b,c), S100A7 (psoriasin) IHC (d,e,f), pStat3 IHC (g,h,i). Bars = 50 µm.
Recommendations and Future Directions
While there are a relatively large number of animal models purported to mimic psoriasis, there are actually only a small number that meet the criteria of resembling psoriasis clinically and pathologically, having a pathogenic mechanism that is known to play a significant role in psoriasis, and having been validated by showing a response to therapies that psoriasis is known to respond to. Only two sets of models currently meet these criteria: the ICR-Prkdcscid/Prkdcscid SCID mouse CD45RBHi CD25− T-cell immune transfer model, and the 2 human psoriatic skin xenotransplantation onto immunodeficient mouse models, CB.17-Prkdcscid/Prkdcscid and AGR129. Of these 2, the human psoriatic skin xenotransplantation models are the most faithful to human psoriasis, but the difficulties in obtaining human psoriatic skin for transplantation limits the widespread utility of these models. The CD45RBHi CD25− immune transfer model has gained relatively widespread use but has several limitations: As is the case for all mouse models, the morphology of lesions in this model does not completely mimic the epidermal changes in psoriasis; in addition, the leukocytic infiltrate lacks CD8+ T cells unless they are specifically added back in.
Despite these limitations, both models are relatively commonly used for evaluation of potential therapeutic agents, as human therapeutics with demonstrated efficacy in psoriasis have also shown efficacy in these models (both small molecules such as cyclosporin A and biologics such as anti-TNFs and anti-CD11a). 9, 43, 55 While both sets of models have been used for efficacy testing of potential therapeutics, the pros and cons of each somewhat balance each other out. The xenotransplantation models are more biologically faithful to psoriasis, but the difficulties in obtaining human psoriatic skin to transplant generally limits the number of animals that can be used in a study. In contrast, the number of SCID CD45RBHi CD25− T-cell immune transfer model mice that can be used in a study can be much greater (we generally try to use 10–12 mice per treatment group), thereby allowing for much more meaningful statistical analysis of differences in treatment effects. In the xenotransplantation models, only the transplanted skin can be evaluated for treatment effects, and generally only histologic evaluation is done (epidermal thickness is the primary measurement that has been used). 9, 55 In contrast, in the SCID CD45RBHi CD25− T-cell immune transfer model treatment effects are evaluated from a number of different sites (ears, nasal planum, paws) and evaluation of both gross appearance (degree of alopecia, scaling, hyperemia) as well as histologic appearance (epidermal thickness, degree of inflammatory infiltration and vascular hyperplasia) 9, 20, 43 can be evaluated (also Khattri and Danilenko, unpublished observations). Therefore, while the xenotransplantation models more faithfully mimic human psoriasis, our laboratory has used the SCID CD45RBHi CD25− T-cell immune transfer model more widely for initial screening, as greater numbers of mice can be used, and more treatment effect parameters can be evaluated. Once a potential therapeutic has shown evidence of efficacy in this model, we will then generally attempt to confirm this effect in one or both of the xenotransplantation models.
While only the 2 sets of models described above are widely used for efficacy testing of potential therapeutics, several recently described genetically engineered mouse models hold promise for studying many aspects of psoriasis pathogenesis and may also eventually turn out to be useful models for efficacy testing (this will only be determined after repeated evaluations). The K5-Stat3 transgenic mouse, Tg(KRT5-stat3∗A661C∗N663C)1Jdg, while currently not meeting the criterion of having been validated via efficacy testing with a therapeutic known to affect psoriasis, nonetheless shows much promise as a potentially useful model of psoriasis, particularly as it appears to link keratinocytes and infiltrating T-lymphocytes in the pathogenesis of psoriatic lesions. 39, 41 In addition, while having the limitations imposed by the differences inherent between mouse and human skin, the morphology of lesions in this model nonetheless appear to relatively faithfully mimic those evident in psoriatic skin. 41 While this model does hold promise pathologically and mechanistically, it still remains to be validated by testing whether its cutaneous lesions will respond to additional therapeutic agents, such as cyclosporin A, anti-LFA-1, anti-TNFs, and others. Another recently described mouse model, the epidermal JunB/c-Jun epidermal inducible double knockout mouse, Tg(Krt1-5cre/ERT)1lpc × Junbtm3Wag × Juntm4Wag , also develops psoriasiform cutaneous inflammation, as well as arthritis in some mice. 55 As is the case for the Stat3 knockout mice, this model has the same limitations imposed by differences between mouse and human skin, and also remains to be validated by testing whether its cutaneous lesions will respond to antipsoriatic therapeutic agents. Still, it is also a valuable model for studying the mechanisms common to the development of both epidermal and joint lesions in psoriasis, as well as for investigating the role that the epidermal keratinocyte plays in the pathogenesis of psoriasis independent of infiltrating activated T cells.
Thus, in summary, new models of psoriasis continue to be developed even as existing ones are refined. While no single model of psoriasis is ideal, many have proven to be extremely valuable in investigating and better understanding the molecular pathogenic mechanisms that underlie the complex interplay between epidermal keratinocytes, the innate and adaptive immune system, and the vascular endothelium in psoriasis.
Footnotes
Acknowledgements
I would like to sincerely thank Roli Khattri, Steve Hurst, and Susan Sa of Genentech for their review of and comments on this manuscript. I would also like to thank Chris Harrison and Heather Abanto (Gardiner-Caldwell Pacific, San Bruno, CA) for their technical expertise in the illustration of ![]() . Portions of this manuscript were presented at the 56th Annual Meeting of the American College of Veterinary Pathologists (December 2005), and were published in extended abstract form on pages 88–91 of the Conference Proceedings.
. Portions of this manuscript were presented at the 56th Annual Meeting of the American College of Veterinary Pathologists (December 2005), and were published in extended abstract form on pages 88–91 of the Conference Proceedings.