
Abstract
A Siamese kitten presented with mild gait dysfunction associated with periodic circling. Pathologic investigation revealed unilateral (right-sided) absence of the corticospinal (pyramidal) tract throughout its normal course. Although an infectious cause cannot be completely ruled out a genetic etiology was suspected.
A 10-month-old female Chocolate Point Siamese cat, 1 of a litter of 4, was presented for investigation of gait abnormalities. At 2 days old, this kitten and 1 littermate exhibited head tilts and were unable to maintain a normal stance. The other affected littermate died after 5 days, and the remaining 2 littermates remained normal. The owner reported having owned similarly affected kittens from previous litters of different parentage.
At presentation the kitten's gait was well coordinated. However, although it could walk in a straight line it would regularly interrupt its forward progress by turning a full anticlockwise circle. Sometimes it would proceed in a series of sequential rotations. On clinical examination the visual system appeared to be normally functional: obstacles were avoided, palpebral reflexes were brisk, the pupils were equal in size and reacted to light normally and the retinas appeared normal. There was no nystagmus, and the head was neither tilted nor rotated. There was no evidence of otitis externa. Muscle tone and flexor reflexes were normal in all 4 limbs, and the patellar reflexes were also normal. The visual placing reflex was slow in the right foreleg but otherwise normal. The tactile placing reflexes were normal. The owner requested the animal be euthanatized, and this was carried out by intravenous injection of pentobarbital.
Immediately after death the body was perfused intravenously with 2.5 liters of buffered 10% formalin solution. Necropsy showed macroscopic abnormalities confined to the central nervous system. Whereas the dorsal aspect of the brain appeared normal, ventrally the right pyramid was small; this was more apparent when the brain was cut transversely.
Transverse blocks of brain and spinal cord were embedded in paraffin wax and histologic sections cut at 7 μm. Sections were stained with hematoxylin and eosin as routine. Other stains and staining methods included methasol fast blue (MFB) and Loyez for myelin and cresyl fast violet for neurons.
The left corticospinal tract (CST) was present throughout its entire normal course. In contrast, the right CST was represented by a substantially reduced number of fibers along its extent from the cerebral peduncle to the decussation in the medulla (Fig. 1). In the spinal cord, caudal to the medullary decussation, the lateral CST was present on the right side but was minimally represented on the left (Fig. 1).
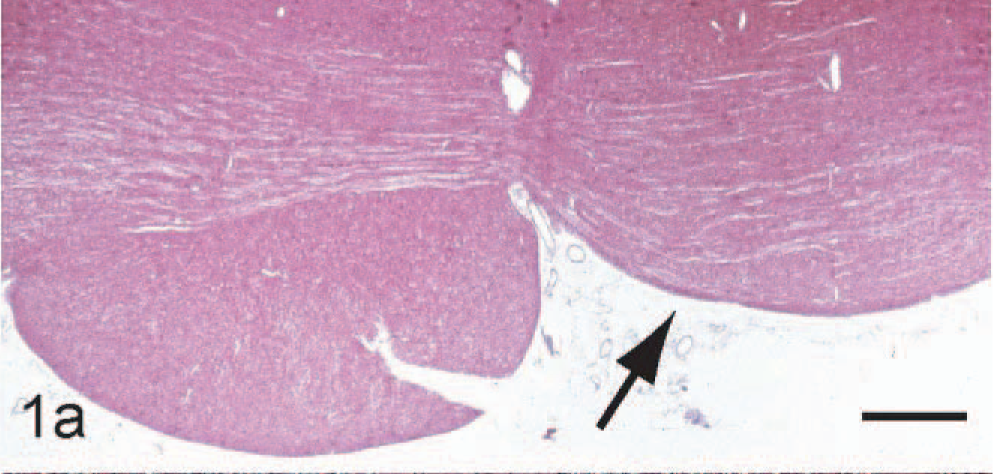
Relative absence of the right corticospinal tract (CST) (arrow) at level of the trapezoid body. HE. Scale bar: 2 mm.
In the brain there was no obvious asymmetry of the hemispheres. Neurons in layer 5 of the motor cortex appeared equal in number in both hemispheres, and the internal capsules appeared to be of equal size. Throughout the central nervous system, including the spinal cord, there was no evidence of a focal pathologic process.
As far as can be ascertained, this is the first report of an animal born with a congenitally absent corticospinal system. This case is especially interesting in that the defect was unilateral. Only one other case report 6 features similar findings—degeneration of a pyramidal tract in a fox—but that case differed in that it occurred secondary to the unilateral destruction of the centrum ovale and internal capsule.
Chambers and Liu 2 reported that the CST of the cat originates from cells of the ectosylvian gyri. Ablation studies showed that there is a large crossed lateral CST in the spinal cord and a small uncrossed ventral CST. A unilateral lesion of the ectosylvian gyrus resulted in a few degenerated fibers in 4 corticospinal pathways, that is, crossed and uncrossed tracts in both the lateral and ventral spinal cord. The preterminal distribution of the CST is chiefly to the dorsal horn and to the intermediate gray matter. Walberg and Brodal 11 had previously shown that in cats some fibers from the motor cortex cross to the opposite hemisphere in the corpus callosum, then descend through the contralateral internal capsule and cerebral peduncle to both lateral and ventral CST. Thus, both groups of authors show that each cerebral hemisphere is represented bilaterally in the coticospinal system in the spinal cord. Such a distribution might account for the minimal clinical deficit exhibited by the animal.
In humans the CST is thought to be responsible for fine motor control, especially of the digits, but its function in nonhuman species is hotly debated. 8 The consequences of unilateral intracranial lesions of the CST (frequently termed the “pyramidal tract”) in humans are profound, including hemiplegia and spasticity. 10 In contrast, profound motor deficits are not generally observed in nonhuman species after damage to the CST, as was apparent in this cat. Indeed, neonatal ablation of the cells of origin of the CST in cats does not produce permanent deficits in skilled motor control. 1 However, the CST in cats does appear to play a role in “skilled” locomotion. 5
The etiology of this condition has not been established, and because of its unusual nature, it is difficult to formulate plausible hypotheses. Despite the absence of inflammatory changes, a viral cause cannot be excluded. If infection arose in utero, residual scarring might be minimal. However, an infectious process would be unlikely to be confined to one side of the brain. The presumed congenital nature of this condition and its manifestation in some but not all of a litter suggest a genetic basis. Gene defects that globally affect the development of the corticospinal system have been described, most extensively mutations in the L1 adhesion molecule. 3, 4 However, these defects are invariably bilateral, and, although phenotypically heterogenous, are often associated with ventricular enlargement and gross cerebellar malformations, 7 none of which was relevant to this case. Moreover, although many developmentally regulated genes are responsible for rostrocaudal, mediolateral, and dorsoventral patterning during neural development, there are none reported that when defective result in asymmetrical development in the neuraxis. It is certainly the case that asymmetry exists outside the nervous system in many organisms, most notably cardiovascular anatomy in mammals, an arrangement generated by so-called binary switching in genes regulating mediolateral patterning such as the nodal cascade. 9 However, such phenomena are not recognized in neural development. Thus, if this condition had a genetic basis it would represent an entirely novel developmental phenomenon of profound biological importance. It is partly for this reason that we report this case, alerting others to importance in identifying future cases.