
Abstract
The significance of p16/Rb tumor suppressor pathway inactivation in T-cell non-Hodgkin's lymphoma (NHL) remains incompletely understood. We used naturally occurring canine NHL to test the hypothesis that p16 inactivation has specific pathologic correlates. Forty-eight samples (22 T-cell NHL and 26 B-cell NHL) were included. As applicable, metaphase- or array-based comparative genomic hybridization, Southern blotting, promoter methylation, and Rb phosphorylation were used to determine the presence, expression, and activity of p16. Fisher's exact test was used to test for significance. Deletion of p16 (or loss of dog chromosome 11) was restricted to high-grade T-cell NHL (lymphoblastic T-cell lymphoma and peripheral T-cell lymphoma, not otherwise specified). These were characterized by a concomitant increase of tumor cells with Rb phosphorylation at canonical CDK4 sites. Rb phosphorylation also was seen in high-grade B-cell NHL (diffuse large B-cell lymphoma and Burkitt-type lymphoma), but in those cases, it appeared to be associated with c-Myc overexpression. The data show that p16 deletion or inactivation occurs almost exclusively in high-grade T-cell NHL; however, alternative pathways can generate functional phenotypes of Rb deficiency in low-grade T-cell NHL and in high-grade B-cell NHL. Both morphologic classification according to World Health Organization criteria and assessment of Rb phosphorylation are prognostically valuable parameters for canine NHL.
Introduction
The significance of inactivation of cyclin-dependent kinase (CDK) inhibitors in hematologic malignancies remains incompletely understood. CDK4 controls the transition of activated T cells from the G0 to the G1 phase and from the G1 to the S phase of the cell cycle, and it is regulated by various transcriptional and post-transcriptional mechanisms. 2, 5 Hence, it has been hypothesized that inactivation of the p16 inhibitor of CDK4 and CDK6 (Ink4-a, Cdkn2, Mts-1) is an important factor in the pathogenesis of T-cell lymphoproliferative diseases. 5, 11, 28 Experimental evidence supports this hypothesis in human T-cell leukemias, especially in pediatric patients; 4, 6, 29, 30 in addition, viral-induced lymphomagenesis in people infected by the human T-lymphotropic virus-I (HTLV-1) may be at least partly due to the inactivation of p16 by HTLV-1 Tax protein. 40 Nevertheless, these findings are confounded by the fact that overlapping reading frames encode another tumor suppressor gene within the INK4 locus (called Arf; p19 in humans and p14 in mice). 21 Although p16 is a regulator of the retinoblastoma (Rb) pathway of cell proliferation, Arf controls p53-dependent pathways that maintain the integrity of the genome; thus, concomitant loss of both genes (for example, by deletion of both INK4 alleles) leads to inactivation of 2 cooperative tumor suppressor pathways with increased risk of tumorigenesis. Mice with a targeted deletion of the INK4 locus have an elevated incidence of lymphoma at an early age and are highly susceptible to carcinogens. 37 However, lymphomas in these INK4-null mice represent almost exclusively B-cell tumors.
Mice with selective inactivation of p16 that retain Arf function still show elevated risk of spontaneous or chemically induced cancers. 38 T cells from these mice are hyper-responsive to mitogenic stimulation in vitro; the animals develop thymic hyperplasia; and when treated with 7,12-dimethylbenzanthracene, they have a higher incidence of thymic lymphoma (21%) compared with wild-type controls (17%), suggesting p16 specifically restrains T-cell proliferation.
We and others have shown that the prevalence of nodal canine T-cell non-Hodgkin's lymphomas (NHL, also known as canine lymphoma, canine malignant lymphoma, and canine lymphosarcoma) are significantly higher in certain dog breeds than in other dogs or humans. 25, 32 Thus, NHL in dogs is a robust model to investigate heritable and sporadic factors that contribute to this group of lymphomas, which is uncommon in other species. Previous results from metaphase-based comparative genomic hybridization (CGH) analysis showed that loss of canine chromosome 11 (CFA 11) was one of the most common numerical abnormalities unique to T-cell NHL. 43 CFA 11 harbors the canine p16 locus (CFA 11q15dist-q16prox) and shares extensive regions of conserved synteny with human chromosome 9 (HSA 9) (R. Thomas et al., submitted for publication). Moreover, the region corresponding to HSA 9p21 (which encodes human p16) lies within the minimal region of loss for CFA 11 in these tumors. For this study, we extended these investigations to analyze the frequency of p16 inactivation (deletion or methylation) and its functional consequences in 4 common types of nodal T-cell NHL, including high-grade or high-proliferation fraction tumors (lymphoblastic T-cell lymphoma [LBT] and peripheral T-cell lymphoma, not otherwise specified [PTCL-NOS]) and indolent tumors (T-zone lymphoma 47 and small lymphocytic T-cell lymphoma [SLL]).
Materials and Methods
Chemicals and reagents
Chemicals and reagents were purchased from Sigma-Aldrich (St. Louis, MO), unless otherwise specified. Tissue culture medium was from Gibco BRL (Grand Island, NY), fetal bovine serum was from Hyclone (Logan, UT), and tissue culture antibiotics (Primocin) were from Invivogen (San Diego, CA). Anti-canine CD3, CD4, CD5, CD8, CD14, CD45, Thy-1, and pan−B-cell marker (CD21) for flow cytometry were from Serotec (Raleigh, NC). Anti-canine CD79a, CD3, and CD20 for immunohistochemistry were from Dako (Carpinteria, CA), Sigma, and Lab Vision Corp. (Fremont, CA). Anti-Rb (G3-245) was from BD-Pharmingen (San Diego, CA), and anti-phospho Rb (S249/T252 and T826) antibodies were from Biosource International (Camarillo, CA).
Tissues and cells
Blood samples from healthy pet dogs and tumors from dogs with NHL were obtained through protocols approved and reviewed by the appropriate institutional animal care committees as described. 17, 18 Peripheral blood lymphocytes were purified over a discontinuous gradient of Ficoll-Hypaque (Histopaque 1.077). 7 Representative sections from sterile biopsy samples were fixed in 10% neutral buffered formalin and processed for histology using routine methods, and single-cell suspensions were prepared from the remaining sample. 17 Human Jurkat and Kit-225 T-cell lymphoma cells were used as controls for Rb phosphorylation analysis. 28 Tumors were classified according to the updated World Health Organization criteria based on morphology and immunophenotype 17, 22, 44, 45 (Table 1).
p16 inactivation and Rb hyperphosphorylation in 48 cases of canine non-Hodgkin's lymphoma.∗
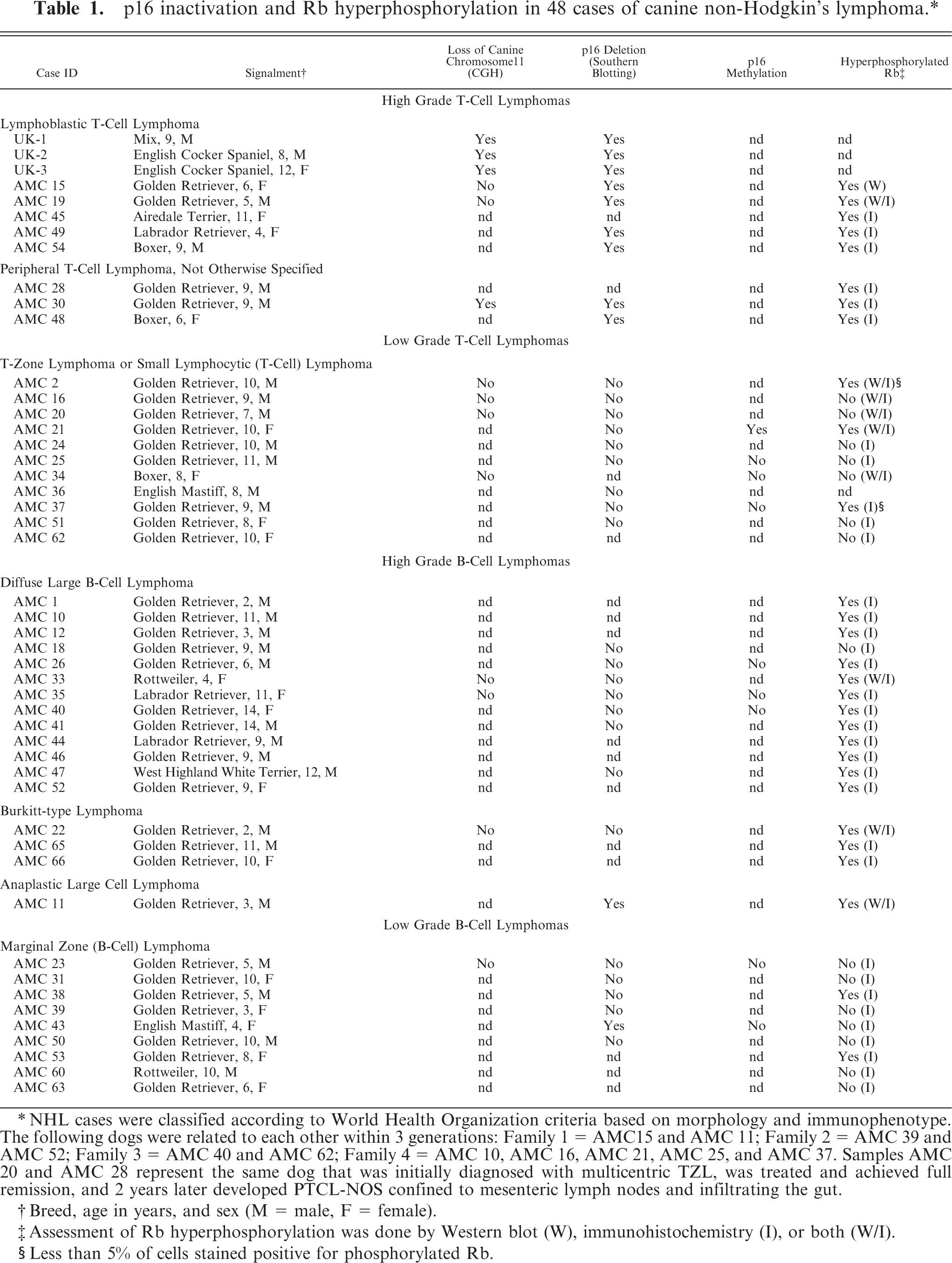
∗NHL cases were classified according to World Health Organization criteria based on morphology and immunophenotype. The following dogs were related to each other within 3 generations: Family 1 = AMC15 and AMC 11; Family 2 = AMC 39 and AMC 52; Family 3 = AMC 40 and AMC 62; Family 4 = AMC 10, AMC 16, AMC 21, AMC 25, and AMC 37. Samples AMC 20 and AMC 28 represent the same dog that was initially diagnosed with multicentric TZL, was treated and achieved full remission, and 2 years later developed PTCL-NOS confined to mesenteric lymph nodes and infiltrating the gut.
†Breed, age in years, and sex (M = male, F = female).
‡Assessment of Rb hyperphosphorylation was done by Western blot (W), immunohistochemistry (I), or both (W/I).
§Less than 5% of cells stained positive for phosphorylated Rb.
Assessment of gene deletion
Metaphase-based or array-based CGH analysis and Southern blotting were used, respectively, to identify gross genomic imbalances and to assess deletion of individual genes as described previously. 27, 42, 43
Gene expression
Gene expression was analyzed by Northern blotting as described 28 using full-length mouse cyclin D2 cDNA and partial canine p16 and ß-actin cDNA as probes. Semiquantitative reverse transcriptase polymerase chain reaction (RT-PCR), which is more sensitive than Northern blotting, was used to confirm results. All RNA samples were subjected to DNase treatment to prevent spurious amplification of genomic DNA sequences. The linear range of amplification for each gene under analysis as a function of variable template inputs and number of PCR cycles was determined in control tissue (canine liver) under identical conditions of primers concentrations, reaction times, and temperature. Every step of the assay was normalized to ensure that p16 expression could be compared across samples: the same number of cells was used at the beginning of the assay, the same input DNase-treated RNA was used for the first-strand cDNA (RT) reaction, the concentration of cDNA was again calibrated for the PCR reaction, and the number of amplification cycles was maintained in the linear range. Quantification was also done in immobilized PCR products by hybridization to radiolabeled probes. Gene expression in tumor cells was examined using these conditions to enable comparisons among samples.
Gene silencing by methylation
Methylation status of p16 was assessed indirectly as described. 20 Briefly, single-cell suspensions from malignant lymph nodes were cultured with or without the inhibitor of methylation 5-aza-2-deoxycytidine (5-aza-C) for 24 hours. At the end of the culture period, RNA was prepared from the cells and RT-PCR was done under limiting conditions for detection as described above.
Rb hyperphosphorylation at canonical CDK4 sites as a measure of p16 inactivation
A functional consequence of p16 inactivation is elevated CDK4 (and/or CDK6) activity resulting in hyperphosphorylation of Rb. We used modification-state antibodies that detect Rb phosphorylation at sites homologous to Ser249/Thr252 and at Thr826 in the human sequence, which are specific CDK4 targets. Blast and Clustal analyses of the human and dog RB-1 genes (Genbank gene IDs 5925 and 476915, respectively) shows that these residues are fully conserved in amino acid sequence between both species.
We first validated the capacity of these antibodies to distinguish the activation status of Rb and to recognize a protein of the correct electrophoretic mobility in canine cells by immunoblotting using the human Jurkat T-cell lymphoma (p16-deleted) and Kit-225 T-cell leukemia (p16 wild-type) cell lines as controls. Jurkat cells have constitutively elevated CDK4 activity and thus show only the hyperphosphorylated forms of Rb, whereas Kit-225 cells can be synchronized by interleukin (IL)-2 deprivation in a “pseudo-G0” state where Rb is largely hypophosphorylated. 28 For immunoblotting, whole-cell lysates were made by disrupting cells in a high salt buffer containing 300 mM sodium chloride, 50 mM Tris, pH 7.6, 0.5% Triton X-100, 1 mM N-ethylmaleimide, 2 μg/ml aprotinin, and 1 μg/ml leupeptin. Insoluble material was removed by centrifugation, and protein concentrations of the cell lysates were determined using the BioRad Protein Assay kit (BioRad, Hercules, CA). Cellular proteins (3−5 μg for human leukemia cells lines) were separated by sodium dodecyl sulphate polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes (Hybond, Amersham, Arlington Heights, IL). Membranes were incubated with primary antibodies for 1 hour at room temperature, followed by secondary antirabbit antibody conjugated to horseradish peroxidase. Detection was performed using the enhanced chemiluminescence system (Amersham) according to the manufacturer's instructions. As shown in Figure 1, phosphorylation at residues Ser249/Thr252 is virtually undetectable in Kit-225 cells deprived of IL-2, and there is also a significant reduction of Rb phosphorylation at residue Thr826. Phosphorylation at these residues is induced upon IL-2 restimulation, and it is clearly detectable in p16-deficient Jurkat cells and in asynchronously growing Kit-225 cells. To validate the antibodies for use in canine samples, we examined their performance in canine peripheral blood lymphocytes (cPBL) and canine TLM-1 melanoma cells that retain normal patterns of Rb phosphorylation. 33 Immunoblots using primary cPBL and lymphoma samples were loaded with 20 μg or protein per sample; canine cell lines were loaded using 5 μg of protein per sample. The antibodies recognized a protein in TLM-1 cells whose electrophoretic mobility was consistent with that for Rb. 33 Resting cPBL had no detectable Rb. As is true for human PBL, Rb increased in canine PBL through the G1 phase and into the S phase. An immunoreactive protein of the correct electrophoretic mobility for Rb was recognized in mitogen-stimulated cPBL by the anti-pRb-Ser249/Thr252 antibody after 48 to 53 hours, concomitant with the rapid increase in steady-state levels of this protein at the G1/S transition, but Rb phosphorylation at residue Thr826 was only detectable in cells cultured longer than 72 hours, suggesting that this event occurred only after cells entered the G2 phase or in cells that had undergone at least 1 round of cell division.
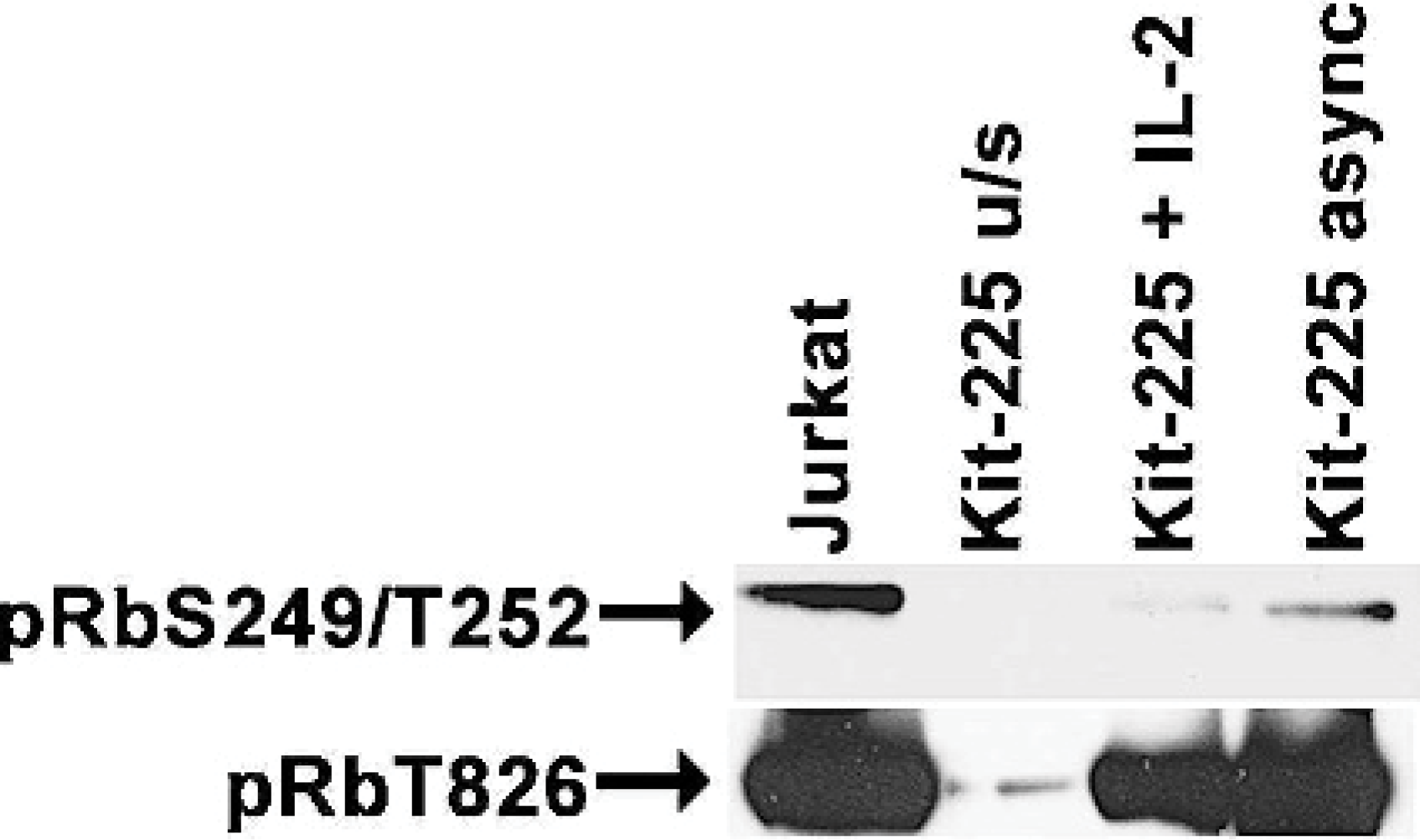
Rb expression and phosphorylation in human lymphoma/leukemia cell lines. Proteins from human Jurkat cells and Kit-225 cells were isolated from cells in asynchronous (async) culture, from Kit-225 cells that were deprived of IL-2 for 48 hr (u/s), or from IL-2−deprived Kit-225 cells after restimulation with IL-2 for 24 hours (+IL-2). Three μg of protein were separated by sodium dodecyl sulphate polyacrylamide gel electrophoresis, transferred to nitrocellulose, and probed with anti-pRbS249/T252, or anti-pRbT826 using enhanced chemiluminescence for detection.
Detection of Rb phosphorylation by immunohistochemistry
For immunohistochemistry, 5-μm serial sections from paraffin-embedded blocks were mounted onto positively charged slides (Probe-on, Fisher Scientific, Pittsburgh, PA) and deparaffinized and hydrated using routine methods. Microwave heat for 6 minutes in a buffer of 0.1 M sodium citrate, pH 6 was used for antigen retrieval. Slides were incubated for 1 hour at room temperature with primary antibodies against phospho-Rb (10 μg/ml), followed by 30 minutes with goat antirabbit IgG conjugated to biotin (Kirkegaard & Perry Laboratories [KPL], Gaithersburg, MD). Phosphorylated Rb species were detected using streptavidin conjugated to alkaline phosphatase with the Histomark red kit (KPL) as previously described. 18, 28, 33
Statistics
Tests for significance were done based on the number of occurrences for 1) loss of CFA 11, 2) p16 deletion, 3) p16 methylation, and 4) Rb hyperphosphorylation using Fisher's exact test with 2 × 2 tables. Various subgroups were collapsed for specific comparisons, and 2-tailed P values are reported despite our a priori hypotheses of increased abnormalities in high-grade T-cell NHL; thus, the P values might be considered conservative.
Results
Loss of CFA 11 and deletion of p16 occurs in high-grade T-cell NHL
Inactivation of p16 has been documented in adult and pediatric sporadic acute T-cell leukemias and in cases of endemic adult T-cell lymphoma secondary to HTLV-1 infection; 4, 6, 29, 30, 40 however, it is unclear if this is an important event in the pathogenesis of nonviral T-cell NHL. To begin to address this, we examined inactivation of p16 in a clinically relevant model of canine NHL. First, we identified chromosomal gains and losses using CGH analysis. We showed previously that loss of CFA 11, which includes the INK4 locus, was common in canine LBT. 43 In the present study, 14 samples (10 T-cell tumors and 4 B-cell tumors) were available to confirm these findings by CGH, including 5 cases classified morphologically as LBT, 1 PTCL-NOS, 3 TZL, 1 SLL, 2 DLBCL, 1 MZL, and 1 BL (Tables 1 and 2). Three LBT cases were analyzed by metaphase-based CGH and were reported previously; 43 the remaining cases were analyzed by array-based CGH with a validated 2-Mb resolution bacterial artificial chromosome array. 42 Four of 6 cases of high-grade T-cell NHL thus analyzed (3/5 LBT and 1/1 PTCL-NOS) had loss of CFA 11; this abnormality was not seen in any other cases analyzed (Table 2), and it was significantly different from the results observed for all other tumor types combined (P = .007), for low-grade T-cell tumors (P = .0476) and for all B-cell tumors combined (P = .0476). The sample sizes were too small to calculate significance against each B-cell group individually.
Distinct mechanisms lead to p16 inactivation in morphologic variants of canine non-Hodgkin's lymphoma.∗
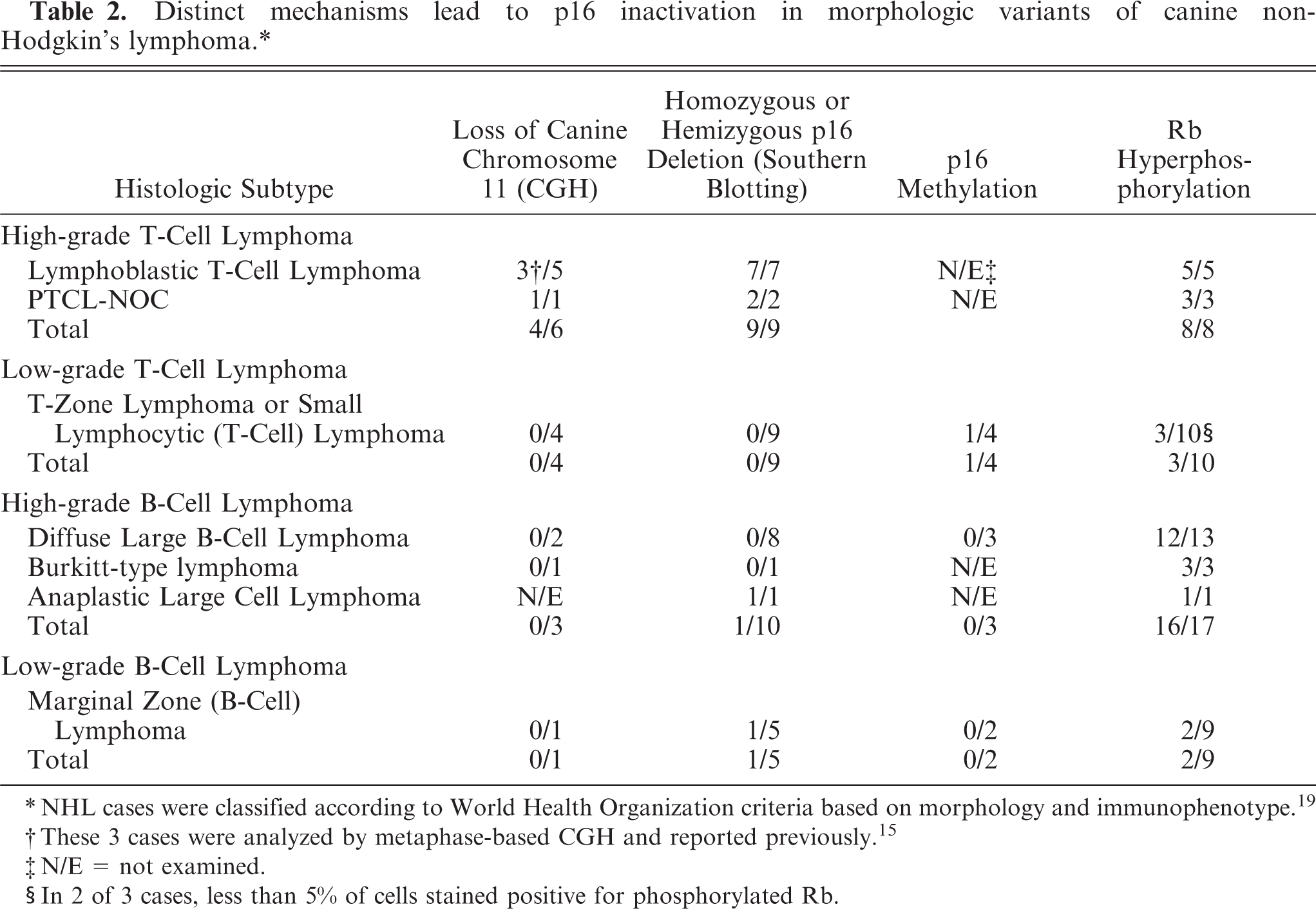
∗NHL cases were classified according to World Health Organization criteria based on morphology and immunophenotype. 19
†These 3 cases were analyzed by metaphase-based CGH and reported previously. 15
‡N/E = not examined.
§In 2 of 3 cases, less than 5% of cells stained positive for phosphorylated Rb.
Assessment of p16 status by Southern blots yielded similar results. Figure 2 shows hemizygous (N = 5) or homozygous (N = 4) deletion of p16 in 9 of 9 cases of high-grade T-cell NHL (7/7 LBT and 2/2 PTCL-NOS) based on comparison to the signal obtained by hybridization to an actin probe; 0/9 cases of low-grade T-cell NHL harbored a deletion of p16, and only 2/15 cases of B-cell NHL analyzed showed deletion of p16 (Table 1). The frequency of p16 deletion was significantly different between the high-grade T-cell group and the other groups combined or individually (P < .01). None of the tumors showed loss of p53 (in CFA 5) by Southern blot analysis, suggesting that loss of p16 in these tumors was a specific recurrent event and not simply due to possible aneuploidy in the tumors.
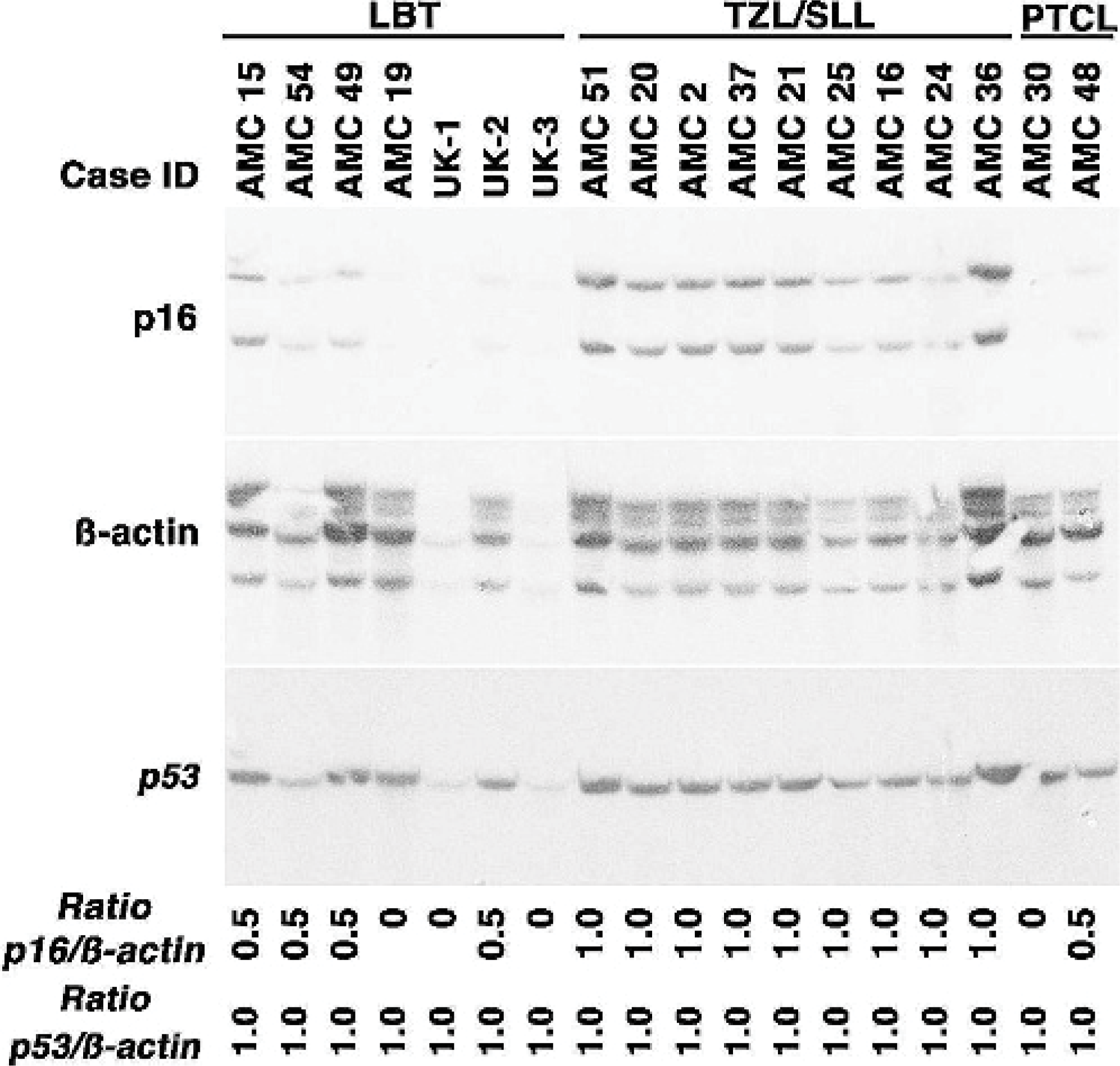
Deletion of p16 in canine high-grade T-cell NHL. The presence of p16 was examined by Southern blotting in 7 cases of LBT, 9 cases of TZL and SLL, and 2 cases of PTCL-NOS. Ten μg of DNA were separated electrophoretically and probed sequentially with p16, ß-actin, and p53 cDNA probes. Autoradiograms were quantified densitometrically using NIH Image software to obtain objective values for band densities that were then used to derive p16/actin and p53/actin ratios. Replicate data were generated for a subset of samples to ensure reproducibility.
Silencing of p16 by methylation is a rare event in canine T-cell NHL
Various mechanisms can lead to functional inactivation of p16 in cancer, including CpG methylation, mutation or deletion of Rb, and overexpression of D-type cyclins. 21, 39 We first examined if there was a correlation between genomic status and gene expression of p16. Northern blots showed comparable expression of ß-actin in 10 sequential samples analyzed, with overexpression of cyclin D2 in 1 of these (case AMC 15). However, p16 gene expression was undetectable by this method in any of the samples. It was shown previously that p16 mRNA is expressed at low levels in normal and transformed lymphocytes, so we used a semiquantitative RT-PCR method to assess p16 gene expression with higher sensitivity. Using this method under limiting conditions, we observed a direct correlation between genomic status and gene expression of p16 in 10 samples for which sufficient RNA could be isolated for this analysis: p16 expression was undetectable in cases AMC 11 and 19 (both of which showed p16 deletion by Southern analysis), and it was present in cases AMC 2, 15, 16, 18, 20, 22, 33, and 34. Case AMC 21, on the other hand, retained 2 copies of p16 (Figure 2) but yielded no p16 amplification products (Figure 3). We examined the possibility that the gene was methylated in this tumor by culturing cells overnight in the presence of 5-aza-C. Case AMC 34 is a representative example in which p16 amplification products were detectable in cells cultured with or without 5-aza-C in the presence or absence of mitogenic stimulation, in contrast to case AMC 21, in which p16 expression was induced in cells after culture with 5-aza-C, a chemical inhibitor of methylation (Figure 3). It is worth noting that the induction of p16 expression in these cells upon culture with 5-aza-C indicates there was at least a small number of spontaneously proliferating cells in the culture, and these cells seemed to downregulate expression of p16 when triggered by mitogenic signals. We cannot definitively conclude that the reduced band intensity in mitogen-stimulated cells from AMC 34 reflects a decrease in p16 mRNA, because these experiments are not quantitative; thus, they can only be interpreted qualitatively.
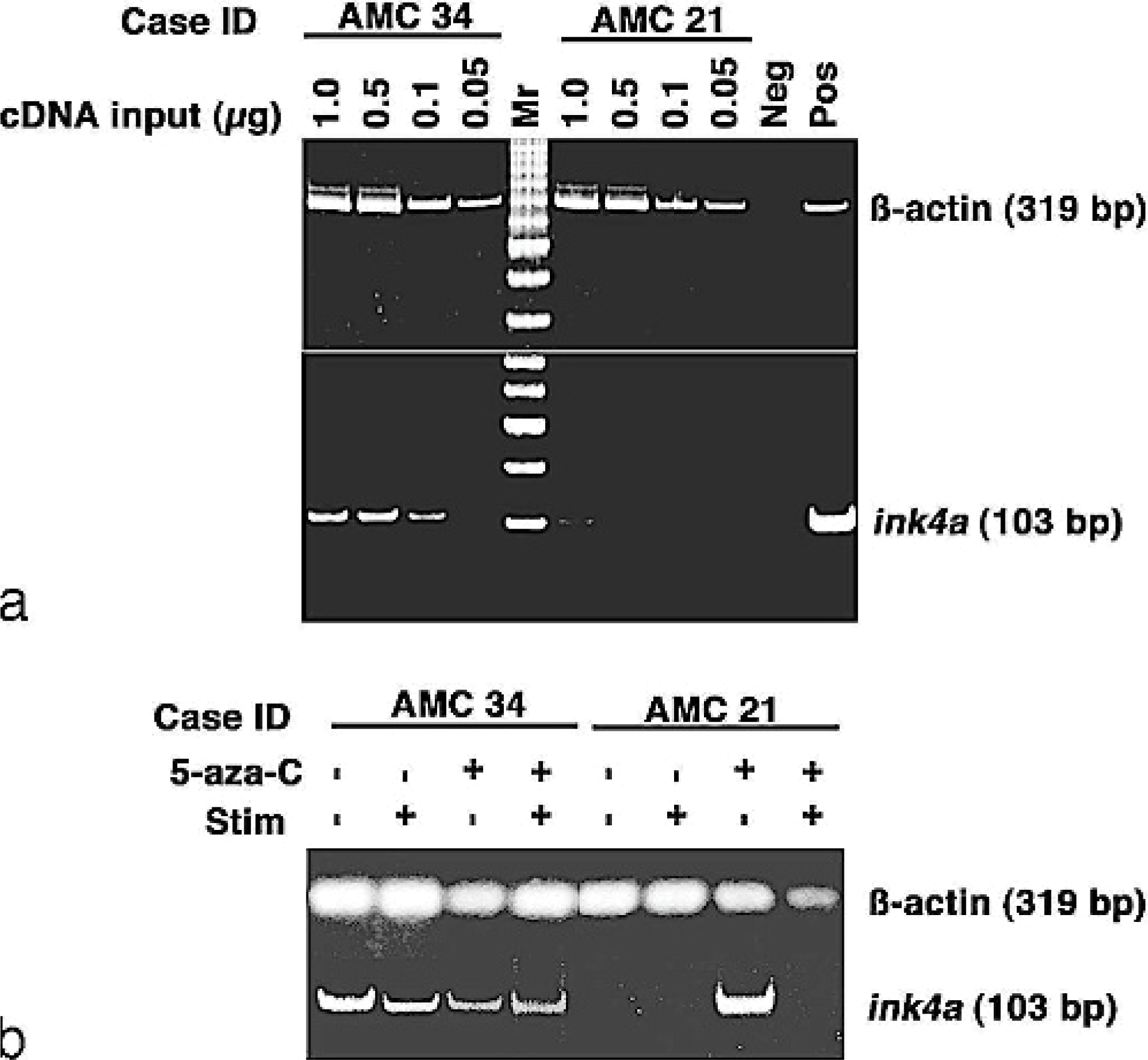
p16 promoter methylation in cNHL. Fig. 3a. Titration of cDNA inputs for RT-PCR with samples from case AMC 34 (p16 positive) and case AMC 21 (p16 negative). Amplification of ß-actin was used to ensure the integrity of starting RNA. The negative control (Neg) used water in place of the cDNA input; the positive controls (Pos) used 100 ng of normal canine liver for ß-actin and 1 μg of a plasmid encoding canine ink-4a (partial cDNA, 18 ) for p16. Fig. 3b. Assessment of p16 expression in the same 2 cases after 24 hours in culture with or without mitogens and 5-aza-C as indicated using 250 ng of cDNA as input for the analysis. Similar data were obtained in 3 independent replicates (using freshly thawed cells from the cryopreserved bank each time).
Inactivation of p16 results in Rb phosphorylation at canonical CDK4 sites
We examined the correlation of Rb phosphorylation status using these antibodies by immunoblotting and IHC in 10 representative samples for which we had both cryopreserved cells and formalin-fixed, paraffin-embedded tissues. The data show that there was a direct correlation between these assays. Six samples (cases AMC 11, 15, 19, 21, 22, and 33) showed phosphorylation of Rb at residues Ser249/Thr252 and/or Thr826 in immunoblots (Figure 4), and each of these had greater than 20 to 30% positive nuclei detectable by the same antibody on IHC staining (Tables 1 and 2 and Figure 5). One additional sample (case AMC 2) showed weak to modest reactivity in the immunoblots and had approximately 5% of positive nuclei on IHC staining. Phosphorylated Rb was undetectable in three samples (cases AMC 16, 20, 34) by either method. Finally, we evaluated Rb phosphorylation by IHC in all the remaining samples for which tissues were available. Table 2 shows a statistically significant difference (P < .005) between high-grade tumors and low-grade tumors.
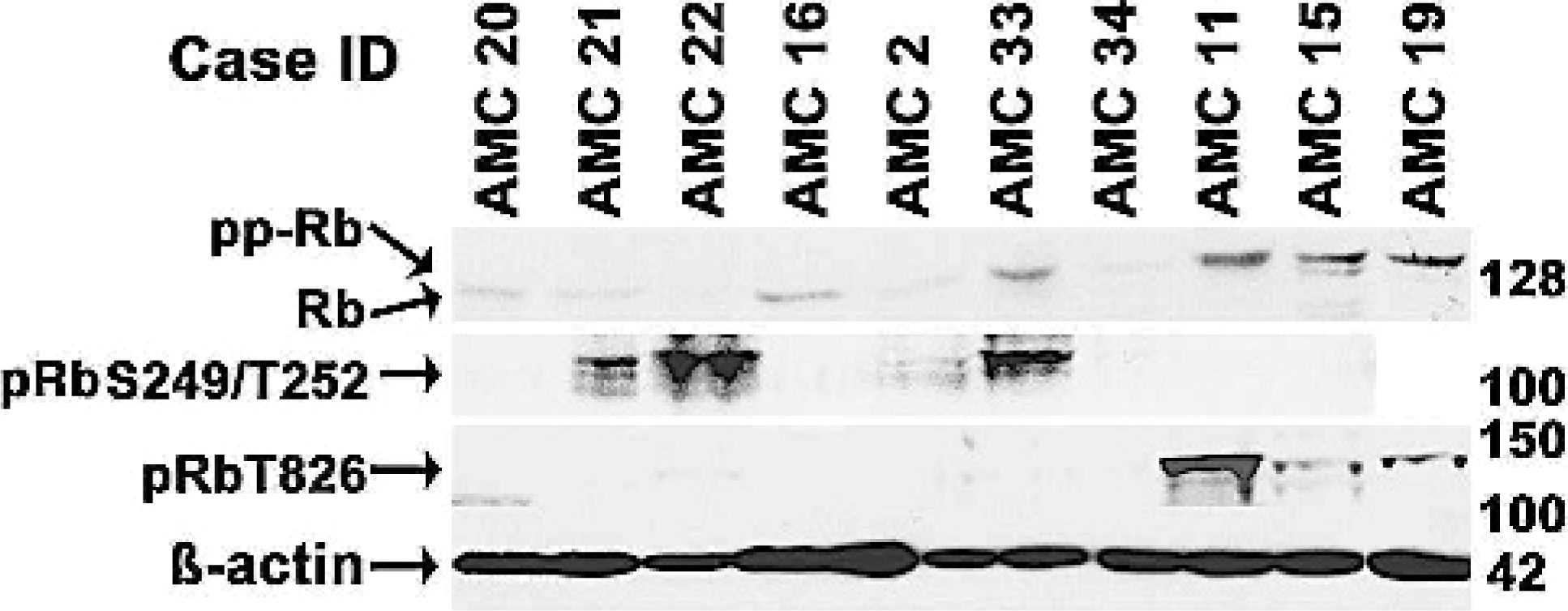
Rb expression and phosphorylation in canine NHL samples. Malignant lymphocytes were enriched from lymph node samples from the indicated cases, and 20 μg of protein were probed with anti-Rb, anti-pRbS249/T252, or anti-pRbT826 as in Figure 1. Material from case AMC 19 was sufficient to load only 3 of the 4 gels, so this sample was not analyzed for pRbS249/T252. ß-actin was used as a loading control.
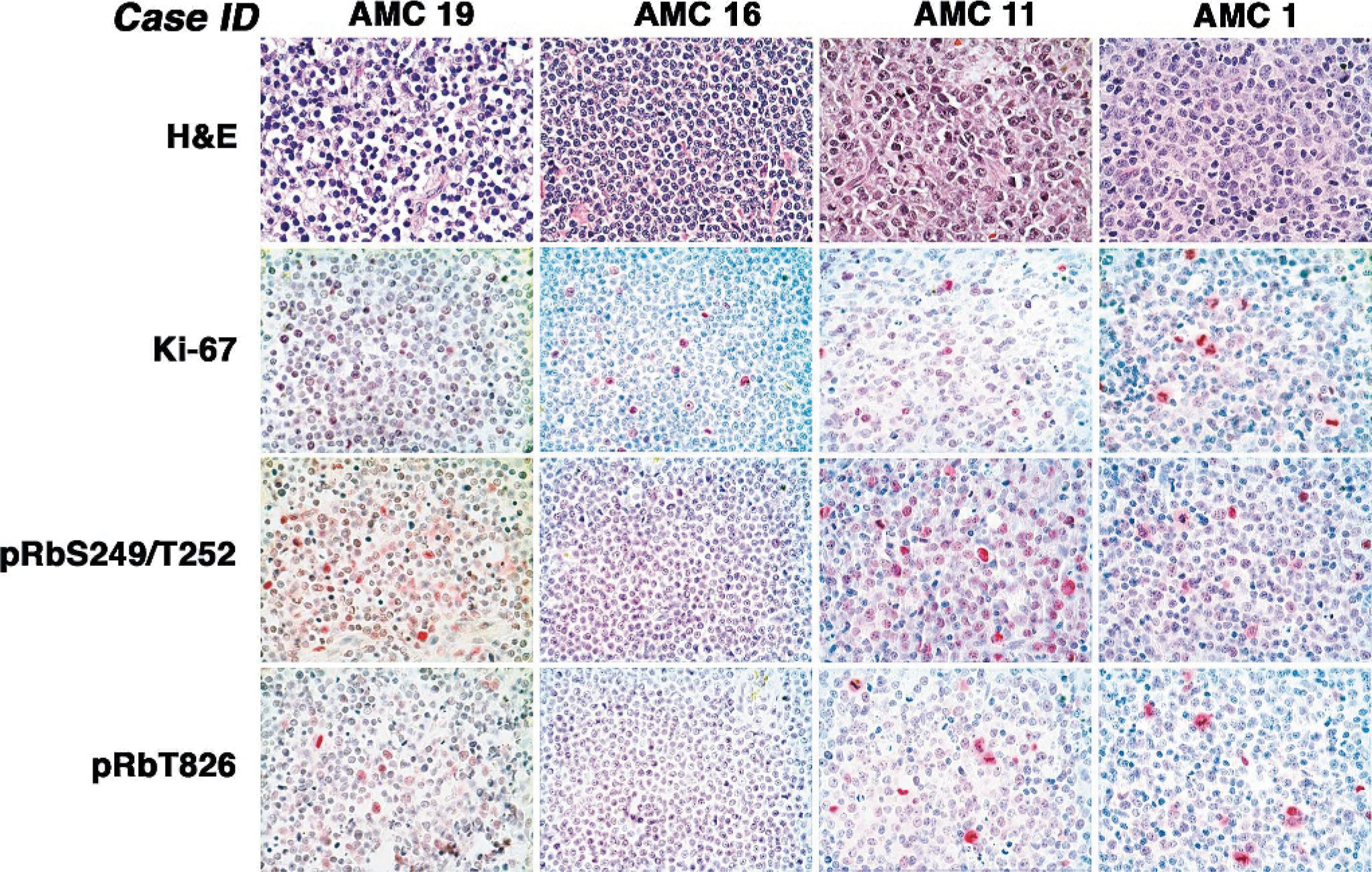
Proliferation and Rb phosphorylation in representative samples of canine NHL. Five-μm sections were cut from formalin-fixed and paraffin-embedded tumors from cases AMC 19, AMC 16, AMC 11, and AMC 1 and stained as indicated with HE, anti-Ki-67, anti-pRbS249/T252, or anti-pRbT826 to assess morphology, proliferation, and Rb phosphorylation at residues Ser249/Thr252 and Thr826, respectively. Immunohistochemical staining was done with a modified streptavidin-biotin-alkaline phosphatase complex reaction. Positive cells are indicated by magenta to red nuclear staining. Magnification = 600 X.
Hyperphosphorylation of Rb at residues Ser249/T252 and/or Thr826 was seen in 100% (8/8 cases) of high-grade T-cell NHL and in 94% (16/17 cases) of high-grade B-cell NHL. In contrast, it was only detectable in approximately 25 to 30% of low-grade NHL of either phenotype (3/10 cases of TZL/SLL and 2/9 cases of MZL). In 2 of the 3 TZLs showing Rb hyperphosphorylation, positive nuclei were restricted to less than 5% of the malignant population. It is noteworthy that phosphorylation of Rb at residue Thr826 was most commonly seen in large cells that may have been near the stage of cell division and in mitotic figures (for example, see Figure 5), consistent with our findings in normal cPBL.
It was possible that phosphorylation of Rb was not due to inactivation of p16 (and increased activity of CDK4) but rather that it simply reflected the proliferative status of these tumors. In other words, high-grade tumors might have had more hyperphosphorylated Rb because of the presence of a larger proliferative fraction. To explore this possibility, we used IHC to stain each tumor for Ki-67, a marker of cellular proliferation. Our results, exemplified in Figure 5, show there was no correlation between Ki-67 staining and Rb phosphorylation, supporting the conclusion that inactivation of p16 and hyperphosphorylation of Rb are causally related and not simply a consequence of rapid cell division in the tumors.
Discussion
Sporadic NHLs that are not associated with viral etiologies occur spontaneously in people and in dogs with similar natural histories and clinical behavior. However, nodal T-cell NHLs are more common in dogs than in people, perhaps because of heritable risk factors that have been firmly established in the derivation of specific breeds. 25 This underscores the suitability of canine NHL to explore the molecular pathogenesis of this group of diseases, as well as to identify biomarkers that are prognostically significant, because outcomes are typically poor. 10, 26
There is abundant evidence that the p16/Rb pathways are important regulators of proliferation in normal lymphocytes, possibly providing mechanisms that enforce quiescence (control the G0 to G1 transition) and that account for replicative senescence of cells that respond to antigen. 9, 19, 24, 28, 35 Yet the precise role of these pathways in the pathogenesis of NHL remains incompletely understood. Deletion or methylation of p16 is a relatively common finding in acute T-cell leukemias, 4, 6, 8, 11, 29, 30, 48 and although p16-null mice do not recapitulate this disease, they show increased susceptibility to develop thymic lymphomas. 38 Deletion or promoter methylation leading to p16 inactivation also is observed in some types of B-cell NHLs; they are common in blastoid mantle cell lymphoma, 31, 41 and they occur less frequently in association with progression from mucosal-associated lymphoid tissue lymphoma or follicular lymphoma to DLBCL 46 and only sporadically in high-grade tumors. 13, 14, 46 The prognosis for human mantle cell lymphoma is generally poor, but the prognostic significance of p16 deletions and Rb inactivation for other NHL subtypes remains uncertain. 14 Cryptic deletions of HSA 9p21 have been documented in primary DLBCL, 12, 23 suggesting this pathway may indeed be pathogenetically important in this disease, although increased susceptibility to develop B-cell tumors in INK4 knockout mice seems to be associated with loss of the Arf gene and the consequent reduced function of p53. 21, 36
Here, we evaluated the hypothesis that abnormalities in the p16/Rb pathways were associated with specific subtypes of T-cell NHL in dogs. We designed our sample collection strategy around a major breed (Golden Retriever) that has approximately equal prevalence of B-cell and T-cell lymphoma 25 and included samples from various other dog breeds to distinguish between abnormalities that are associated with heritable risk (breed specific) and those that are associated with disease pathogenesis (not breed specific). We have previously reported specific recurrent cytogenetic abnormalities that are significantly more frequent in Golden Retrievers than in other dogs, 25 as well as others that occur in lymphoid tumors of dogs irrespective of breed. 43 The results from this latter study suggested loss of CFA 11 was in the latter group: it was seen exclusively in T-cell lymphoblastic tumors, but it did not appear to be associated with a single breed. However, the sample size in that project was too small to definitively conclude whether loss of CFA 11 was associated with specific breeds. Here, we analyzed samples from 48 cases (8 breeds) that were approximately equally distributed among 4 groups: high-grade or high-proliferation fraction T-cell NHL, high-grade B-cell NHL, low-grade T-cell NHL, and low-grade B-cell NHL. Our results show that inactivation of p16/Rb occurs almost universally in high-proliferation fraction tumors but is relatively rare in the indolent tumors. In addition, distinct mechanisms appear to account for the patterns of p16/Rb inactivation in each disease, suggesting that these are not pathogenetically redundant; in other words, B-cell and T-cell precursors are each susceptible to discrete mutations that lead to Rb hypofunction. The lack of breed, age, or sex associations (Table 1) suggests that the contribution of these pathways to the progression of sporadic tumors is not necessarily related to heritable risk factors and is instead inherently associated with their role in lymphocyte development and activation. Specifically, deletion of p16 appears to be the most common means of inactivating Rb in high-grade precursor (LBT) or mature (PTCL-NOS) T-cell tumors. This can occur through gross deletions of CFA 11, although it also may occur through more cryptic events that are detectable by specifically probing for p16 but that lie beyond the resolution limit of CGH (see cases AMC 15 and AMC 19). It is important to note that deletion of CFA 11 would lead to deletion of both p16 (ink-4a) and the closely related gene p15 (ink-4b). The p16 probe we used for Southern blotting is complementary to the ankyrin domains in exon 2, 18 which are highly homologous in both genes. Also, this sequence does not have an internal Bam HI site to explain the 2 bands present in the p16 Southern blots, suggesting that these likely represent both p16 and p15. The loss of signal strength in the Southern blots was equivalent for both bands in each case in which we documented deletion of p16, suggesting that loss of genomic material in each case resulted in deletion of both genes.
At least one other mechanism may create a similar phenotype. Case AMC 15 met the criteria for classification as LBT and appeared to have a cryptic deletion of p16 by Southern analysis. This case also showed overexpression of cyclin D2 and hyperphosphorylation of Rb, suggesting that deletion of p16 with or without deregulation of D-type cyclins can result in a similar morphologic phenotype. Invariably, cases of high-grade T-cell NHL responded poorly to standard of care therapy, 26 indicating that complete characterization of the disease has predictive value.
In contrast to these findings in T-cell NHL, deletion of p16 was rather uncommon in high-grade B-cell NHL. Among 15 cases examined, only 1 (ALCL) showed a hemizygous deletion of p16, and none had evidence of silencing by methylation. However, we showed previously that DLBCLs generally have detectable gain of CFA 13 43 with consequent amplification of c-Myc, and BL in dogs has an homologous translocation to that seen in human BL that juxtaposes c-Myc to the IGH enhancer, also leading to Myc overexpression (M. Breen and J. F. Modiano, in preparation). In light of the fact that Myc increases CDK4 expression and decreases the expression of CDK inhibitors, 1, 3, 15 one would predict that Myc amplification would result in net activation of CDK4 and hyperphosphorylation of Rb, likely accounting for the phenotype observed in the cases of DLBCL and BL.
Inactivation of the p16/Rb pathway was less common in low-grade tumors. Silencing of p16 by methylation was documented in a single case of TZL, and among low-grade T-cell tumors, this case was the only one that showed Rb hyperphosphorylation in a significant number of malignant cells. Only 1 case of hemizygous loss of p16 was documented in MZL, but this case did not show extensive hyperphosphorylation of Rb. Finally, 2 cases of MZL showed hyperphosphorylation of Rb. It is worth noting that it is sometimes difficult to distinguish late phase MZL from DLBCL, because in the later stages of the disease, MZL acquires a more aggressive pattern of behavior and might actually acquire genetic abnormalities that lead to “transformation” to DLBCL. 45, 46
Together, this could be interpreted to mean that other mechanisms account for the pathogenesis of indolent NHL and that inactivation of Rb is not necessary in tumors that do not show aggressive proliferation. This notion is supported by the observation that indolent (e.g., follicular) lymphomas in people are characterized by gain of function of antiapoptotic genes, such as Bcl-2, 16, 34 so genotypes that promote survival, rather than proliferation, may lead to low-grade phenotypes in both B-cell and T-cell NHL. Nevertheless, analysis of outcomes for dogs in this study treated with standard of care (CHOP-based chemotherapy) shows that both p16 inactivation and Rb phosphorylation at S249/T252 and/or T826 are predictive for poor clinical response and shorter overall survival time. 26
In summary, we used a naturally occurring model of canine NHL to examine the frequency of p16/Rb inactivation in high-grade and low-grade morphologic subtypes. The data indicate that inactivation of this pathway can occur by various distinct and mutually exclusive mechanisms, each associated with peculiar morphologic phenotypes; however, different events lead to similar or overlapping morphology in some cases (for example, LBT might result from deletion of p16 and/or cyclin D2 amplification). Further characterization of homologous structural and functional features that occur in human and canine lymphoid tumors will enhance our ability to classify NHL subtypes, as well as illustrate pathogenetically significant events that contribute to the origin and progression of these diseases and that can be targeted in the course of therapeutic development.
Footnotes
Acknowledgements
We thank the owners and veterinarians who contributed cases, assisted with recruitment, and provided diligent follow-up. This work was supported in part by grants 1626, 2254, and 2214 from the AKC Canine Health Foundation (to JFM and MB), charitable donations from individuals, the Starlight Fund, the Kate Koogler Canine Cancer Research Fund, and the Monfort Family Foundation (to JFM) and by retention funds from the Integrated Department of Immunology and the University of Colorado Cancer Center (JFM).