
Abstract
We examined the presence of phosphatase and tensin homolog deleted from chromosome 10 (PTEN) abnormalities that could contribute to the origin or progression of naturally occurring canine endothelial tumors (hemangiosarcoma). Our results document somatic point mutations or deletions encompassing the PTEN C-terminal domain in canine hemangiosarcoma that might provide cells a survival advantage within their microenvironment. This represents the first characterization of a naturally occurring, highly metastatic tumor with biologically significant mutations of PTEN in the C-terminal domain.
Phosphatase and tensin homolog deleted from chromosome 10 (PTEN) is a tumor-suppressor gene located on human chromosome 10q23.3 that encodes a bi-functional phospholipid and protein tyrosine phosphatase. This gene is frequently inactivated in human cancers, with mutations commonly found in endometrial and prostate carcinomas, glioblastomas, and melanomas. 31 In fact, it has been reported that the frequency of PTEN mutations in human tumors may rival the number observed in the DNA damage sensor p53. 6
As somatic deletions or mutations of PTEN are documented at both early and advanced stages of tumor growth, it is possible that PTEN participates in the onset of tumorigenesis in some cancers (e.g., endometrial), but may play a role in progression and metastatic spread in others (e.g., glioblastoma and melanoma). 44 Mutations may lead to inactivation of the phosphatase activity of PTEN as well as the complete or partial loss of expression of mRNA and/or the PTEN protein.
PTEN inhibits cell proliferation and activates the cell suicide program; however, the tumor-suppressive activity of PTEN appears to be largely dependent on its lipid phosphatase activity. 33 PTEN-dependent de-phosphorylation of phosphatidylinositol (3,4,5)-tris-phosphate (PIP-3) antagonizes the activity of phosphatidylinositol 3-kinase (PI3K), which is responsible for the generation of PIP-3. Reduced levels of PIP-3, in turn, prevent activation of the survival factor Akt 21 as well as downstream effects including inactivation or degradation of BAD, 13 caspase 9, 7 and p27 protein. 19 The significance of PI3K in tumor progression is underscored by the observations that expression of a dominant-negative form of PI3K or the addition of the selective PI3K inhibitor LY294002 restores normal growth control pathways and can reverse malignant phenotypes. 9, 21, 25 Conversely, loss of PTEN function results in increased levels of PIP-3 and Akt hyperactivation that lead to protection from various apoptotic stimuli 8, 25, 46 and which can be reversed by overexpression of PTEN. 30 A recent report also shows that PTEN inhibits cellular migration in a wound-healing assay (a model of motility and metastatic invasion), 34 which appears to be mediated by intermolecular interactions between the protein phosphatase domain and the C2 domain, and which is independent of the phospholipid phosphatase activity of the protein.
PTEN also inhibits angiogenesis. Several proangiogenic growth factors, such as vascular endothelial cell growth factor-A (VEGF), transmit signals through the PI3K pathway, 18 and PTEN may modulate angiogenesis by regulating VEGF gene expression. 24 Because of its antiangiogenic activity, PTEN also may play a critical role in regulating growth of endothelial tumors, where production of proangiogenic factors, such as VEGF, may establish autocrine growth loops. 1 As a result, maintenance of PTEN expression and activity may be essential to preserve and regulate normal endothelial cell growth and survival.
For this study, we sought to determine the role of PTEN in the origin or progression of canine hemangiosarcoma (HSA), a cancer of primitive endothelial cells. 1, 15 Because of the aggressive nature of the disease, its endothelial cell origin, and the production of various growth-promoting and proangiogenic factors by these tumors, 1 we hypothesized that many of the biological properties of canine HSA could be explained by aberrations of PTEN. Our results indicate that mutations of the C-terminal domain of PTEN occur frequently in these tumors, offering a possible mechanism for their high metastatic potential.
Materials and Methods
Tissues and cell lines
Tumor samples were obtained from pet dogs with primary (spleen, right atrium, skin) or metastatic (liver) lesions with gross features of visceral or cutaneous HSA (Table 1). Participation in the study required the dogs' owners to sign an informed consent form indicating they understood the goals and procedures for this study. Protocols and procedures were reviewed by the Institutional Review Board and Institutional Animal Care and Use Committee of AMC Cancer Center and by the Institutional Animal Care and Use Committee of the University of Wisconsin. Cell lines were established from viable samples and have been described elsewhere. 15 Each of the cell lines was derived from the primary tumor, except CHAD-P9, which was derived from a liver lesion that was presumed to be metastatic, because a spleen tumor was present concurrently. Paired lines from the primary tumor and metastatic foci were not available for any of the dogs. We were unable to establish immortalized cell lines from two tumor samples (CHAD-G3 and CHAD-G5). The morphology of cells that grew out from these tumors during the first three passages resembled that of the lines that were successfully immortalized from other tumors, 15 but neither CHAD-G3 nor CHAD-G5 survived beyond five to eight passages. Cryopreserved cells from these early passages were used to determine expression and sequence of PTEN in these samples. The same strategy was used to obtain cells from splenic hematomas, which routinely died out before the sixth passage. Only formalin-fixed tissues were available for examination from dog Nos. 12–18.
Expression of CD31, PTEN, VEGF, p27, Akt, and p-Akt in canine hemangiosarcoma.
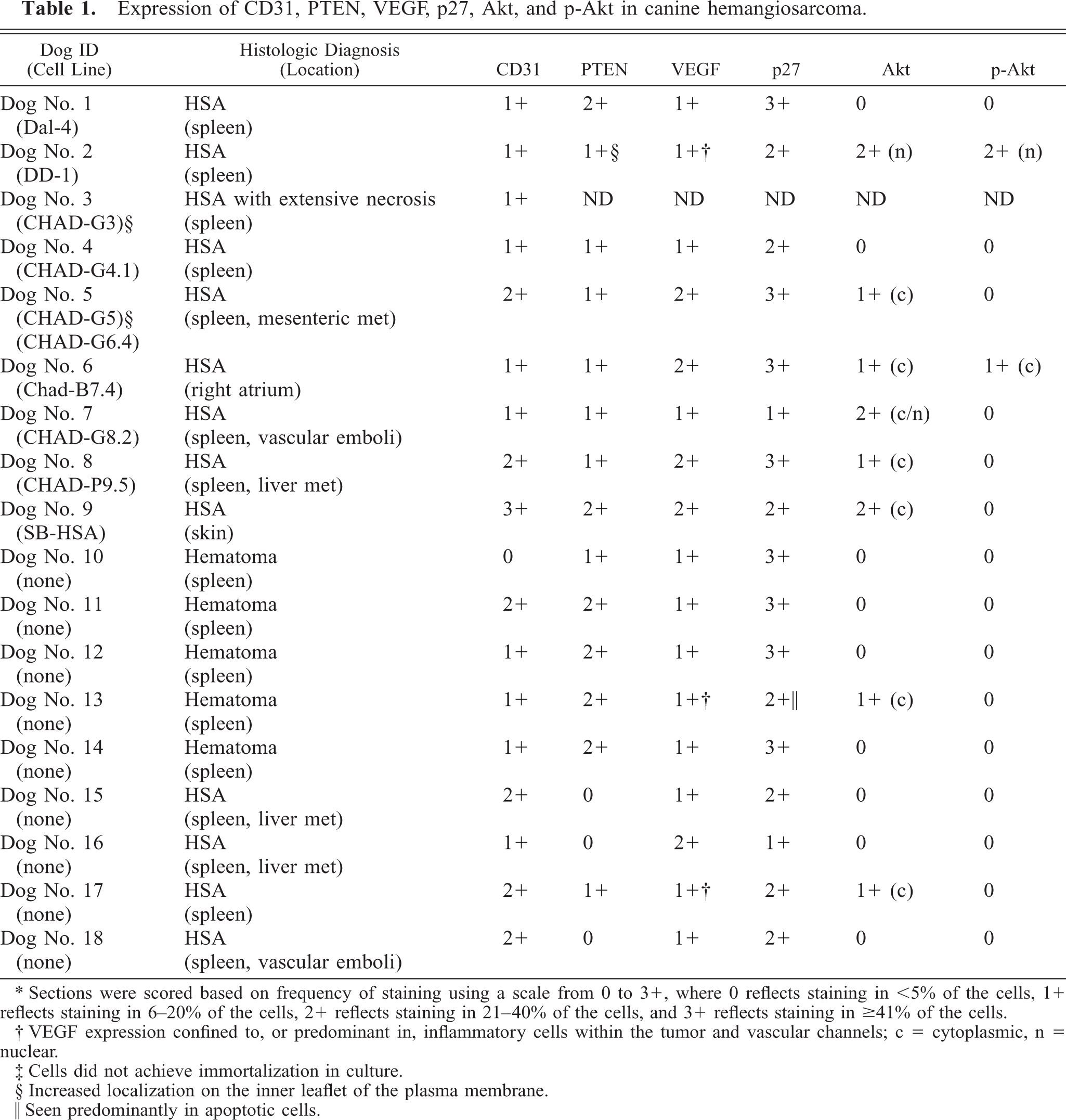
∗ Sections were scored based on frequency of staining using a scale from 0 to 3+, where 0 reflects staining in <5% of the cells, 1+ reflects staining in 6–20% of the cells, 2+ reflects staining in 21–40% of the cells, and 3+ reflects staining in ≥41% of the cells.
† VEGF expression confined to, or predominant in, inflammatory cells within the tumor and vascular channels; c = cytoplasmic, n = nuclear.
‡ Cells did not achieve immortalization in culture.
§ Increased localization on the inner leaflet of the plasma membrane.
‖ Seen predominantly in apoptotic cells.
Chemicals and reagents
Tissue culture materials were obtained from VWR Scientific (South Plainfield, NJ) and chemicals from Sigma-Aldrich (St. Louis, MO), unless otherwise specified. Monoclonal anti-CD31 (PECAM-1) antibody (clone JC/70A) was from Dako Cytomation (Carpinteria, CA); monoclonal anti-PTEN antibody (A2B1) and rabbit anti-p27 polyclonal antibody (N-20) were from Santa Cruz Biotechnology (Santa Cruz, CA); polyclonal anti-VEGF antibody (Z-CVF3) was from Zymed Laboratories (South San Francisco, CA); rabbit anti-Akt and anti-p-Akt (S473) polyclonal antibodies were from Cell Signaling Technology (Beverly, MA); and monoclonal anti-ß-actin antibody was from Sigma-Aldrich.
Pathology and immunohistochemistry
The histologic diagnosis for each of the 18 dogs included in this study is shown in Table 1. Sections from each sample were fixed in 10% neutral buffered formalin for 24 hours. Diagnoses were made from routine HE-stained slides using standard histopathologic criteria. 15 The HSAs were characterized by poorly demarcated and nonencapsulated proliferation of atypical ovoid-to-spindyloid cells. The cells proliferated as solid sheets but often broke apart to form rudimentary and tortuous vascular channels. The cells also were markedly invasive into adjacent parenchyma. Individual cells were characterized by scant-to-moderate eosinophilic cytoplasm and moderately pleomorphic, euchromatic nuclei with medium-sized nucleoli. There usually were one to two mitotic figures per high-power field, and there was significant organizing hemorrhage in the adjacent, noninvolved parenchyma. Splenic hematoma specimens were characterized by poorly demarcated and nonencapsulated hemorrhage throughout the splenic parenchyma. There was early peripheral proliferation of well-differentiated fibroblasts (fibroblastic organization), accompanied by significant hemosiderin deposition. Within the lesion, there was variable proliferation of stromal fibroblasts, fibrohistiocytic cells, and vascular endothelial cells, indicating chronicity in all cases. The adjacent, noninvolved splenic parenchyma was usually normal. It is reported that splenic hematomas often form adjacent to regions of nodular lymphoid hyperplasia, which may be a predisposing factor for this lesion. 38 These samples were acquired because HSA was considered in the differential diagnoses and led to biopsy or splenectomy, and their availability provided a control population that would include endothelial cell proliferation without overt malignant transformation. Samples submitted for the study were limited to representative sections of the lesions, so normal (unaffected) spleen samples were not available for these cases. One sample from normal spleen was available, 23 but because this was dominated by lymphoid elements, we felt it was not a valid control for this sample population. One sample (No. 3) showed extensive necrosis throughout the tumor and surrounding tissue and was excluded from the immunohistochemical analyses. Immunostaining also was not done on the primary tumor from dog No. 9, but rather, it was done on a tumor derived from the primary tumor, which was passaged in an immunocompromised murine host. 1 Immunostaining was performed on 5-μm serial sections from paraffin-embedded blocks using a modified streptavidin-biotin complex method as described. 23 Slides were incubated for 1 hour at room temperature with primary antibodies against CD31, PTEN, VEGF, Akt, or p-Akt, or for 2 days at 4°C with anti-p27 antibody. Sections obtained from canine tissues (liver, kidney, brain, spleen, gut, skin) served as positive controls for the immunostains. Glomerular cells and small vessels in the gut, spleen, and skin showed distinct staining using CD31 and PTEN. Renal glomeruli were routinely used as positive controls for these stains (Fig. 1). The pattern of reactivity for these antibodies was described previously. 15, 23 VEGF is detectable using immunohistochemistry (IHC) in various types of malignant canine tumors (J. W. Wojcieszyn, unpublished), but in normal tissues, VEGF expression was restricted to cells of the reticuloendothelial system and was clearly evident in Kupffer cells of the liver (Fig. 1). The anti-human p27 antibody used recognized a 27-kd protein in human, murine, and canine samples (S. P. Osmire and J. F. Modiano, unpublished 16, 32 ). Validation of the antibody for IHC was done by evaluation of lymphoma samples that expressed p27 on immunoblots. IHC and immunoblotting results showed full concordance, with the p27 variably expressed in nuclear and cytoplasmic compartments as previously reported for this protein and for p21. 2, 14, 35 The anti-Akt and anti-pAkt antibodies were derived by immunizing rabbits with corresponding synthetic peptides (keyhole limpet hemocyanin [KLH] coupled) derived from mouse Akt sequences. The manufacturer reports the antibodies cross-react with Akt from human, mouse, rat, and hamster origin. We validated the reactivity of these antibodies in canine samples using the same strategy as for p27. Sections were examined using an Olympus BX60 microscope with an Olympus Microfire S79809 cooled digital camera (Scientific Instrument Company, Aurora, CO). Transmitted light images were captured using a gain of 1.0 and automatic exposure and white balance settings. Brightness and contrast were adjusted to equivalent settings using Photoshop 7.0 (Adobe, San Jose, CA). Sections were graded on a scale of 0 to 3+ for intensity and percentage of positive cells averaged from five fields at 100×, 200×, and 400× magnification. 23 Samples were called 0 if <5% of tumor cells were positive or if the intensity of staining was no greater than that seen in negative controls. Specific staining in 6–20% of tumor cells was graded as 1+, 21–40% as 2+, and ≥41% as 3+.
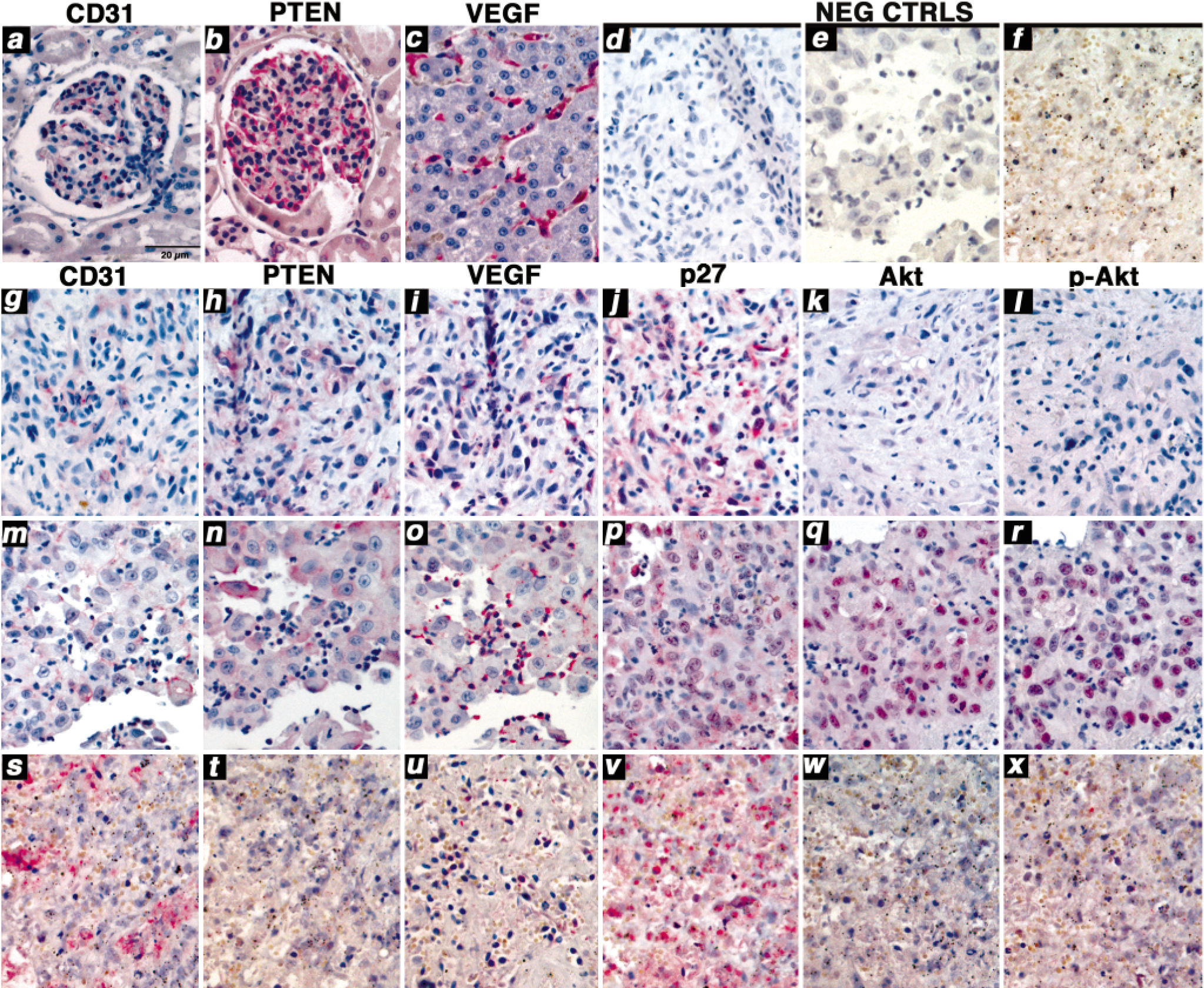
Canine kidney, liver, and spleen (hemangiosarcoma). Serial 5-μm sections from
paraffin-embedded tissues were stained as described in Materials and Methods.
Photomicrographs represent similar or contiguous regions within each tissue at
200× magnification.
Reverse-transcriptase polymerase chain reaction
PTEN gene expression was analyzed by reverse-transcriptase polymerase chain reaction (RT-PCR) as described. 23 Briefly, RNA was isolated using the RNAWiz kit (Ambion, Austin, TX) as recommended by the manufacturer. cDNA synthesis was done using 1 μg of total RNA with the First-Strand cDNA Synthesis kit from Roche Diagnostics (Chicago, IL) followed by PCR amplification using the primers designed from the canine PTEN sequence (Genbank accession U92435) shown in Table 2. Amplifications were done using equimolar amounts of cDNA with the Expand High Fidelity PCR kit (Roche Diagnostics). Amplification products were separated on 0.8–1% agarose gels for purification and sequencing or by loading equivalent amounts from each reaction into nondenaturing 8% polyacrylamide gels for multiplex analysis. ß-actin expression was used to control for integrity of the RNA. 23 Amplification products were gel-purified and sequenced by the core facility of the University of Colorado Cancer Center. Predicted amino acid sequences were derived from the nucleotide sequences using Mac-Vector 6.0 (Oxford Molecular Group/Accelrys, Cambridge, UK) and compared with the wild-type sequence in Genbank (accession NP_001003192).
PTEN primer pairs for PCR amplification.
Immunoblotting
The steady-state levels of protein accumulation were examined as described. 23 The same antibodies against PTEN, p27, Akt, and p-Akt used for IHC were used for immuno-blotting.
Generation of recombinant constructs
For reconstitution of wild-type PTEN, we cloned a fragment including the complete PTEN coding domains (nucleotides 109–1317) into a green fluorescent protein (GFP) expression vector as described. 16 The resultant wtPTEN-GFP fusion protein was delivered into PTEN-null CML-13, CHAD-G6, and SB-HSA cells, and into PTEN-mutant DD-1 cells through cationic, liposome-mediated transfection. 16, 35
Results
PTEN expression in primary canine HSA
Inactivation of PTEN can result from mutations or epigenetic events that lead to reduced expression or loss of function of the protein. To begin to test the hypothesis that aberrations of PTEN contribute to the malignant phenotype of canine HSA, we used IHC to examine expression of PTEN in 12 cases of HSA and in five cases of benign splenic hematomas (Table 1). 15 Endothelial origin was confirmed by assessment of CD31 expression. Virtually 100% of normal, nonproliferating capillary endothelial cells or glomerular cells of the kidney used as controls had abundant levels of CD31 and PTEN (see red staining in Fig. 1, panels a and b). Expression of VEGF was undetectable in normal endothelial cells and glomerular cells of the kidney but was highly expressed in normal cells of the reticuloendothelial system such as Kupffer cells of the liver (Fig. 1, panel c). Endothelial cells in vessels from splenic hematomas retained expression of CD31 and PTEN, but these lesions also showed increased expression of VEGF in both endothelial cells and inflammatory cells, possibly contributing to the process of endothelial proliferation and repair (data not shown). The tumors showed consistent expression of CD31 (Fig. 1, compare negative control panels d–f to panels g, m, and s); expression of PTEN encompassed as many as 40% of the cells or as few as <5% of the cells (Fig. 1, panels h, n, and t) and was localized to the cytoplasm in normal endothelial cells, splenic hematoma cells, and most tumors. In one case (dog No. 2), expression of PTEN was predominantly localized to the inner leaflet of the plasma membrane (Fig. 1, panel n). We anticipated that the HSAs that showed reduced PTEN expression might show increased VEGF expression, but this was only partially the case (Table 1). Eight out of 12 HSAs examined showed modest-to-moderate VEGF in the malignant cells (see, for example, Fig. 1, panels i and u); in the other four, VEGF expression in the malignant cells was low to undetectable, but it was present in abundance in inflammatory cells within the lesions (see, for example, Fig. 1, panel o). To further explore if reduction of PTEN was functionally significant, we examined malignant cells for altered levels of p27 (Kip1), Akt, or constitutively phosphorylated Akt. Most cells within the tumors expressed p27, and although differences in intensity were modest, six of nine tumors that showed PTEN expression in <20% of cells also had reduced levels of p27 expression compared with the levels seen in glomerular endothelial cells and vascular cells in the splenic hematomas (Table 1; compare, for example, Fig. 1, panels j, p, and v). There was no correlation between the levels of p27 and its location to the nucleus (see Fig. 1, panel v) or the cytoplasm (see Fig. 1 panels j and p). Akt expression was only seen in seven of the tumors. Some tumors that expressed low levels of PTEN also showed low levels of Akt (see for example Fig. 1, panel w), but this did not represent a trend (Table 1; see for example Fig. 1, panel k and q). In addition, active Akt (constitutively phosphorylated at serine 473) was detectable only in the tumor showing membrane localization of PTEN (compare Fig. 1, panels l, r, and x). In this tumor, active Akt also was confined to the nucleus. The immunohistochemical results shown here can only be interpreted qualitatively, but they were consistent with the possibility that endothelial tumors harbor abnormalities that might interfere with the normal function of PTEN. To explore this in more detail, we used cell lines established from these tumors to examine patterns and mechanisms that may account for the observed abnormalities in PTEN expression.
Mutations in the C-terminal domain of PTEN are common in HSA
We showed previously that the cell lines examined here originated from malignant endothelial cells and not from benign stromal cells. 1, 15 Specifically, each of the cell lines expressed CD117 (c-Kit, a marker of immature endothelial precursors), showed extended survival in cell culture, and was capable of anchorage-independent growth. 15 Moreover, the SB-HSA cell line forms tumors when implanted into susceptible mice 1 (in vivo tumorigenic capacity has not been determined for the other cell lines used in this study). Yet, there is concern that genetic drift and artificial selection conditions can limit the value of cell lines as models to study cancer pathogenesis. In this case, the observation that PTEN expression in DD-1 (dog No. 2) cells was preferentially localized to the inner leaflet of the plasma membrane, as it was in the primary tumor (Fig. 2a), supported the notion that the cell lines were representative models to examine PTEN abnormalities found in tumors from which they were derived.
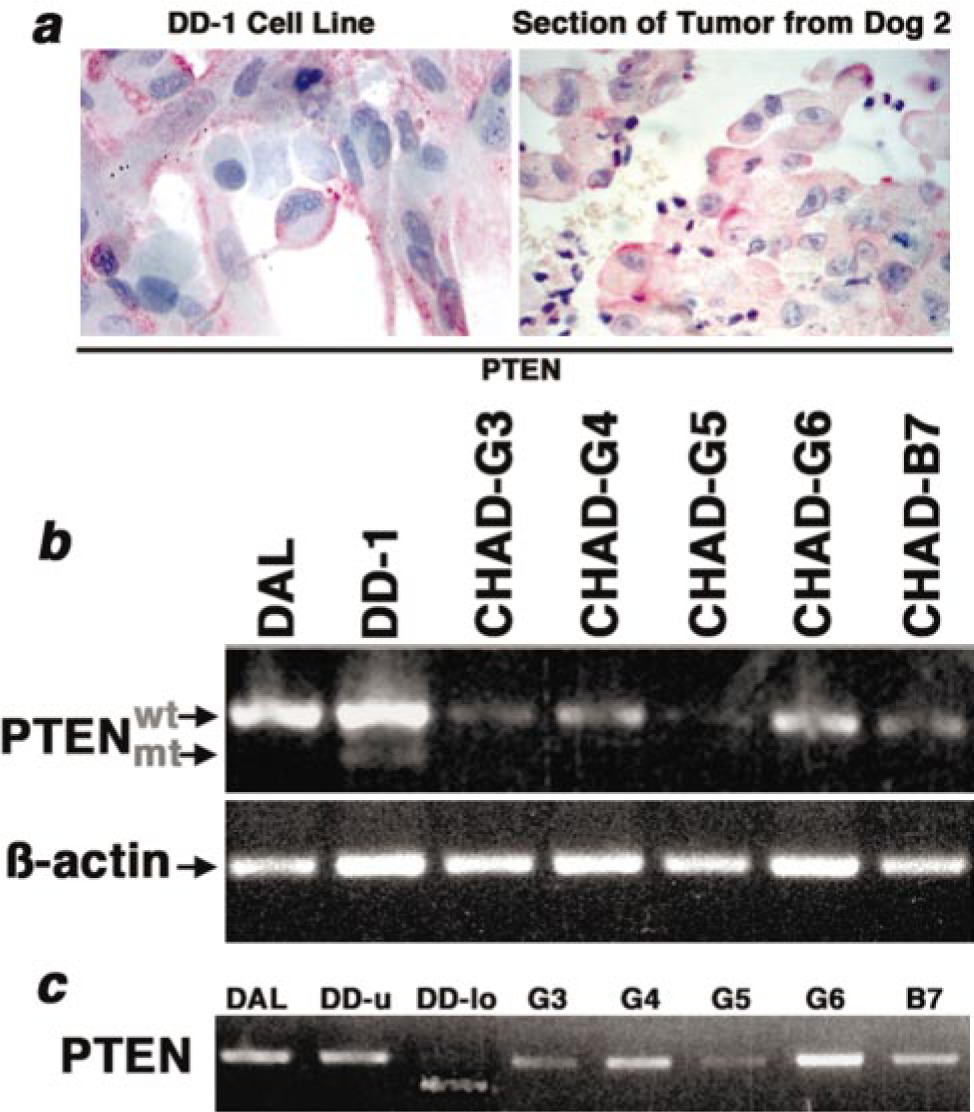
Canine hemangiosarcoma immunostaining and detection of PTEN mRNA.
We examined the frequency and consistency of mutations in the coding domains of PTEN in the corresponding cell lines. RNA was isolated from seven cell lines and reverse-transcribed to cDNA to form templates for PCR (Fig. 2b). Primers encompassing the complete coding domain for canine PTEN were used along with high-fidelity Taq polymerase to avoid the incorporation of pseudomutations in the PCR reactions. Amplification of a 319-bp fragment from ß-actin was used to verify the integrity of the RNA and as a loading control for the reaction (Fig. 2b, lower panel). Tissue controls included liver, spleen, and kidney from normal dogs. 23 Using primers specific for PTEN, a single product representing the predicted size of the complete coding domain (1,208 bp) was amplified in each of the seven cell lines except for DD-1, which generated two distinct amplification products of ∼1,200 bp and ∼900 bp, respectively (Fig. 2b, upper panel). The levels of PTEN mRNA (and protein) in Dal-4 cells were reproducibly comparable with those seen in normal liver and kidney samples (data not shown), allowing us to use Dal-4 cells as a standard to determine alterations in PTEN expression. Among the cell lines and early-passage cells from the tumors we tested, CHAD-G3, CHAD-G5, and CHAD-B7 consistently had lower levels of PTEN message than Dal-4. It is noteworthy that neither CHAD-G3 nor CHAD-G5 achieved immortalization in cell culture, so the reduced expression of PTEN in these samples might be a reflection of fragility or impaired survival by these cells in culture, whereas other mechanisms would more likely affect the stability of the PTEN message in CHAD-B7. The PTEN products from each of the samples were then isolated and gel-purified for sequencing. Small amounts of each purified product were separated on agarose to confirm their purity. As shown in Fig. 2c, two distinct bands were generated by PCR amplification of DD-1 products: the upper band (DD-u), corresponding to the wild-type PTEN sequence and the lower band (DD-lo), corresponding to a putative deletion mutant PTEN sequence.
Nucleic acid sequences for each of these products were translated to the predicted amino acid sequence and these were compared with the wild-type canine PTEN (Genbank accession AAC48709). This analysis revealed that deletions or point mutations were clustered in the C-terminal domain of PTEN in five of the seven samples (Fig. 3). Dal-4 cells retained wild-type PTEN, whereas the products isolated from DD-1 cells included wild-type PTEN (DD-u) and a truncated protein that included the N-terminal and phosphatase domains but had lost most of the C-terminal domain (DD-lo). The sequence for this product was obtained using the 5′ primer (F–K9P1OU109, Table 2) for the forward sequencing reaction. Only two of the lines showed mutations in the phosphatase domain, and Southern blotting and fluorescence in situ hybridization confirmed that, unlike humans, 11, 40 dogs do not harbor a PTEN pseudogene that might give rise to the mutant transcripts (R. Thomas, M. Breen, S. P. Fosmire, J. F. Modiano, unpublished).

Canine hemangiosarcoma—predicted PTEN amino acid sequences. PTEN amplification products from the reactions shown in Fig. 2 were sequenced in both directions. Wild-type sequence shown for comparison was from Genbank (accession AAC48709). Sequence data were verified by sequencing additional products from the reactions shown in Fig. 4. Mutations are highlighted in gray; single amino acid deletions are noted by - and termination of the sequence by deletion or truncation is noted by ∧. The dash at the end of the sequences denotes the translation stop codon.
To increase the throughput efficiency of analysis, we designed multiple PCR primers spanning the PTEN coding domains and used these in six independent PCR reactions to generate distinct amplification products (1,208 bp, 852 bp, 744 bp, 529 bp, 465 bp, and 393 bp). We used these multiple primer sets to generate PTEN amplification products in cDNA samples from eight HSA cell lines (DD-1, Dal-4, CHAD-G4, CHAD-G6, CHAD-B7, CHAD-G8, CHAD-P9, and SB-HSA), early passage cultured cells from samples CHAD-G3 and CHAD-G5, and two samples derived from hematomas of dog Nos. 10 and 11. To identify regions in the sequence harboring abnormalities, equivalent aliquots from each reaction were separated on a multiplex polyacrylamide gel that also included a ß-actin loading control. Aberrant amplification patterns were seen in seven of the 12 samples (CHAD-G3, CHAD-G4, CHAD-G5, CHAD-B7, CHAD-G8, SB-HSA, and dog No. 10; see Fig. 4). A 1,208-bp amplification product representing the complete coding domains of PTEN was detectable in each of the 12 samples, but the levels of this product appeared diminished in five of the 10 tumor lines (CHAD-G3, CHAD-G5, CHAD-B7, CHAD-G8, and SB-HSA) and in the sample from dog No. 10 (also, see below). In addition, the 852-bp product was absent from CHAD-G3 and CHAD-G4; the 744-bp product was undetectable in SB-HSA cells; the 529-bp product was not observed in CHAD-G4, CHAD-G5, or CHAD-B7; the 465-bp product was absent in CHAD-G8; and the 393-bp product was diminished in SB-HSA cells. The 529-bp and the 465-bp amplification products also were undetectable in the sample from dog No. 10, and the levels of the 393-bp product were notably decreased. Intriguingly, larger PCR products were amplified in samples described above that had reduced levels of smaller PCR products, despite what would be expected if a deletion were present within the coding domains (for example, CHAD-G4 had detectable 1,208-bp and 744-bp amplification products, but not 852-bp or 529-bp amplification products). There are several possible explanations for these findings. One possibility is that the early passage samples used (routinely less than passage 7 to minimize in vitro artifacts resulting from the selection) contained a mixture of malignant cells and normal stromal elements, and the stromal elements accounted for the presence of wild-type sequences. Another possibility is that the cells retained a wild-type allele and a mutant allele, as was demonstrated for the DD-1 cells above. Alternatively, the observed deletions might be small enough (<25 bp) that the apparent size of the larger products was not altered using this multiplex gel analysis. In the case of absent products, mutations at or near the target sites for the primers that were designed as exact matches for the wild-type sequence might have prevented recognition or altered the melting temperature of the primer pairs, thus interfering with the efficiency of the PCR reaction. This could account for the presence of a single major sequence in six of the lines tested (Fig. 3). Several common amino acid substitutions were present in two or more of the CHAD-G3, CHAD-G4, CHAD-G5, and CHAD-B7 samples (A359G, S362A, T363M/I/T, S364C, Y377C, S380C, S385F D386V, and I400F). Even when these mutations were not present, other mutations or deletions were present at the same or nearby amino acids (Fig. 3). However, there was not a perfect correlation between these mutations and the pattern of amplification products detectable in the multiplex RT-PCR assay. The products that were most commonly reduced or absent span the C-terminal domain; the mutations identified in Fig. 3 might thus affect the relative stability of the mRNA, the efficiency of the amplification reaction, or both, because each contributes to the sensitivity to detect PTEN mRNA by RT-PCR. Nevertheless, the results indicate that alterations of PTEN mRNA were present in six of 10 HSA samples using this technique. The observation that both Dal-4 and DD-1 cells had detectable products for each reaction was predictable based on the fact that these lines retained at least one wild-type allele. However, the fact that CHAD-G6 similarly generated amplification products for each reaction despite the presence of various mutations indicates that other mechanisms probably contribute to PTEN mRNA stability. As may have been true for samples CHAD-G3 and CHAD-G5, the absence of amplification products in the sample from dog No. 10 could reflect poor viability of the cells in culture, but we cannot fully exclude the possibility that this mass contained malignant cells within the grossly visible hematoma. 15
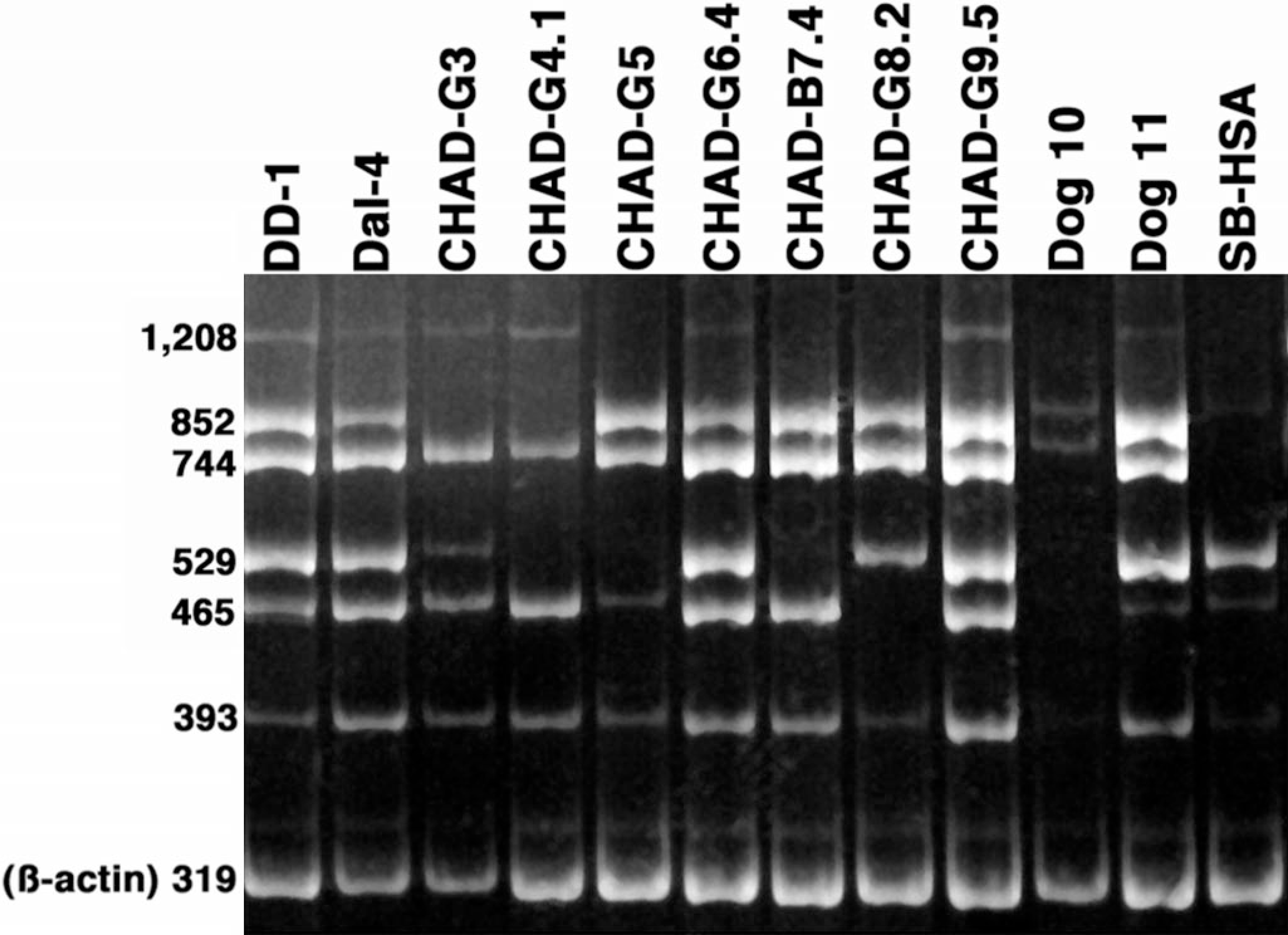
Canine hemangiosarcoma multiplex RT-PCR gels for analysis of PTEN mRNA expression. RNA was used to make cDNA from the indicated cell lines or from early passage, cultured cells from HSA (CHAD-G3, CHAD-G5) or hematoma samples (dog No. 10, dog No. 11). Equimolar amounts of each cDNA were used in six independent PCR reactions with primers spanning the PTEN coding domains. The predicted amplification products for each reaction were 1,208 bp (109–1,317, full length); 852 bp (465–1,317); 744 bp (465–1,209); 529 bp (788–1,317); 465 bp (852–1,317); and 393 bp (207–600). The amplification products were then separated on a multiplex gel along with amplification products from ß-actin (319 bp). PCR amplification products stained with ethidium bromide were visualized under UV illumination.
Functional significance of PTEN abnormalities in canine HSA cell lines
We predicted that the anomalies of PTEN seen in the canine HSA lines might result in reduced levels of p27 and accumulation of constitutively phosphorylated Akt (p-Akt). We compared the steady-state levels of PTEN and p27 in seven HSA cell lines near log growth and in two hematoma samples by immunoblotting (Fig. 5). The levels of expression for PTEN, Akt, and p27 in Dal-4 cells were similar to those seen in normal liver samples and in the Cf2th line 23 (an immortalized cell line derived from fetal thymic epithelial cells). Although two different transcripts were present in DD-1 cells, only the full-length PTEN protein was detectable by immunoblotting (Fig. 5). This is likely because the C-terminal epitope recognized by the anti-PTEN antibody used was deleted from the mutant protein. Consequently, we cannot presently determine if the putative protein with a C-terminal truncation or internal deletion was expressed and stable because antibodies recognizing the N-terminal end of the protein failed to recognize wild-type canine PTEN.
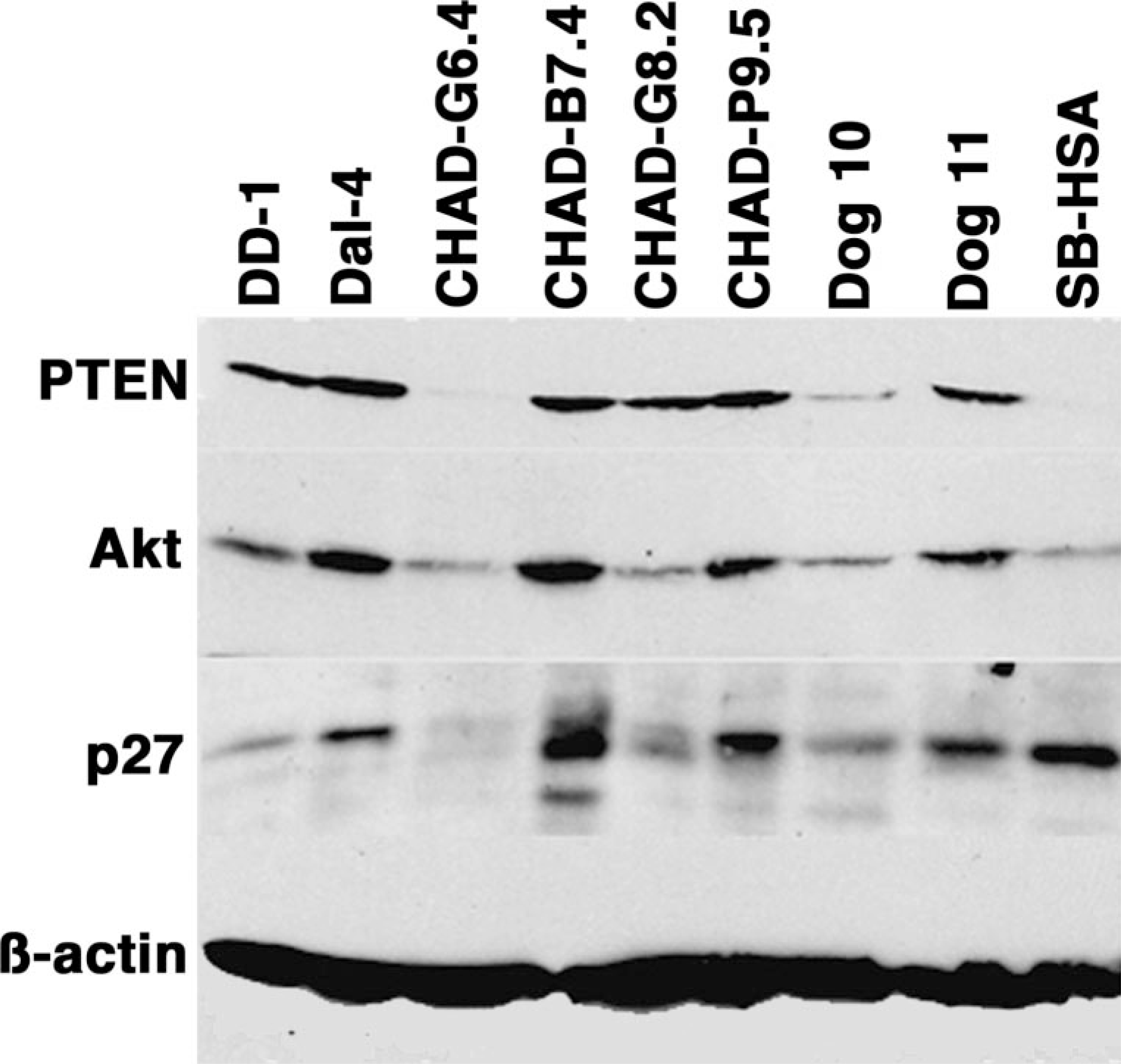
Canine hemangiosarcoma—immunoblots for PTEN, Akt, and p27. Expression of PTEN and p27 was examined in HSA cell lines and in early passage, cultured cells from HSA (CHAD-G3, CHAD-G5) or hematoma samples (dog No. 10, dog No. 11). Accumulation of the relevant proteins was examined in whole-cell lysates from cells near log growth by immunoblotting. Expression of ß-actin was used to control for differences in loading or sample integrity. Immunoblots for PTEN, Akt, and p27 were done in separate membranes prepared under the same conditions.
When compared with the levels seen in Dal-4, PTEN protein was slightly reduced in DD-1 and CHAD-G8 cells, and it was undetectable in CHAD-G6 and SB-HSA cells. Consistent with the mRNA findings, PTEN also was decreased in the sample from dog No. 10. Immunoblot analyses showed a similar trend between the levels of PTEN expression and the levels of p27 in each of the cell lines except SB-HSA (Fig. 5). This result supports the notion that reduced PTEN would lead to degradation of p27, 4, 27, 36 but the expression of p27 in SB-HSA cells suggests that, in some cases, other factors can override the degradation of this protein in cells that are deficient in PTEN function.
As observed for p27, the expression of Akt also paralleled expression of PTEN in these cells. In other cell types, loss of PTEN function has been shown to render Akt phosphorylation resistant to stimuli that promote growth arrest, such as serum withdrawal. 29 We examined how serum deprivation and restimulation affected the levels of Akt and p-Akt (S473) levels in each of the immortalized HSA lines. Results from four representative lines are shown in Fig. 6. Subconfluent cells were incubated overnight in growth factor–deficient media supplemented with 0.5% fetal bovine serum (FBS), followed by stimulation in the same growth factor–deficient media (“− serum”) or media supplemented with 10% FBS (“+ serum”) for 30 minutes. Commercially available lysates of 3T3 murine fibroblasts were used as positive controls for PTEN regulation of Akt signaling. The levels of total Akt and p-Akt in CML-13 canine melanoma cells that do not express PTEN 23 were refractory to serum withdrawal (Fig. 6), indicating that total loss of PTEN function leads to constitutive activation of Akt. These cells also had no detectable p27 under all conditions tested (not shown). Among the HSA lines, only DD-1 cells retained detectable, albeit decreased, levels of p-Akt after serum withdrawal (Fig. 6) with no change in the levels of total Akt. Thus, despite the presence of full-length protein in these cells, the observation that Akt is constitutively activated suggests that PTEN may be hypofunctional in these cells. In contrast, 30 minutes of serum restimulation not only led to detectable levels of p-Akt but also was sufficient to increase expression of total Akt in the remaining cell lines. The results in CHAD-B7.4, CHAD-G8.2, and CHAD-P9.5 cells were similar to those shown for CHAD-G4.1 and CHAD-G6.4 cells, suggesting that the accumulation of Akt in these cells might be controlled at least partly by nontranscriptional mechanisms. Because some cell lines (e.g., CHAD-G6.4) retained Akt serum-responsiveness in the absence of PTEN, other signaling pathways and participants may affect the expression and phosphorylation of Akt along with PTEN. In addition, lower levels of Akt expression suggest that, in these cells, there may be feedback mechanisms that prevent Akt overexpression under conditions of reduced PTEN function.
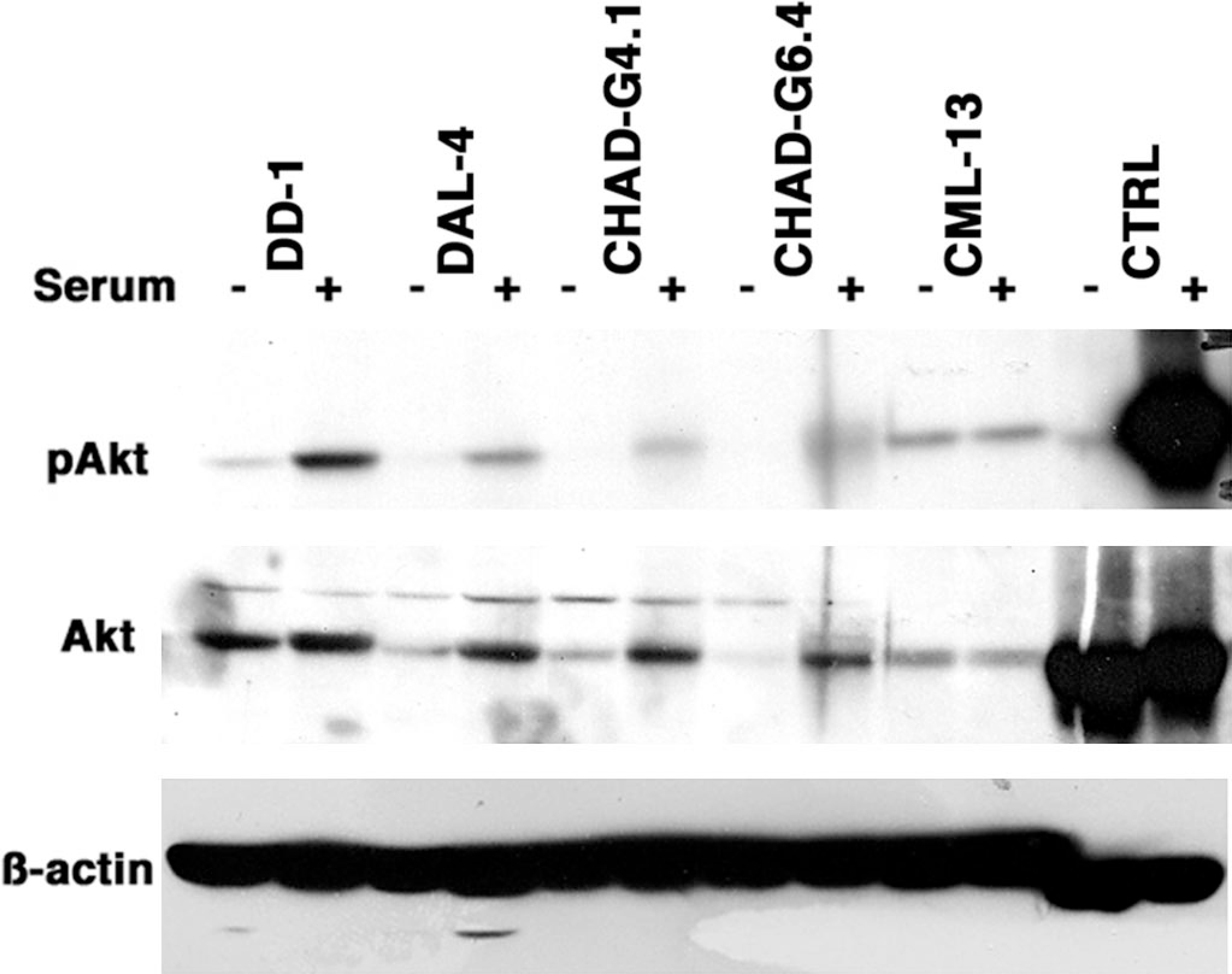
Canine hemangiosarcoma, Akt phosphorylation. Subconfluent cell lines were passed at a density of 1 × 105/ml and allowed to adhere to plastic culture plates. Cells were incubated in growth factor–deficient media (F12K supplemented with 0.5% FBS) overnight, followed by stimulation in the same growth factor–deficient media (− serum) or media supplemented with 10% FBS (+ serum) for 30 minutes. Akt phosphorylation was examined by immunoblotting using phosphospecific anti-Akt antibodies. Akt levels were subsequently examined in the same blots as were levels of ß-actin. CML-13 is a PTEN-deficient, canine, melanoma cell line. Commercially available lysates of 3T3 murine fibroblasts were used as controls.
To begin to verify that loss of PTEN was biologically significant, we examined the effect of PTEN reconstitution in representative PTEN-null or PTEN-mutant lines, as well as PTEN depletion in cells with wild-type PTEN. We engineered a fusion protein consisting of the full-length canine PTEN with a GFP tag at the N-terminus to retain the function of the C-terminal end that was mutated in HSA lines. PTEN-null CML-13, CHAD-G6.4, SB-HSA cells, and PTEN-mutant DD-1 cells were transfected with the fusion protein or the unmodified GFP vector and followed in culture for 3 days. Initial transfection efficiencies for the experimental (wtPTEN-GFP) and control (GFP) plasmids were similar for CML-13, DD-1, and CHAD-G6, measuring 35–50% during the first 6 hours. The transfection efficiency for SB-HSA cells was between 5–15%. Despite the similarity in the initial transfection efficiencies between both plasmids, the frequency of cells expressing the wtPTEN-GFP fusion protein was noticeably lower after 1 day of culture, and most of the transfected cells were small, rounded, and had pyk-notic nuclei suggestive of cells undergoing apoptosis (Fig. 7). By the 2nd day in culture, the frequency of cells expressing the wtPTEN-GFP fusion protein was significantly decreased (P < 0.05) in each of the cell lines, and cells expressing this construct were virtually undetectable by the 3rd day (Fig. 7). The observation that reconstitution of PTEN inhibited growth of PTEN-null cells was predictable. More interesting was the observation that this resulted in growth inhibition or a competitive disadvantage of DD-1 cells, which retained one wild-type allele. The distribution of the fusion protein did not appear to be restricted to the membrane in these cells, as was seen for the endogenous PTEN, although this could be the result of rapid induction of apoptosis in these cells. However, we cannot exclude the possibility that the N-terminal GFP altered the localization of PTEN in these cells. Along with the observation that active (phosphorylated) Akt was present in DD-1 cells deprived of trophic growth factors, this effect to suppress growth or, more likely, survival, supports the conclusion that the endogenous PTEN protein was hypofunctional in DD-1 cells.
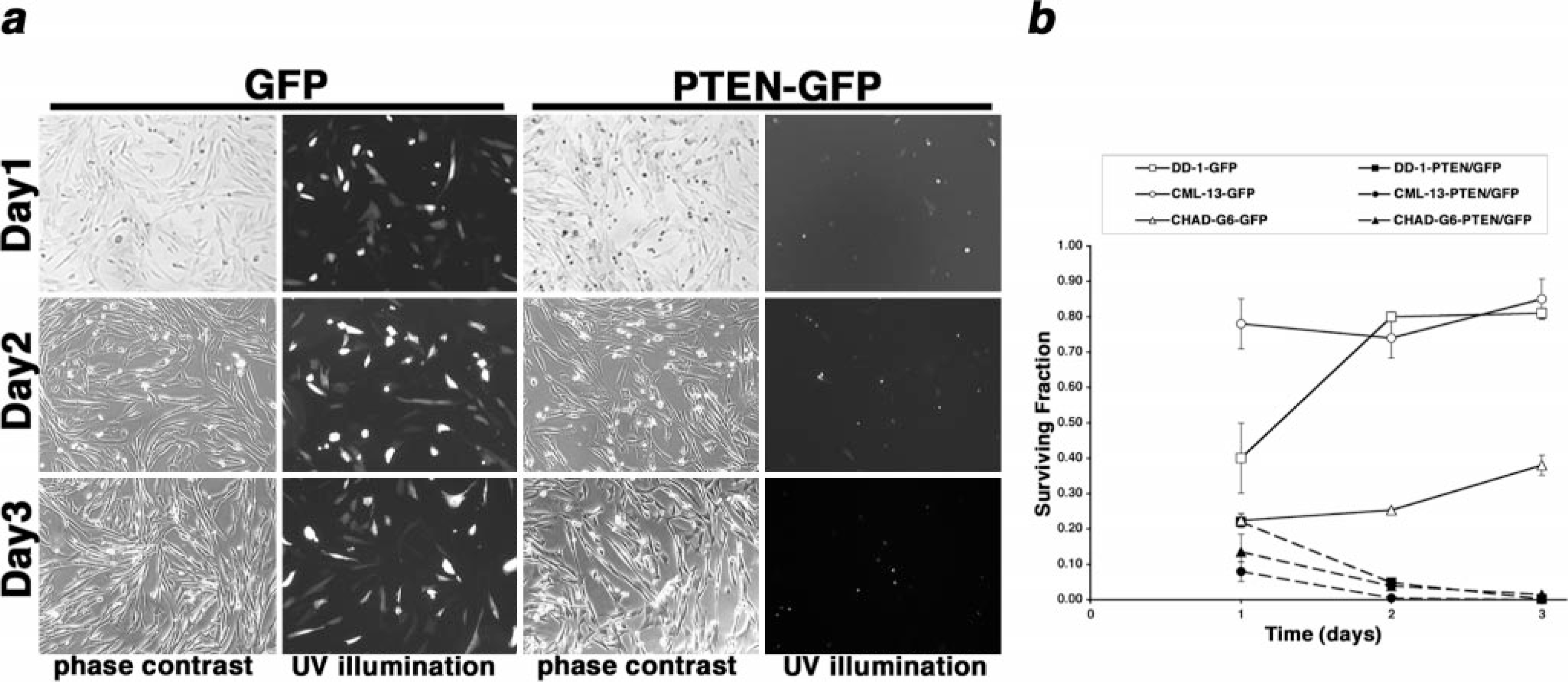
Canine hemangiosarcoma effects of PTEN reconstitution of PTEN-deficient or
PTEN-mutant cells. Constructs encoding GFP or a wtPTEN–GFP fusion protein were
transfected into CML-13 melanoma cells, DD-1 cells, or CHAD-G6 cells using
cationic liposomes.
Discussion
Signals that activate PI3K promote growth and survival of endothelial cells. 18 In normal cells, PI3K-dependent signals are antagonized by the product of the PTEN tumor-suppressor gene. 20 Constitutive activation of PI3K or loss of PTEN function might, therefore, establish autocrine growth loops that promote autonomous growth and transformation of endothelial cells. 6, 22, 45 For this study, we sought to test the hypothesis that PTEN inactivation contributes to the origin and progression of vascular endothelial tumors using naturally occurring canine HSA as a model. 1, 15
PTEN is composed of an N-terminal phosphatase domain (approximately 180 amino acids), a C2 domain (approximately 165 amino acids), and a C-terminal tail (approximately 50 amino acids). Although PTEN mutations in human tumors tend to cluster around the phosphatase domain, mutations have been documented along the entire length of the protein, suggesting that each of the three domains of the protein serve important functions. 26, 31 IHC analysis showed that PTEN expression in tumor sections was variable, but generally it seemed lower in the malignant cells from 12 of 13 cases of HSA than in normal endothelial cells or in endothelial cells from four of five splenic hematomas. The variable PTEN expression in the tumors may reflect the coexistence of cells that retain wild-type PTEN with cells that have different PTEN mutants. However, the antibody cannot distinguish between wild-type and mutant PTEN, making it difficult to ascertain whether any one form was predominant in the tumors. We began to address this by using eight cell lines or early passage cells derived from canine HSA samples as a more homogeneous source to investigate mutations of PTEN. We initially identified mutations clustered within the C-terminal 55 amino acids, and a large truncation (∼80 aa) of the C-terminal domain in five of seven samples where PTEN amplification products were amenable for sequencing. Despite the identification of mutations across the complete coding domains of PTEN in human tumors, the significance of mutations outside the phosphatase domain is unclear; thus, these somewhat unexpected results could be interpreted as preliminary evidence for PTEN-dependent regulation of biochemical pathways that contribute to tumor progression through mechanisms unrelated to its phosphatase activity.
Recruitment of PTEN to the membrane and subsequent activation relies on the phosphatase and C2 domains. 12, 17, 26, 43 However, the mechanisms that control translocation also appear to involve phosphorylation of PTEN on serine and threonine residues in the C-terminal domain (S370, S380, T383, and S385) by casein kinase 2 (CK2). 41, 43 Constitutive phosphorylation of these residues may lead to conformational changes that mask recruitment domains 42 or, alternatively, may interfere with electrostatic interactions required for membrane binding. 12 In addition, the ability of PTEN to inhibit migration of glioblastoma cells requires de-phosphorylation of T383 through intermolecular interactions in trans with other PTEN molecules. 34 This is especially significant in canine HSA, a tumor with high metastatic potential, 5, 10, 37 and particularly in the HSA explanted cells, which we have shown previously are anchorage-independent (grow in soft agar) and have invasive phenotypes when grown in fibronectin-based matrix (Matrigel). 15 Although T383 was retained by all the PTEN mutants except the two with deletions of the full C-terminal tail, structural changes might prevent T383 dephosphorylation. Intriguingly, CHAD-G3 and CHAD-G4.1, which both had S→C amino acid substitutions at residue 380, respectively, failed to immortalize (G3) and showed slow kinetics of growth (G4). 15 CHAD–G6.4 cells, which had a S→F amino acid substitution at residue 380, had no detectable PTEN protein, suggesting this may have affected protein stability in the manner described by Das et al. 12 CHAD–G5 and CHAD–G6.4 represent samples derived from splenic biopsies obtained at different times from the same dog (a diagnostic biopsy and a therapeutic splenectomy obtained 2 months apart). Cells from the first biopsy sample, referred to as CHAD–G5, failed to immortalize even though the tissue was diagnostic for HSA. Cells derived from the second sample, which was isolated after splenectomy, gave rise to the CHAD–G6 cell line. The differences noted between these samples at low passage (Figs. 2–4) suggest that the mutations in the C-terminal domain of PTEN probably represent events that occurred in vivo and that populations with different PTEN abnormalities may indeed coexist within a tumor. In addition, loss of a single PTEN allele with the preservation of the remaining wild-type allele is common in human tumors, suggesting that a single predominant abnormality in this tumor-suppressor gene can contribute to tumor progression. 39 The similarities between the sample from dog No. 10 and the dogs with confirmed HSA are particularly intriguing. This dog had reduced levels of PTEN, p27, and Akt, which made it more akin to the HSA samples than to the other hematoma sample. This could be the result of the presence of a population of malignant endothelial cells within the heterogeneous cell population. The inability of those cells to survive in culture would not be unique because samples from two other dogs with confirmed histological diagnoses of HSA (dog Nos. 3 and 5) similarly did not give rise to immortalized cells in vitro. Follow-up for dog No. 10 at 3 months after diagnosis did not detect any recurrence or metastatic disease. There remained no evidence of hemangiosarcoma when this dog was euthanized due to lumbosacral disease 2 years later.
In the primary tumor from dog No. 2 and the derivative DD-1 cells, constitutive localization of PTEN to the plasma membrane was associated with nuclear localization and constitutive activation of Akt. PTEN in these cells had similar migration to wild-type PTEN in one-dimensional gel electrophoresis, but the presence of a product from the mutant allele could not be detected using immunological methods because the antibody used recognizes an epitope within the carboxyl terminus (amino acids 388–400 of the human sequence). Whereas it could be predicted that membrane localization might decrease PTEN stability, the possible coexistence of wild-type and mutant PTEN product in these cells could explain its persistence. If the mutant PTEN product were similarly localized to the membrane because of the absence of the C-terminal tail, it would be rapidly degraded, which, in turn, could act as a competitive step that prevented degradation of the wild-type PTEN and caused it to “buildup” at the plasma membrane. Alternatively, the deleted or truncated allele may be highly expressed and encode a protein that acts as a dominant, negative repressor of PTEN in these cells, or DD-1 cells could harbor abnormalities in other proteins that regulate the stability and subcellular localization of PTEN, increasing the localization of the wild-type product to the membrane but not its rate of degradation. When we overexpressed a wtPTEN–GFP fusion protein in these cells, the green fluorescent signal was seen largely in the cytoplasmic compartment in the early stage of expression (which is where wild-type PTEN tends to localize), but cells underwent rapid apoptosis thereafter, making it difficult to ascertain whether the PTEN fusion protein might eventually accumulate in the inner leaflet of the plasma membrane. In each cell line tested that had mutant or deleted PTEN, reconstitution of the wild-type protein (by transfection of the wtPTEN–GFP fusion protein) led to a competitive disadvantage when compared with untransfected cells in the cultures. Moreover, the cells attained morphological characteristics reminiscent of apoptosis. In contrast, the observation that cells expressing the GFP protein alone grew unhindered suggests that PTEN reconstitution impeded growth and inhibited survival. Results from preliminary experiments using small hairpin RNA (shRNA) constructs to knock down PTEN in Dal-4 HSA cells and TLM-1 melanoma cells are consistent with data obtained from the cells with spontaneous PTEN mutations. Depletion of PTEN did not appear to produce an obvious growth advantage. Rather, cells showed visible alterations in their morphologic phenotype, becoming round and loosely adherent. These changes do not seem to be the result of anoikis or apoptosis, as nuclear condensation was not evident and the cells retained this morphology over more than three passages in culture. Nevertheless, additional work is required to define specific PTEN domains that might mediate these morphologic changes.
In DD-1, CHAD-G6, and CHAD-G8 cells, as well as in the samples from dog No. 10, reduced expression of PTEN coincided with reduced total levels of p27 and Akt. However, consistent with the absence of mutations in the phospholipid phosphatase domain that regulates PI3K, only DD-1 cells had altered Akt regulation among the cell lines examined, suggesting that the abnormalities in the C-terminal domain of PTEN did not necessarily affect its ability to regulate Akt-dependent signaling. SB-HSA cells, which had no detectable PTEN protein, also had decreased levels of Akt, but they retained p27 expression. This suggests that additional proteins influence the biochemical signals that link PTEN activity with the pathways that maintain expression of p27. In fact, retained expression of p27 may be essential for tumor cell survival. Cell migration of p27-null fibroblasts is decreased when compared with wild-type cells. 2, 3 This appears to be because of an interaction between p27 and the RhoA GTPase, which contributes to formation of actin stress fibers. Hence, in some circumstances p27 expression may actually promote enhanced tumor cell motility and invasion. Our observation that Akt was rapidly induced on restimulation of serum-deprived cells suggests that posttranscriptional mechanisms affecting Akt stability may be similarly important to support endothelial cell growth and survival.
In summary, canine HSA may represent the first naturally occurring, highly metastatic tumor model with mutations of PTEN in the C-terminal domain that may affect the subcellular localization and stability of the protein. Recent evidence has identified novel pathways where proteins, such as PDK-1 and PKN, which are modulated by PTEN, regulate cellular migration. 28 Future experiments to define the precise effect of PTEN mutations on endothelial cell survival, as well as on the canonical, downstream effectors Akt and p27 and on novel effectors, such as RhoA, PDK-1, and PKN, will provide additional insights into the origin or progression of canine HSA.
Footnotes
Acknowledgements
The authors wish to thank owners and veterinarians who contributed cases, and especially Drs. Michelle Ritt and Frank Coons for diligent follow-up information, Dr. Shairaz Baksh for assistance with construct design, Drs. Baksh, Robert Strange, Robert Sclafani, Michelle Ritt, and Robert Weiss for review of the manuscript and helpful discussions. This work was supported in part by grants 1626, 2025, and 2254 from the AKC Canine Health Foundation (to JFM, SCH, MB), 1R55CA86432 from the NCI (to JFM), and a grant from the Monfort Family Foundation (to the University of Colorado Cancer Center).
1Present address: Georgia Tech/IBB, Atlanta, GA.
2Present address: Animal Hospital Center, Highlands Ranch, CO.