
Abstract
End-stage hypertrophic cardiomyopathy (ES-HCM), affecting 5-10% of human hypertrophic cardiomyopathy (HCM) patients, is characterized by relative thinning of the ventricular walls and septum with dilation of the ventricular lumen, decreased fractional shortening, and progression to heart failure. C. J. Baty and others recently documented similar progressive changes to ES-HCM in a family of four cats through serial echocardiograms. At the time of heart failure, these cats exhibited changes similar to those exhibited by human ES-HCM patients. Our objectives were to describe the pathologic alterations associated with ES-HCM and investigate the pathogenesis in three of the four cats. Grossly, there was left atrial dilation with relative thinning of the interventricular septum (IVS) and left ventricular free wall (LVFW). The left atrium contained large thrombi in two of the three cats, and all three cats died following thromboembolization of the aortic bifurcation. Histologically, all three cats had subendocardial and myocardial fibrosis, predominantly of the IVS and LVFW, and one cat had acute, multifocal, myocardial infarcts with mononuclear inflammatory cell infiltrates. The pathogenesis of ES-HCM is uncertain, but theories implicate occlusion of the coronary blood flow by thickening of the coronary vessels, coronary vascular thromboembolism or coronary vessel spasm, apoptosis of myocytes, and myocardial hypertrophy beyond the ability of the vasculature to supply blood. Apoptosis assays did not reveal any apoptotic myocytes. Considering the hypercoagulative state of these cats, coronary vascular thromboembolism could be a major contributing factor. We cannot exclude apoptosis or coronary vessel spasm on the basis of the data presented.
HCM is a relatively common cardiac disease seen in many species, including cats and humans. The disease is characterized by ventricular hypertrophy that is not attributable to other cardiac or vascular disease, including systemic hypertension. It is a heterogeneous disease with many phenotypes, including the dilated phase or end stage, which we propose to call end-stage hypertrophic cardiomyopathy (ES-HCM). The changes defining ES-HCM are relative dilation of the ventricular chamber, relative thinning of the ventricular wall, and decreased ventricular contractility manifested as a decrease in fractional shortening, usually resulting in marked left atrial dilation. 7, 12, 22, 27, 33
Descriptions of the histologic lesions of ES-HCM in human patients refer to myocardial scarring, which is thought to be the result of myocardial infarction. 12, 22, 24 The myocardial scars are variably sized, focally extensive regions of mature fibrous tissue within the myocardium that replace myocytes. 22 Their location is highly variable and can be subendocardial, sub-epicardial, or transmural and focal or multifocal. 21 The areas of fibrosis more commonly occur in the ventricular septum and anterior wall, although they can occur anywhere in the ventricular myocardium. 12, 21
The cause of ES-HCM is not known. Data gathered by medical researchers of HCM suggest several mechanisms for myocardial infarction. The most widely accepted mechanism is myocardial ischemia as a result of coronary vessel hypertrophy with resultant luminal narrowing. 22 Supporting this theory is the close association of myocardial scarring with abnormal coronary vessels. 12, 27 Coronary vessel spasm, increase of cardiac muscle mass beyond the ability of the vasculature to supply blood, and thromboembolism of the coronary arteries have also been suggested. 3 Ino et al. 11 have recently suggested apoptosis as a primary cause of myocardial loss and subsequent fibrosis. Several recent papers suggest mutations in genes encoding various sarcomeric proteins and mitochondrial genes as underlying causes of ES-HCM. 5, 6, 14, 29
The outcome of HCM in humans and cats is highly variable, but human and feline HCM patients often survive for many years. However, the development of ES-HCM is often closely followed by congestive heart failure and, thus, is associated with a poor prognosis. 22 Approximately 10–15% of human patients develop ES-HCM, which typically occurs in midlife, although it has been described in children. 12, 22 Diagnosis of ES-HCM is based on serial echocardiograms in which progression from a thickened ventricular wall with a narrowed ventricular lumen to a relatively thin wall and dilated lumen and a decrease in myocardial contractility indices (e.g., fractional shortening) can be documented. 27
ES-HCM has long been suspected to occur in feline HCM patients, but this manifestation has only recently been documented in cats by Baty et al. 2 and thus has not been described in cats. The purpose of this paper is to describe and document the gross and histologic changes associated with ES-HCM in these cats, to discuss the possible pathologic mechanisms underlying these changes, and to compare these changes to those seen in cases of feline primary restrictive cardiomyopathy.
Materials and Methods
Animals
Three sibling domestic shorthair cats, a female (cat No. 2) and two males (cat Nos. 3 and 4), were examined. The cats were queened by a stray cat (cat No. 1) shortly after her adoption. All four cats were diagnosed at a young age with hypertrophic cardiomyopathy at North Carolina State University's Veterinary Teaching Hospital by echocardiography (Table 1).
Chronologic progression of echocardiographic measurements in this family of cats.∗
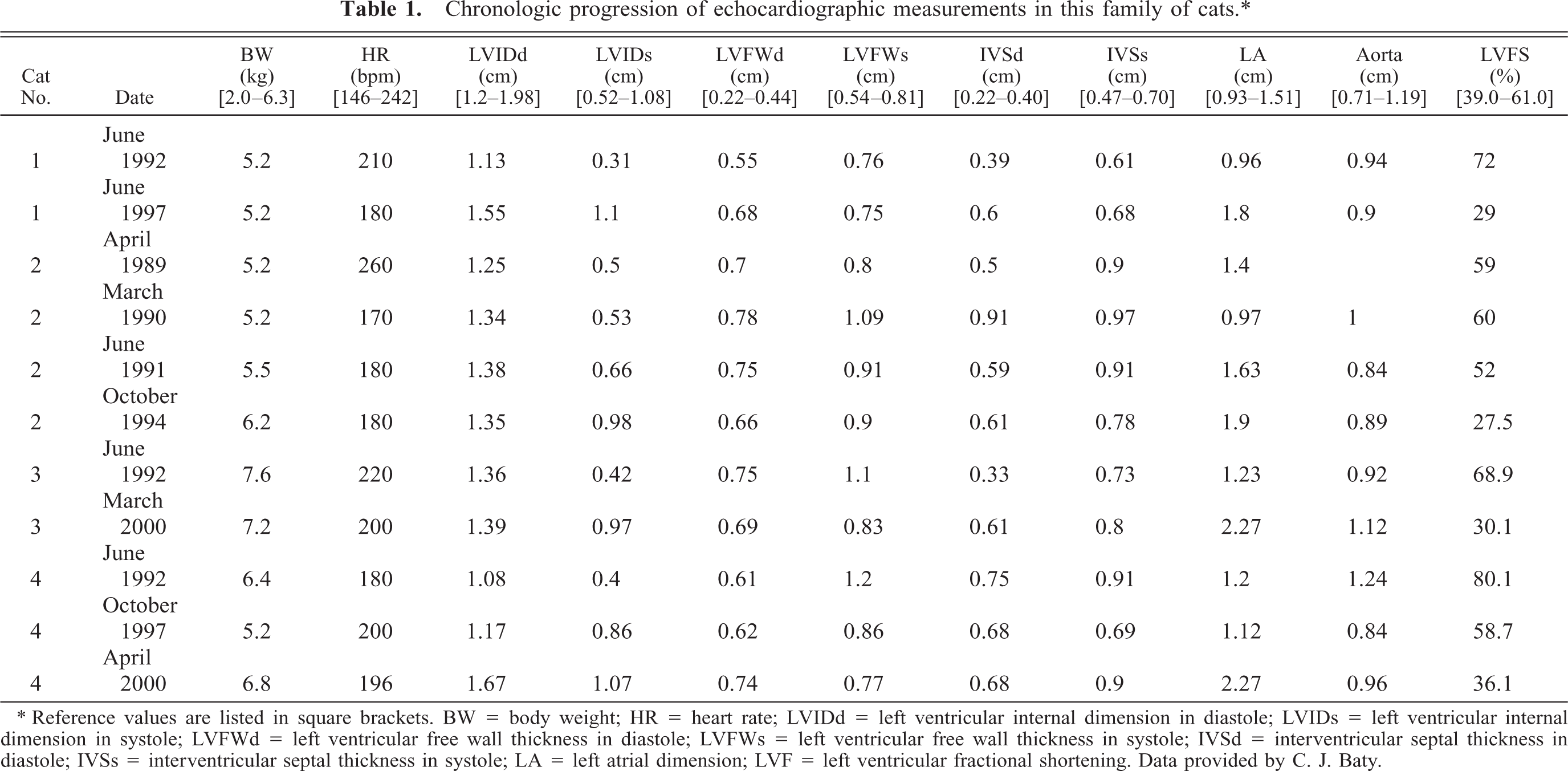
∗ Reference values are listed in square brackets. BW = body weight; HR = heart rate; LVIDd = left ventricular internal dimension in diastole; LVIDs = left ventricular internal dimension in systole; LVFWd = left ventricular free wall thickness in diastole; LVFWs = left ventricular free wall thickness in systole; IVSd = interventricular septal thickness in diastole; IVSs = interventricular septal thickness in systole; LA = left atrial dimension; LVF = left ventricular fractional shortening. Data provided by C. J. Baty.
Postmortem histological examination
A full necropsy was performed on cat Nos. 2 and 4. A postmortem examination of the heart only was performed on cat No. 3, and no postmortem examination was performed on cat No. 1. The entire heart and representative samples of other major organs from cat Nos. 2 and 4 and the heart only from cat No. 3 were fixed in 10% neutral buffered formalin. The tissues were routinely processed for paraffin embedding, and 5-μm sections were stained with hematoxylin and eosin. Eleven transverse sections of the heart from cat No. 4 from the following locations were examined: the left ventricular free wall (LVFW) 1 and 1.5 cm below the mitral valve (section from 1.5 cm below the mitral valve includes the papillary muscle) and 1 cm above the heart apex, the interventricular septum (IVS) 1 and 1.5 cm below the mitral valve, the right ventricular free wall (RVFW) 1 cm below the tricuspid valve, the junction of the LVFW and the IVS 1 cm below the mitral valve, the interatrial septum, and the left and right atrial walls. Two sections of left ventricle and one section each from the right ventricle and IVS from cat No. 3 and one section each from the left ventricle and IVS from cat No. 2 were evaluated. Additionally, 5-μm sections of the LVFW, RVFW, and IVS from cat Nos. 3 and 4 were stained with Gomori's trichrome for evaluation of myocardial fibrosis.
TUNEL (CardioTACS) protocol
Paraffin-embedded tissue was sectioned at 5 μm, floated onto slides, and air dried. Sections were deparaffinized in xylene and a decreasing ethanol series to 1× phosphate-buffered saline (PBS). Apoptosis was evaluated with the CardioTACS (R&D Systems, cat No. TA5353 lot No. 4078F1) apoptosis detection kit. Briefly, sections were incubated with 2% proteinase K in dH2O for 15 minutes at 25°C, rinsed in dH2O, then treated with 3% H2O2 in methanol for 5 minutes, rinsed, and immersed in TdT Labeling Buffer for 5 minutes at 25°C. Sections were treated with Labeling Reaction Mix (TdT dNTP Mix, Mn2+, TdT enzyme, TdT Labeling Buffer) for 1 hour at 37°C in a humid chamber. Sections were immersed in TdT Stop Buffer, rinsed, and treated with 0.1% Streptavidin-HRP for 10 minutes at 25°C, rinsed, and treated with TACS Blue Label. Sections were rinsed in dH2O, counterstained with Nuclear Fast Red, rinsed in dH2O, coverslipped, and photographed. Positive controls (sections of heart from a cat with no clinical, gross, or histologic signs of heart disease) were pretreated with nuclease solution for 10 minutes at 37°C, and negative controls were treated with Reaction Mix lacking TdT enzyme.
IHC for active caspase-3
Paraffin-embedded tissue was sectioned at 5 μm, floated onto slides, and air dried. Sections were deparaffinized in xylene and a decreasing ethanol series to 1× PBS. Sections were permeabilized with 0.1% saponin buffer for 10 minutes at 25°C, then treated with peroxide block for 5 minutes at 25°C, rinsed, and treated with goat serum block for 20 minutes at 25°C. Sections were incubated overnight at 4°C in rabbit polyclonal antibody to the p17-kd active subunit of caspase-3 (Promega No. G-748A lot No. 11551302). Sections were rinsed and treated with Biogenex kit components Multilink (biotinylated anti-rabbit IgG) then Label (peroxidase-conjugated streptavidin). Sections were rinsed and exposed to chromiate solution for 5 minutes, then rinsed, counterstained with hematoxylin, coverslipped, and photographed.
Results
Case history
At 20 months of age, cat No. 2 was diagnosed with asymptomatic HCM via echocardiography after auscultation of a grade II/VI heart murmur. Cat No. 4 was diagnosed with asymptomatic HCM at 4 years of age after auscultation of a grade II–III/VI heart murmur prior to routine dental prophylaxis, also via echocardiography. After diagnosis of HCM in the second offspring, the other two cats (cat Nos. 1 and 3) were screened (via echocardiography) for HCM. Cat No. 1 was borderline for HCM and cat No. 3 was diagnosed with asymptomatic HCM. Serum T4 concentrations were within normal limits in all four cats. Systolic arterial blood pressure was elevated in cat No. 3 (170 mm Hg) at the time that HCM was diagnosed, but this was attributed to anxiety secondary to the examination.
Follow-up examinations, including echocardiograms, were performed several times during their lives. All four cats eventually developed signs consistent with aortic thromboembolism (ATE) and died or were euthanized—cat 1 at 5 years after initial diagnosis (age unknown), cat 2 at 8 years of age, cat 3 at 13 years of age, and cat 4 at 14 years of age. At the time of death, cat 1 had signs of congestive heart failure and cat 3 was in renal failure (ATE at the level of the renal arteries was suspected). Echocardiograms performed just before death in cat Nos. 2 and 3 revealed relative left ventricular chamber dilation (compared with measurements obtained earlier in the course of disease), relative LVFW or IVS or both, left atrial chamber dilation, and a marked decrease in left ventricular fractional shortening. Just before death, cat No. 1 was found to have decreased left ventricular fractional shortening, left ventricular chamber dilation, and left atrial dilation. An echocardiogram was not obtained just before death for cat No. 4. The echocardiographic measurements are presented in Table 1.
Postmortem and histologic examination
The gross lesions in cat No. 4 consisted of a markedly dilated left atrium with intra-auricular thrombi. The thicknesses of the IVS and LVFW were variable, measuring from 4 to 6 mm and 2 (apex) to 7 mm, respectively (Fig. 1). Multifocal foci of pallor were apparent in the subendocardial myocardium and typically coincided with areas of LVFW and IVS thinning. The heart weight was 31.34 g, and heart weight in relation to body weight was 5.7 g/kg (normal = 4.8 ± 0.1 g/kg 19 ). The pericardial sac contained approximately 5 ml of serosanguinous fluid. Within the right proximal ileac artery was a 4–5-mm-long thrombus lodged approximately 3 cm distal to the aortic bifurcation. An area of roughening was apparent on the intimal surface of the left proximal ileac artery extending from approximately 0.5 cm distal to the aortic bifurcation to approximately 2.5 cm distal to the bifurcation. The liver and lungs were moderately congested, and the liver had a mildly enhanced lobular pattern. There was severe atrophy and, histologically, chronic interstitial nephritis of the right kidney, which measured 2.5 cm in length. The left kidney, measuring 4.7 cm, was mildly enlarged, which was interpreted as compensatory hypertrophy.
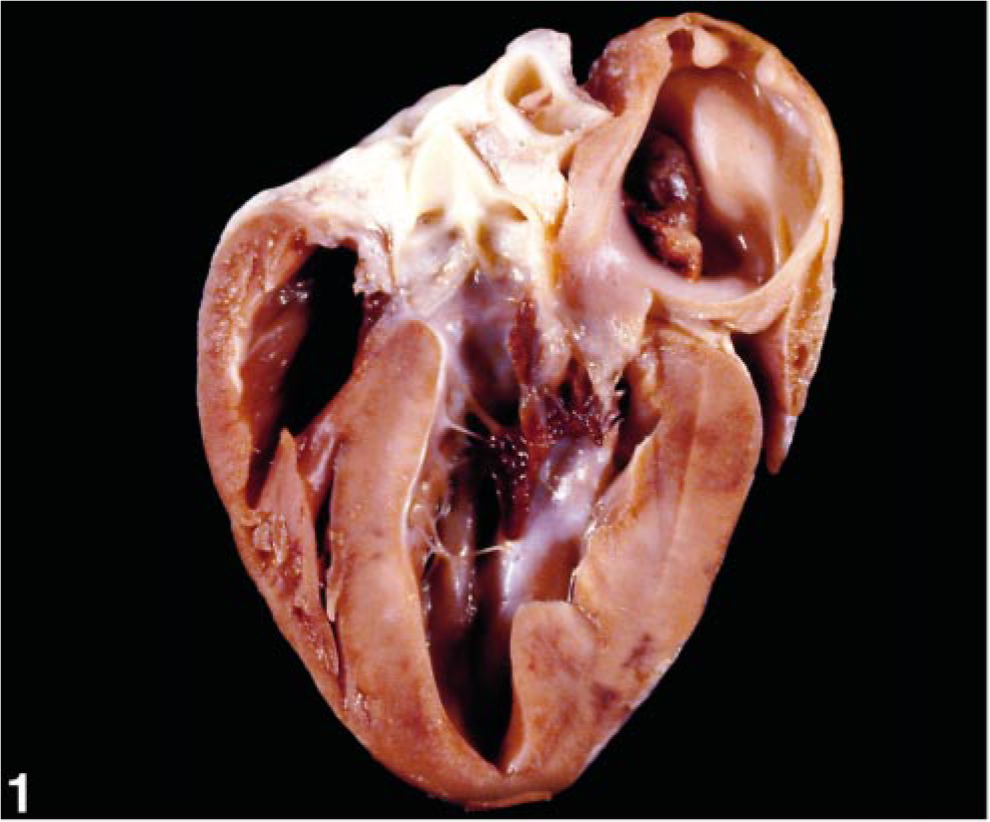
Heart; cat No. 4. The ventricular walls are very mildly hypertrophied (LVFW, 7 mm maximally; IVS, 4–6 mm). There is a thrombus in the left atrium.
The gross cardiac lesions from cat No. 2 were similar and included left atrial thrombosis and thromboembolization at the aortic bifurcation. The heart was moderately enlarged, weighing 28 g, and the heart weight in relation to body weight was 4.3 g/kg (normal = 4.8 ± 0.1 g/kg 19 ), although the cat was obese. The LVFW and IVS measured 9 mm thick. There was an acute infarct in the cortex of the left kidney.
On postmortem examination of the heart from cat No. 3, the LVFW had a maximal thickness of 0.8 cm, the IVS measured 0.6 cm in thickness maximally, and the left atrium was 2.4 cm in diameter. Although there was clinical evidence of aortic thromboembolization, an atrial thrombus was not identified.
Histologic examination revealed similar changes in all cats examined, with varying degrees of chronicity and severity. In all three cats, large, multifocal areas of mature fibrosis were within the myocardium and endocardium Figs. 2–6). There was mild to moderate anisokaryosis of the remaining myocytes, with many mildly to moderately enlarged round nuclei and there were scattered Anitschkow cells. There were occasional binucleated myocytes, and some of the myocytes had variably sized, clear, cytoplasmic vacuoles. Disorganization of the myofibers was moderate to marked, with disarray apparent in approximately 10–25% of longitudinally oriented fibers in the sections of IVS examined and, to a lesser extent, in the LVFW (Fig. 5). The disarray in cat No. 4 was most abundant in the IVS 1 cm and 1.5 cm below the mitral valve. Occasional coronary arteries in cat Nos. 2 and 3 had mildly thickened, often hyalinized tunica media with mild luminal stenosis, whereas those of cat No. 4 appeared normal. The sections examined from all three cats had large regions of fibrosis, with the most fibrosis in cat No. 4. Cat No. 4 had the most severe histopathologic changes characterized by extensive, multifocal, coalescing areas of edema and fibrosis within the myocardium (comprising >50% of some sections of LVFW), sometimes surrounding islands of necrotic myocytes (myocardial infarcts; Figs. 2, 3). The infarcts were more extensive in the LVFW compared with the IVS and were occasionally noted in the right ventricle. Scattered lymphocytes, plasma cells, and macrophages infiltrated the areas of fibrosis around the infarcts, which had projections of fibrous tissue dissecting between myofibers extending away from the infarcts. Occasional necrotic myofibers with loss of striations were at the edges of these areas. Occasionally there were also large regions of mature fibrosis (scars), particularly in subendocardial regions and extensive regions of the endocardium, including around moderator bands (Figs. 4, 5). In the regions surrounding the infarcts and fibrosis were occasional scattered shrunken cells with pyknotic nuclei consistent with nonviable myocytes. These results are summarized in Table 2.
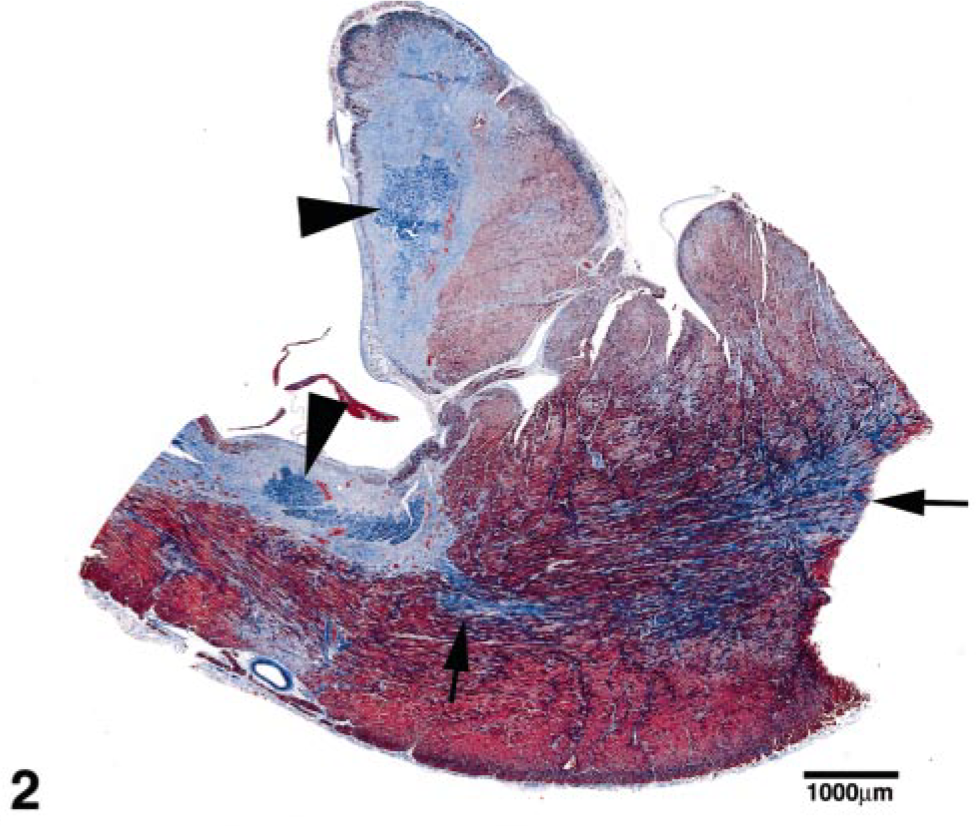
Left ventricular free wall with papillary muscle; cat No. 4. Multiple myocardial infarcts (arrowheads) are surrounded by immature fibrosis and regions of mature fibrosis (myocardial scars, arrows). Gomori's Trichrome stain. Bar = 1,000 μm.
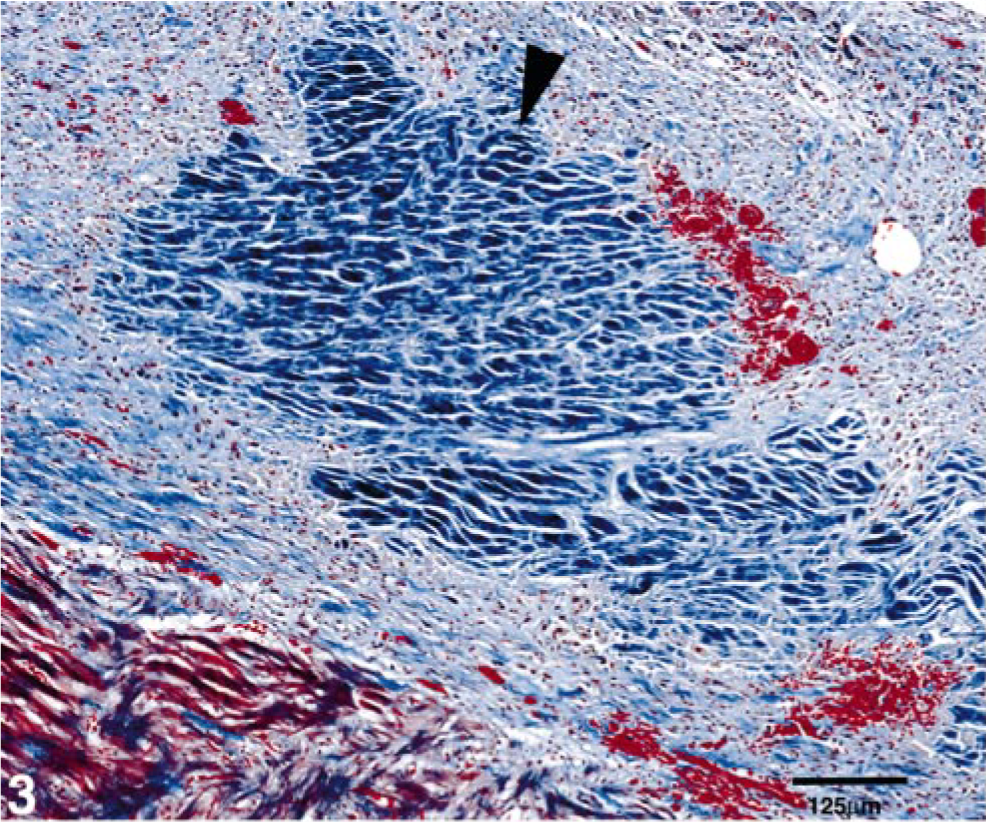
Left ventricular free wall; cat No. 4. Higher magnification of one of the myocardial infarcts in Fig. 2 (arrowhead). Gomori's Trichrome stain. Bar = 125 μm.
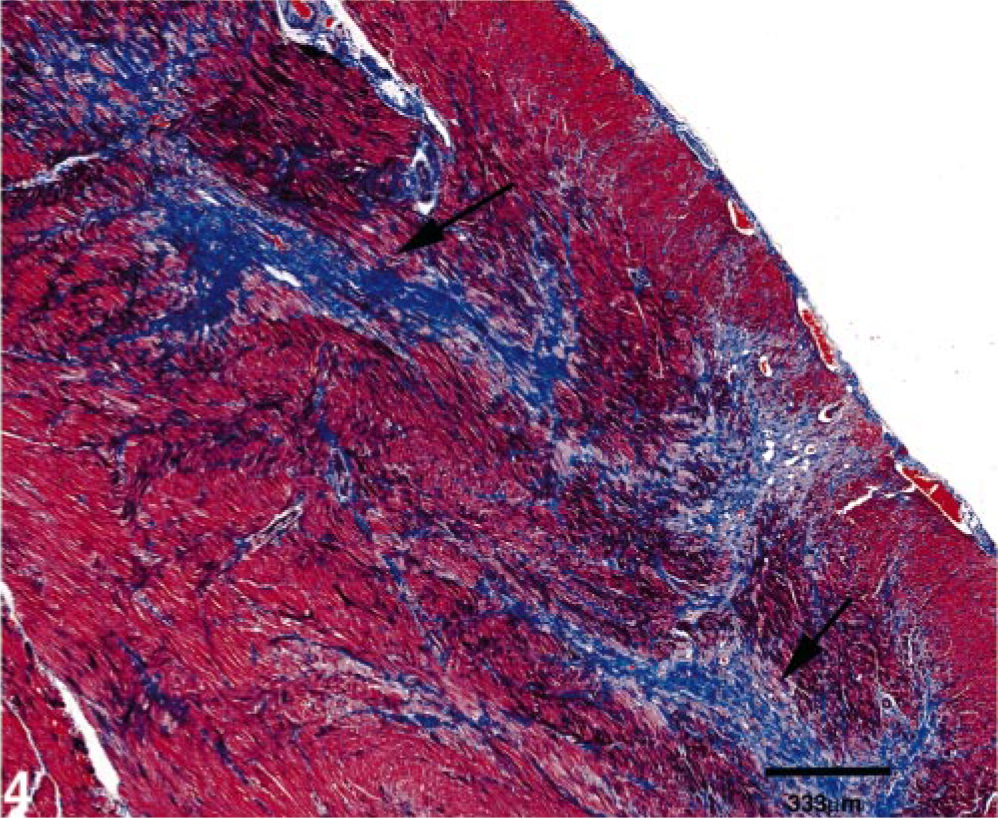
Left ventricular free wall; cat No. 4. Higher magnification of the myocardial scars in Fig. 2 (arrows). Gomori's Trichrome stain. Bar = 339 μm.
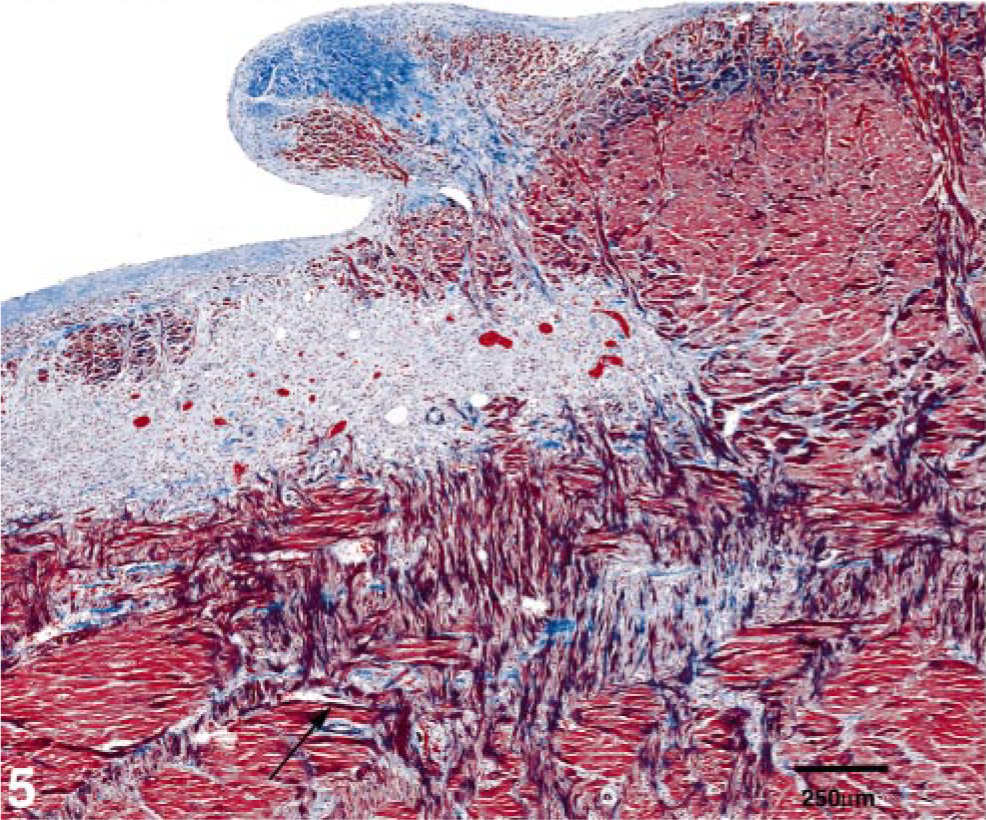
Left ventricular free wall with papillary muscle; cat No. 4. Myofiber disarray (arrow) and mature fibrosis within the papillary muscle (top of photo) and endocardium. A region of less mature fibrosis also appears in the subendocardial region. Gomori's Trichrome stain. Bar = 250 μm.
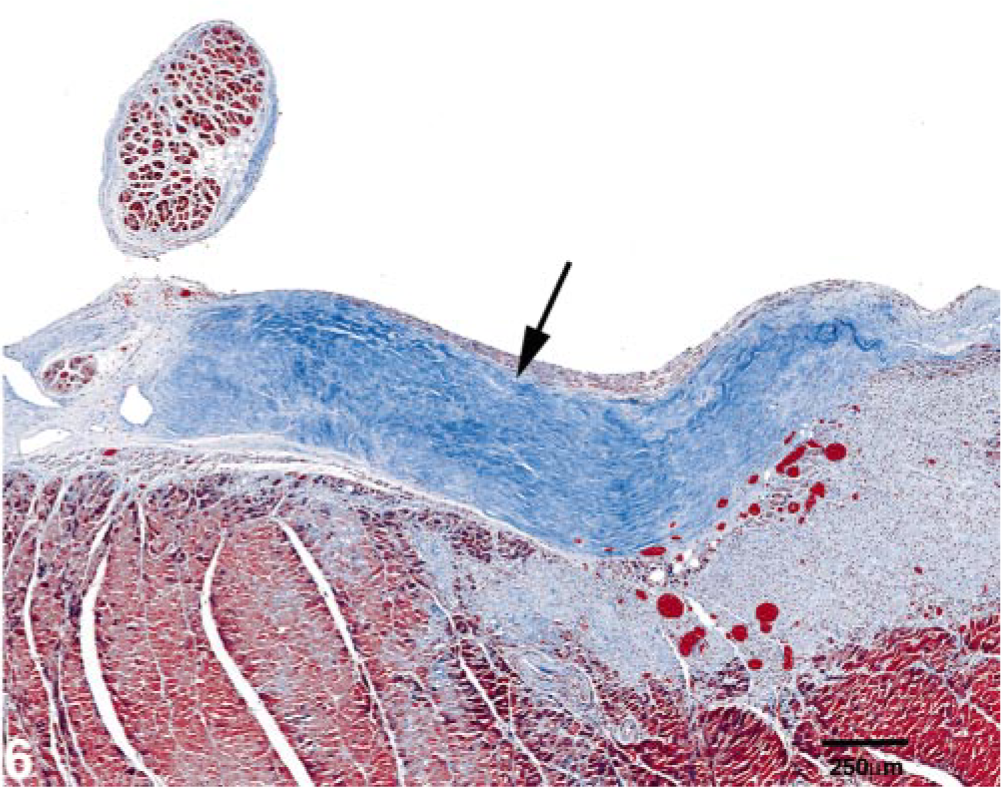
Interventricular septum; cat No. 4. There is a region of mature endocardial and subendocardial fibrosis (arrow) and a subjacent region of less mature fibrosis with scattered lymphocytes, plasma cells, macrophages, and blood vessels. Gomori's Trichrome stain. Bar = 250 μm.
Chronologic progression of disease with gross and histologic findings in the three cats examined.∗
∗ nd = data not available; HW = heart weight; LVFW = left ventricular free wall; IVS = interventricular septum.
Apoptosis assays
TUNEL assays for fragmented DNA and IHC for activated caspase-3 did not reveal any apoptotic cells within the sections examined. The results of positive and negative controls were as expected.
Discussion
To our knowledge, this is the first description of the gross and histopathologic changes associated with ES-HCM in cats, a terminal stage of HCM. The lesions indicative of ES-HCM in these cats include loss of myocytes with replacement fibrosis (scarring) associated with relative thinning of previously hypertrophied left ventricular and intraventricular myocardial walls, relative dilation of a previously narrowed left ventricular lumen, and a sudden increase in left atrial size. These lesions are often accompanied by left atrial thrombosis, ATE, and regionally extensive endocardial fibrosis. The gross and histologic changes in these cats are similar to those described in humans with ES-HCM. 12, 21, 22 Likewise, the rapid development of cardiac failure following diagnosis of ES-HCM in these cats (see Table 2) is similar to that seen in human patients. 7, 10, 22 The progression to ES-HCM is poorly understood and could be related to coronary vessel occlusion (via intimal and medial hypertrophy, thromboembolism, or arteriospasm), increased cardiac muscle mass beyond the ability of the vasculature to provide blood flow, apoptosis, hereditary genetic mutations, or a combination of these factors.
It must be emphasized that the myocardial wall thinning and ventricular chamber dilation associated with ES-HCM in both human and feline HCM patients are relative changes. During the course of HCM, the myocardial walls thicken and, as a result, the ventricular chambers become narrowed. Detection of ventricular wall thinning and ventricular chamber dilation requires that at least one clinical measurement of the ventricular walls be made at an earlier stage of the disease, most commonly by echocardiography. The postmortem measurements can then be compared to the ante-mortem measurements, thus documenting the relative thinning of the ventricular walls and dilation of the ventricular chamber(s). With ES-HCM, the myocardial walls become thinner and the ventricular chambers dilate relative to previous measurements but might or might not reach the dimensions present before onset of the asymptomatic phase of the disease (i.e., “normal dimensions”). For example, the echocardiographic systolic measurement of the IVS was 9 mm 11 months before death in cat No. 4, 8 mm on March 2000 in cat No. 3, and 9.1 mm 3 years before death in cat No. 2—all above normal range by echocardiography (Table 1). At the time of necropsy, the IVS dimensions in two of three cats were decreased to normal thickness and reduced in the third cat, indicating that the IVS decreased in thickness during the later portions of the disease process. The postmortem IVS dimension in cat No. 2 was 9 mm, which is mildly thickened relative to normal, and was 6 mm in cat Nos. 3 and 4, considered to be within normal limits (Table 2). The sudden increase in left atrial size is associated with a marked decrease in myocardial contractility manifested as a decrease in myocardial contractility indices such as fractional shortening. Detection of decreased myocardial contractility requires serial echocardiographic measurements (for a summary of serial echocardiographic measurements in these cats, see Table 1).
A hallmark histologic feature of feline ES-HCM, as in human ES-HCM, is multifocal myocardial scarring (large regions of replacement fibrosis) for which myocardial infarction is considered the most likely cause. 12, 22, 27 Many mechanisms have been proposed for myocardial infarction in human HCM patients, including increased oxygen demand with decreased supply because of narrowing of the intramural coronary arteries related to atherosclerosis, coronary vessel spasm, intimal or medial hypertrophy of intramural coronary vessels, coronary vessel thromboembolism, or an increase in cardiac muscle mass beyond the ability of the coronary vessels to provide adequate blood flow. 3, 21 This last mechanism is far less likely as a primary cause because the echocardiographically measured IVS and LVFW dimensions are only moderately elevated and therefore would not be expected to impair the coronary vessels' ability to supply blood, although it might play a contributory role. Coronary vessel spasm has also been suggested as a cause of myocardial infarction in humans, but such a mechanism is difficult to demonstrate or disprove. 7, 21, 24
Intimal and medial hypertrophy of the coronary vessels has been described in feline HCM patients, although it is more common in humans with HCM. 19 These abnormal coronary vessels typically have narrowed lumina and have been implicated as a cause of myocardial infarction. 3, 21, 22 These abnormal vessels are often more numerous in areas of fibrosis, suggesting a relationship between the two, although the nature of that relationship has not been identified. 12 Maron and Spirito 22 have proposed a mechanism for ES-HCM in human HCM patients in which the abnormal coronary vessels result in alterations of blood flow leading to myocardial ischemia, which causes myocardial cell death and subsequent replacement fibrosis. In this chain of events, the myocardial infarction and fibrosis is the cause of IVS and LVFW thinning. The wall thinning results in left ventricular remodeling leading to systolic dysfunction (i.e., decreased fractional shortening). 22 This theory is supported in this study by the two siblings with the most severe LVFW thinning who also had the most severe fibrosis and disarray and by the coincidence of the infarcts with the areas of wall thinning in these cats. However, cases of end-stage remodeling without intramural coronary arterial changes have been reported in human HCM patients. 21 In fact, Varnava et al. 32 have been unable to establish a relationship between fibrosis and coronary arterial disease and suggest that myocardial disarray predisposes to ischemia. Iida et al. 10 suggest that myocardial disarray might be related to the myocardial fibrosis. In this study, the coronary vessels examined in cat Nos. 2 and 3 were rarely thickened, and those in cat No. 4 were within normal limits, suggesting another cause for the fibrosis. Additionally, myofiber disarray was observed in all three cats examined, suggesting a relationship between disarray and fibrosis.
Although no thrombi were seen in the coronary vessels, thromboembolism is considered a possible cause of myocardial infarction resulting in ES-HCM, particularly considering the presence of atrial and aortic thrombi, which indicate a propensity for thromboembolization in these cats. In fact, thrombosis might be more common in ES-HCM patients (human and feline) than in those with HCM. All four cats in this family were either confirmed to have ATE (n = 2) or have clinical signs consistent with ATE (n = 2). Two cats also had left atrial thrombosis. In a previous study of feline HCM, ATE was found to occur at a rate of 39% (9 of 21 cats). 4 In one human study, four of seven patients with ES-HCM had thrombosis (57%) compared with no detectable thrombosis in HCM patients. 12 An alternative, and perhaps more attractive scenario, is that ES-HCM (i.e., ventricular wall thinning, decreased myocardial contractility, and left atrial enlargement) predisposes to the formation of left atrial thrombosis, with coronary vascular thromboembolism and the resultant myocardial infarcts being a secondary chronic “embolic showering” event over time. In cat Nos. 2 and 3, the maturity of the fibrotic regions and the lack of acute infarcts suggest that the fibrosis occurred some time before ATE. Also, these cats had ventricular wall thinning, chamber dilation, and decreased systolic function (as shown by the decreased left ventricular fractional shortening) in the absence of acute myocardial infarction. In cat No. 4, the concurrent presence of mature scars and acute myocardial infarcts suggests that some scarring occurred before development of the infarcts. Likewise, the development of ES-HCM in human HCM patients is thought to be a slow process in many cases, and thromboemboli are rarely seen in association with the myocardial scars. 21, 22 It is therefore reasonable to assume that coronary vessel thromboembolism caused the myocardial infarcts in cat No. 4, which contributed to congestive heart failure and death, but that it is a secondary event and not solely responsible for the wall thinning and chamber dilation.
Ino et al. have suggested that myocyte apoptosis might play a role in end-stage wall thinning in humans on the basis of apoptosis assays performed on myocardial tissue from one patient with ES-HCM. 11 Additionally, the morphology of occasional nonviable scattered myocytes in these cats was suggestive of apoptosis on light microscopic examination. Therefore, two assays for apoptosis were performed on paraffin-embedded tissue from cat Nos. 3 and 4. These assays failed to reveal any apoptotic cells. This negative finding does not entirely exclude apoptotic myocyte loss in these cats. Apoptosis could have occurred earlier in the course of the disease or at a frequency below the detection limits of the assays.
Given the clinical and postmortem similarities in all four cases and the familial relationship between the cats, it is likely that genetics played a major role in the development of ES-HCM in these cats. Familial HCM in humans and Maine Coon cats is inherited as an autosomal dominant trait. 13, 20, 25 To date, over 125 mutations in at least nine genes encoding sarcomeric proteins (β-myosin heavy chain, α-tropomyosin, troponin I, troponin T, myosin light chains, myosin binding protein C, α-cardiac actin, and titin) have been associated with familial HCM in humans. 8, 20, 25, 30, 31 Recently, it has been proposed that specific mutations in genes encoding sarcomeric proteins are associated with the ES-HCM phenotype: two in the gene encoding cardiac troponin T, one in the gene encoding cardiac troponin I, one in the gene encoding myosin binding protein C, and one in the mitochondrial gene encoding tRNALys. 5, 6, 14, 15, 29 Although highly suspected, a specific genetic mutation has not been associated with HCM in cats; however, Meurs et al. 23 have found that levels of myomesin, another sarcomeric protein, are reduced in Maine Coon cats with familial HCM. In these same cats, there is an alteration in the β-myosin heavy chain. 23 No genetic analyses were performed, so we cannot confirm the role of genetic mutations on the phenotypic outcome in these three cats.
The numerous gross and histologic similarities between ES-HCM and primary or idiopathic restrictive cardiomyopathy (RCM) and, often, a failure to diagnose cats with HCM before the onset of heart failure suggest that some cases of RCM could, in fact, be misdiagnosed cases of ES-HCM. Primary or idiopathic RCM is defined as heart muscle disease resulting in decreased cardiac compliance and impaired ventricular filling of unknown cause. 16 Cardiac contractility is usually normal, at least early in the disease. 16 It is the rarest of the cardiomyopathies and few comprehensive published reports describe the histological findings in cats. Sisson and Thomas 26 describe interstitial and subendocardial fibrosis in hearts with no or mild myocardial hypertrophy. Liu et al. 17 describe a form of restrictive cardiomyopathy in cats in which excessive moderator bands restrict ventricular expansion with subendocardial fibroplasia. Some of these cats had thinner than normal ventricular walls, with edema and fibrous tissue separating myofibers, but they lacked extensive myocardial scars. 17 The cats in our study had normal numbers of moderator bands. The lesions in the cats also resembled the lesions of feline endomyocarditis/left ventricular endocardial fibrosis (another form of restrictive cardiomyopathy), as described by Stalis et al., 28 in which interstitial endocardial and subendocardial fibrosis was present with or without infiltrates of inflammatory cells. The hearts were typically enlarged, and no infarcts or myofiber disarray were described. 28 However, this does not preclude the possibility that endomyocarditis/left ventricular endocardial fibrosis and ES-HCM represent the same disease because not all cases of HCM exhibit myofiber disarray. In one study, only 30% of hearts from cats diagnosed with HCM exhibited myofiber disarray. 18 Additionally, the fibrosis/inflammation seen in endomyocarditis/left ventricular endocardial fibrosis might represent an intermediate stage of scarring in which the necrotic myocytes have been resorbed but the lesion has not yet progressed to mature fibrosis. Two reviews of RCM in humans describe some degree of fibrosis in all cases. 1, 9 They also describe mild LVFW or IVS thickening or both, mild myocyte hypertrophy, and myofiber disarray in some, but not all, of the cases. 1, 9 These findings are also common in ES-HCM (a comparison of the gross and histologic findings of RCM and ES-HCM is presented in Table 3).
Comparison of pathologic findings in RCM and ES-HCM.∗

∗ ES-HCM = end-stage hypertrophic cardiomyopathy; RCM = restrictive cardiomyopathy; N = normal; ↑ = increased; ↓ = decreased.
ES-HCM is an uncommon manifestation of HCM in cats and humans and is associated with a poor prognosis. The wall thinning seen in these cats and in the majority of human patients with ES-HCM is considered to be caused by myocardial infarction and/or ischemia and resultant fibrosis (scarring). The cause of the ischemia and infarction is unknown but is likely multifactorial, related to abnormalities in coronary vessels, coronary vessel thromboembolism, coronary vessel spasm, apoptosis of myocytes, or phenotypic expression of genetic mutations. The lack of significant numbers of abnormal intramural coronary vessels and apoptotic cells suggests that these play a minor role or are not involved in the disease process. The high occurrence of atrial thrombosis and ATE in these cases suggests that thrombosis of coronary vessels is likely a major contributing factor, the effects of which could be exacerbated by the metabolic demands of the hypertrophied myocardium. On the basis of recent findings in humans and some cats, genetic mutations might underly many of the phenotypic features of ES-HCM. The similarities in the gross and histologic appearance between ES-HCM and RCM suggest that some cases of RCM are misdiagnosed cases of ES-HCM. Because the development of ES-HCM is a dynamic process, definitive diagnosis cannot be made solely on the basis of postmortem findings; at least one antemortem echocardiogram is required to document the ventricular wall thinning and ventricular chamber dilation, and preferably two antemortem echocardiograms are needed to document the decrease in myocardial contractility. One might, however, suspect ES-HCM in cats dying of congestive heart failure if there is extensive myocardial or subendocardial scarring, or both, especially in cases complicated by ATE.
Footnotes
Acknowledgements
We thank Gautam Ghatnekar for performing the TUNEL and activated caspase-3 staining and for his help in interpreting them. The expertise of Sandra Horton and Monica Mattmuller in tissue processing and histologic preparation is also gratefully acknowledged.