
Abstract
We evaluated gene expression and antimicrobial responses of bovine monocyte—derived macrophages incubated with Mycobacterium avium subsp. paratuberculosis (M. a. ptb), the causative agent of Johne's disease. Gene expression was evaluated by the use of human noncompetitive high-density oligonucleotide microarrays. Bovine messenger RNA hybridized with 14.2–18.2% of the 12,600 oligonucleotide probe sets. When macrophages incubated with M. a. ptb were compared with nonactivated control macrophages, macrophages activated by addition of interferon-γ and lipopolysaccharide, and macrophages incubated with Mycobacterium avium subspecies avium (M. a. a), 47, 79, and 27 genes, respectively, were differentially expressed. Differential expression of six of these genes was confirmed using reverse transcriptase polymerase chain reaction. Several functional assays were performed to evaluate the potential relevance of differentially expressed genes to host defense. Macrophages phagocytizing M. a. a had a greater capacity to kill the organisms and to acidify phagosomes and a greater degree of apoptosis than did macrophages incubated with M. a. ptb. The results of these studies indicate that multiple genes and metabolic pathways are differentially expressed by macrophages ingesting mycobacterial organisms. Although the intracellular fate of mycobacterial organisms appears to be dependent on a complex interaction between macrophage and organism, phagosome acidification and apoptosis may play central roles in organism survival.
Mycobacterium avium subspecies paratuberculosis (M. a. ptb) is the causative agent of Johne's disease, a chronic granulomatous enteritis of wild and domestic ruminants. 5,16 However, M. a. ptb also infects foxes, stoats, weasels, crows, rooks, jackdaws, rats, wood mice, rabbits, and badgers and is considered to be a possible cause of Crohn's disease in human beings. 2,10,18,21,25,26 In cattle, infection probably occurs in the first few months of life, primarily through the fecal-oral route. Lesions develop in the intestinal lamina propria over several months, but clinical disease does not occur for several years. 5,6 Lesions are characterized by loose aggregates of epitheloid macrophages and giant cells. 3,5 Few lymphocytes are present within lesions, and tubercle formation and necrosis, which are characteristic of other mycobacterial infections, do not occur. 5
A major gap exists in our understanding of the events that determine whether cattle eliminate the infection or become permanently infected. In vitro studies have shown that M. a. ptb organisms proliferate within bovine macrophages. 37–39 This may be, in part, due to inhibition of phagosome acidification and phagosome-lysosome fusion. 4 Cytokines appear to modulate macrophage killing of the organisms. Pretreatment of bovine monocytes with interferon-γ (IFN-γ), granulocyte macrophage colony–stimulating factor, or high doses of tumor necrosis factor-α (TNF-α) restricted M. a. ptb growth in vitro. 37–39
The purpose of the study reported here was to characterize the response of bovine monocyte–derived macrophages to infection by M. a. ptb by evaluating macrophage gene expression and antimicrobial responses. DNA microarray technology was used to rapidly screen for differential expression of thousands of genes. 15 To identify genes of importance to resistance to M. a. ptb infection, we compared genes expressed by M. a. ptb–infected macrophages with those of inactivated control macrophages, macrophages activated by incubation with IFN-γ and lipopolysaccharide (LPS), and macrophages incubated with Mycobacterium avium subsp. avium (M. a. a). M. a. ptb and M. a. a are genetically and antigenically similar, but M. a. a causes only a transient infection in cattle. 7,22
Materials and Methods
Bacteria
M. a. ptb strain 19698 and M. a. a strain 35716 were obtained from the American Type Culture Collection. These strains were isolated from naturally infected cows. The organisms were grown to approximately 1 × 108 per milliliter, washed, and resuspended in 7H9 broth containing OADC (Difco Labs, Detroit, MI), Tween 80, mycobactin J (Allied Labs, Ames, IA), and 5% fetal bovine serum. Both organisms were stored at 4 C for up to 3 months. Viability of organisms was assessed at least once a month by use of standard colony-counting assays. Just before addition to macrophage cultures, organisms were washed and resuspended in Dulbecco's phosphate-buffered saline.
Monocyte isolation
Blood for monocyte isolation was collected from six adult Holstein dairy cows that tested negative for Johne's disease as determined by fecal culture and serum enzyme-linked immunosorbent assay tests. Mononuclear cells were isolated by Percoll (Sigma Chemical Co, St. Louis, MO) density gradient (58%) centrifugation. 33 Isolated cells were washed in Dulbecco's phosphate-buffered saline and resuspended at 3 × 107 mononuclear cells per milliliter in Roswell Park Memorial Institute (RPMI) medium containing 10% fetal bovine serum. 33 For preparation of monocyte-derived macrophages, 3 × 107 mononuclear cells were allowed to adhere to 60- × 15-mm tissue culture plates for 90 minutes, and nonadherent cells were removed by extensive washing with RPMI medium. Adherent cells were cultured for 7 days, at 37 C, 5% CO2, in RPMI medium supplemented with 10% fetal bovine serum. Approximately 10 plates each were incubated with vehicle (nonactivated control), bovine IFN-γ (100 U/ml; a gift from Novartis Animal Health Inc., Basel, Switzerland), and LPS derived from Escherichia coli serotype 026:B6 (10 µg/ml; Sigma Chemical Co; activated control) or with M. a. ptb or M. a. a (10 bacilli per macrophage) on day 7 of culture. After 2 hours of incubation, uningested organisms were washed off. RNA was harvested after 16 hours of incubation. In separate experiments macrophages were treated as described above, and two plates each were incubated for an additional 2 hours (for phagosome acidification), 24 and 48 hours (for apoptosis), or 96 hours (for organism killing).
Microarray technique
Messenger RNA (mRNA) was harvested from 10 plates using the guanidunium isothiocyanate–phenol-chloroform procedure (Triazol, Biotex Lab, Houston, TX). The integrity of RNA preparations was assessed with RNA agarose gel electrophoresis and northern blotting, and RNA was frozen at − 80 C until analysis.
Human noncompetitive high-density oligonucleotide arrays (Gene-chip® U95Av2, Affymetrix Inc., Santa Clara, CA) were used to analyze bovine macrophage gene expression. These microarrays were chosen for several reasons. First, bovine microarrays were not available when this study was undertaken. Second, these microarrays provide a highly controlled approach to determine whether hybridization to a particular set of oligonucleotides has occurred. The presence or absence of hybridization for each gene is determined by evaluation of the relative hybridization to 16 pairs of match and mismatch probes. 15 An algorithm then determines whether hybridization has occurred and reports gene expression as absent or present. This approach substantially decreases the occurrence of false-positive results. Third, these microarrays permit screening of over 12,000 genes. The major problem with the use of human oligonucleotide microarrays for evaluation of bovine gene expression is that only genes that cross-hybridize can be evaluated.
mRNAs obtained from six cows were combined into three groups of two cows each to obtain adequate mRNA for the study and to reduce the effects of responses of individual cows on the results. Total RNA was converted into “target” suitable for hybridization according to the protocol provided by the manufacturer. Briefly, 10 µg of total RNA was used to prepare double-stranded complementary DNA (cDNA) using T7-(dT)24 primer (GENSET Corp, La Jolla, CA) and Superscript II reverse transcriptase (RT) (GIBCO-BRL, Gaithersburg, MD). Approximately half of the generated cDNA was subjected to in vitro transcription in the presence of biotinylated uridine triphosphate and cytidine triphosphate (CTP) using the Enzo BioArray high-yield RNA transcript labeling kit (Enzo Diagnostics Inc., Farmingdale, NY). Labeled complementary RNA (cRNA) was purified using the Qiagen RNeasy kit (Qiagen Inc., Valencia, CA) and fragmented to < 200 nucleotides long by incubating at 94 C for 35 minutes in the presence of 40 mM Tris-acetate, pH 8.1, 100 mM potassium acetate, and 30 mM magnesium acetate.
Hybridization to the U95Av2 oligonucleotide arrays and scanning was performed as described in the protocol provided by the manufacturer. The hybridization mixture, consisting of 15 µg biotinylated and fragmented cRNA targets and appropriate controls and spikes, was heated to 99 C for 5 minutes followed by incubation at 45 C for 5 minutes before injection of the sample into the probe array cartridge. All hybridizations were carried out at 45 C for 16 hours with constant rotation at 60 rpm. After hybridization the solutions were removed, and subsequent washing and staining of the arrays was carried out using the microarray fluidics station protocol EukGE_WS2 provided by the manufacturer. Probe arrays were scanned twice at 3-µm resolution, and hybridization intensities for each of the genes/transcripts were collected from the scanned images.
Scanned-image files were inspected visually for artifacts, and gene expression data were analyzed using the microarray expression analysis software (Affymetrix Microarray Suite 4.0, Affymetrix Inc.). To minimize discrepancies due to variables such as sample preparation and hybridization/staining conditions, the intensity values of each image were scaled to an average hybridization intensity of 1,000. As a result, data from different experiments or different samples could be compared directly. An average fold change was calculated within groups, and differences between groups were determined by a Student's two-tailed t-test. 6
Semiquantitative RT polymerase chain reaction
A semiquantitative RT polymerase chain reaction (PCR) method with limited amplification of specific cDNA products was used for characterization of transforming growth factor-β (TGF-β), thrombospondin-1, BCL2L1, STAT-induced STAT 3 inhibitor, protein tyrosine phosphatase, cyclic adenosine 3′: 5′monophosphate (cAMP)–dependent protein kinase regulatory subunit, two Src kinases, and β-adrenergic receptor as described. 34 Because bovine sequences were not available, human sequences were used to generate primers for both Src kinases, STAT-induced STAT 3 inhibitor, and protein tyrosine phosphatase. The mRNA used in these experiments was the same as that used for the microarray studies. The RT reaction contained 0.74 µg random hexamer primers, 3.0 µg total RNA, 1× first-strand buffer, 10 mM dithiothretrol, and 0.5 mM deoxynucleoside triphosphate (dNTP). Samples were heated to 42 C for 2 minutes, and 200 units of Superscript II RT (GIBCO-BRL) was added to bring the final volume to 20 µl. The samples were further incubated at 42 C for 50 minutes, followed by incubation at 70 C for 15 minutes. The PCR reaction contained 1× PCR buffer, 0.2 mM dNTPs, 1 µM of cytokine-specific forward and reverse primers (Table 1), 0.4 µCi [32P]deoxycytidine triphosphate (3,000 Ci/mmol), 1.25 units of Amplitaq polymerase (Perkin–Elmer, Foster City, CA), and 2 µl of first-strand cDNA template. Thermocycler conditions included 94 C for 2 minutes, 54 C for 30 seconds, and 72 C for 1 minute. In preliminary studies cDNA was diluted serially to show that the amount of cDNA product directly correlated with the amount of cDNA template (data not shown). Additionally, all reactions were initially analyzed at 22, 25, and 28 cycles to demonstrate that amplification increased in a linear manner (data not shown). All samples were amplified separately for 25 cycles. The products were separated on 5% nondenaturing acrylamide gels, and specific products were detected using a phosphoimaging system (Molecular Dynamics, Foster City, CA). Primers for β-actin were used to normalize the amount of cDNA product in each sample. β-Actin was amplified for 21 cycles.
Primers used for semiquantitative RT-PCR.

Ingestion and intracellular survival of mycobacterial organisms
Macrophages from each experiment were stained with Ziehl-Neelsen carbolfuchsin stain, which specifically stains mycobacterial organisms. 34 The percentage of macrophages containing one or more organisms was determined by light microscopy.
Organism survival was assessed by a standard colony-counting technique after 96 hours of incubation. 34,39 Macrophages were lysed by addition of saponin (1 mg/ml final concentration), washed, diluted serially, and cultured on 7H9 agar plates supplemented with OADC, Tween 80, and mycobactin J. Colony counts were performed after 4–6 weeks for M. a. a and 8–12 weeks for M. a. ptb.
Phagosome acidification
For phagosome acidification studies mycobacterial organisms were labeled with fluorescein, and Lysotracker red (Molecular Probes, Eugene, OR) was added during the last 60 minutes of incubation. 17 Lysotracker red is taken up by acidified phagosomes, where it is modified to become fluorescent. 17 Bovine monocyte–derived macrophages were grown on 22- × 22-mm coverslips, and labeled mycobacteria (10 bacilli per macrophage) were added for 2 hours. Thereafter, coverslips were washed and inverted onto a glass slide. The preparation was evaluated immediately on a confocal microscope (Leica Microsystems, Exton, PA) using an excitation wavelength of 568 mm and an emission wavelength of 633 mm. Data were analyzed by a computer program (Image-Pro Plus, Media Cybernetics, Silver Springs, MD). Images were merged, and green and red fluorescence intensities of at least 100 phagosomes containing mycobacteria were quantified and the results reported as the colocalization coefficient. Colocalization coefficient was defined as the density of red fluorescence divided by the density of green fluorescence.
Apoptosis
Bovine monocyte–derived macrophages were grown on 22-× 22-mm coverslips and incubated with and without M. a. ptb or M. a. a organisms (10 bacilli per macrophage) for 24 or 48 hours. The slides were then washed and incubated with Hoechst 33342 stain (Molecular Probes) for approximately 30 minutes. 1 Coverslips were then inverted onto glass slides and examined using a fluorescence microscope (Nikon E800, Brooklyn, NY) with excitation at 350 mm and emission at 461 mm. At least 200 cells were counted on each of two slides, and the percentage of fluorescent cells was determined.
Statistical analysis
Data for mycobacterial digestion and killing, phagosome acidification, and apoptosis were analyzed by analysis of variance. Means of interest were compared using the Bonferroni-Dunn F-test.
Results
Gene expression by M. a. ptb–infected macrophages compared with inactivated control macrophages
Nonactivated control macrophage mRNA hybridized with 14.6–15.5% of 12,600 oligonucleotide probe sets on the U95Av2 chips. Bovine macrophages exposed to M. a. ptb hybridized with 14.1–14.8% of the oligonucleotide probe sets. Statistically significant differences were observed for 47 genes (Table 2). These genes were classified as inflammation/stress related (n = 13), cell membrane receptor related (n = 6), cell signaling related (n = 4), transcription/translation related (n = 13), cell metabolism related (n = 3), and of unknown function (n = 8). Of the inflammation/stress–related genes, four cytokines, including TGF-β, interleukin-6 (IL-6), thrombospondin-1, and macrophage inflammatory protein-1-β (MIP-1β)–like protein, had greater expression in M. a. ptb–infected macrophages. Six members of the metallothionein family of heavy-metal–binding proteins had greater expression in M. a. ptb–infected macrophages. Integrin receptor, TNF-β receptor, major histocompatibility complex (MHC) class II DQ-β, and immunoglobulin superfamily member 4, all had lower expression in M. a. ptb–infected macrophages. Of the cell-signaling genes, two protein kinase genes in the mitogen-activated protein (MAP) kinase pathway (MAP kinase and MAP kinase kinase) had lower expression in M. a. ptb–infected macrophages. Of the 13 genes involved in transcription/translation, four were Poll II genes, and three others were involved in Poll II regulation (data not shown).
Selected genes that were differentially expressed by bovine monocyte–derived macrophages incubated with Mycobacterium avium subsp. paratuberculosis compared with nonactivated control macrophages.∗.
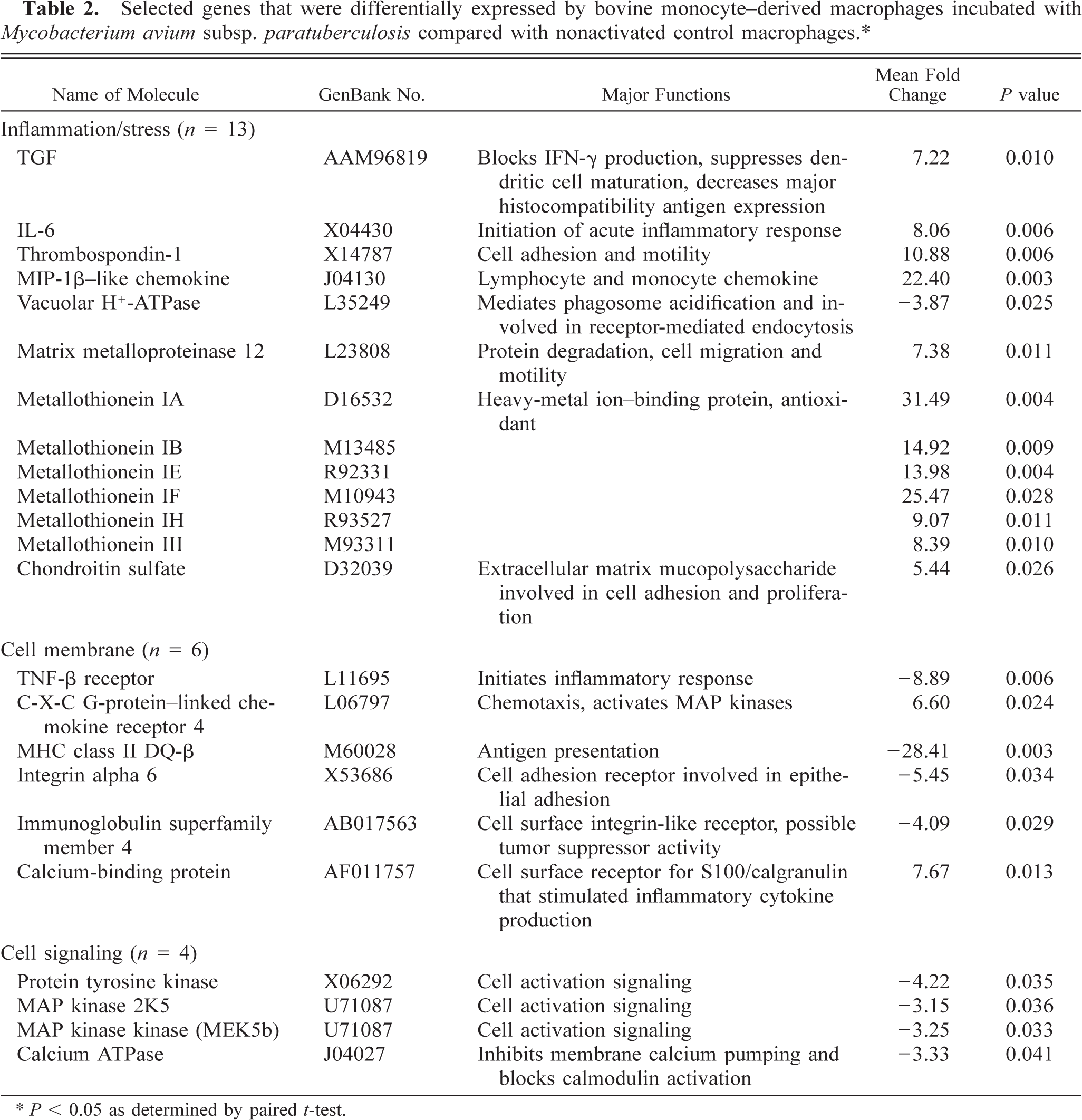
∗ P < 0.05 as determined by paired t-test.
Gene expression by M. a. ptb–infected macrophages compared with activated control macrophages
mRNA from macrophages activated with IFN-γ and LPS hybridized with 16.1–18.2% of 12,600 oligonucleotide probe sets on the U95Av2 chips. When compared with macrophages incubated with M. a. ptb organisms, 79 genes were differentially expressed. Differentially expressed genes were categorized as inflammation/stress related (n = 8), cell membrane receptor related (n = 5), cell signaling related (n = 7), apoptosis related (n = 1), transcription/translation related (n = 15), cell metabolism related (n = 12), and of unknown function (n = 31). Differentially expressed cytokines included thrombospondin-1 and two chemokines (Table 3). Five metallothionein molecules had greater expression by M. a. ptb–infected macrophages.
Selected genes that were differentially expressed by bovine monocyte–derived macrophages incubated with Mycobacterium avium subsp. paratuberculosis compared with macrophages activated by addition of interferon-γ and lipopolysaccharide.∗.
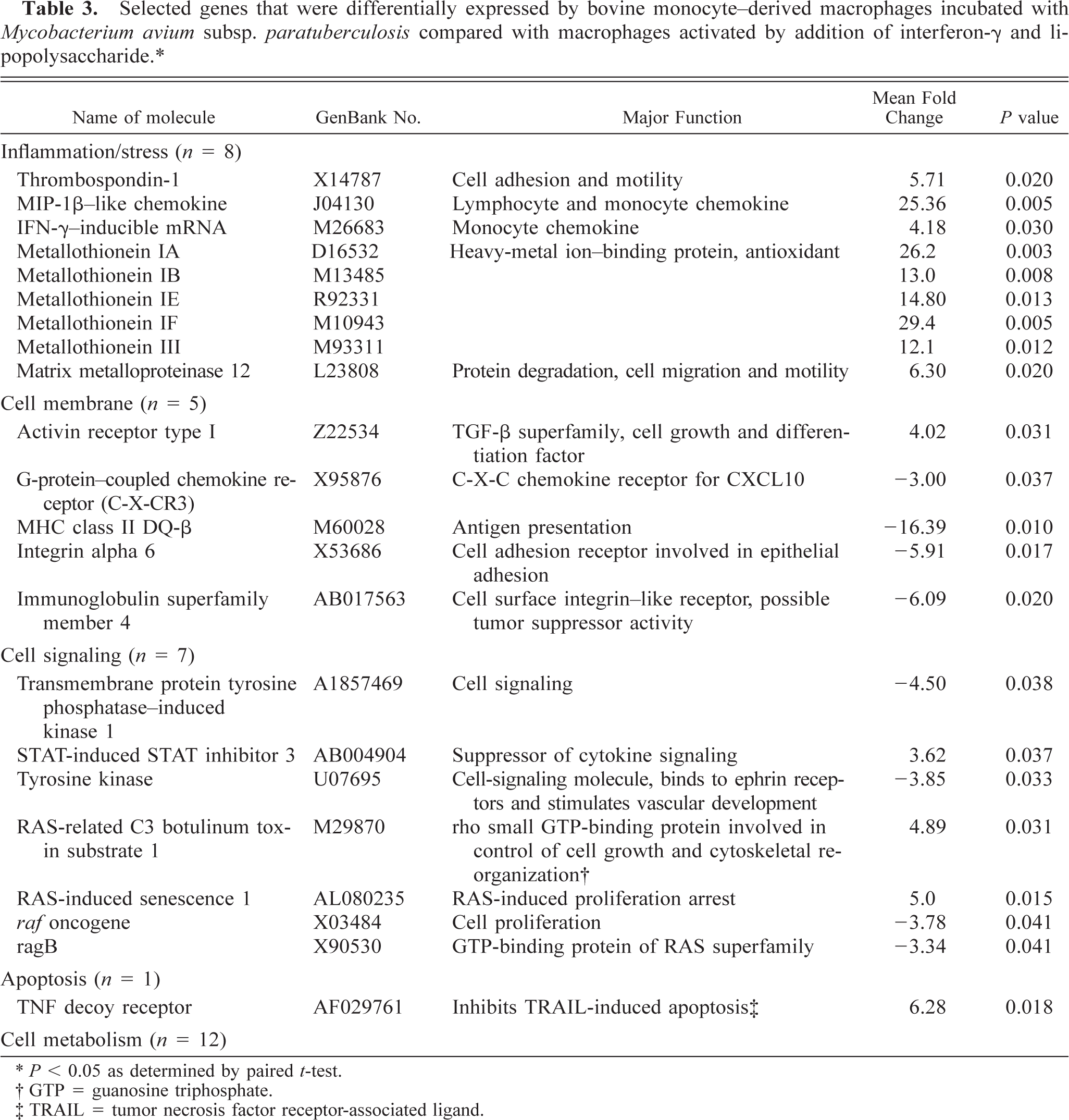
∗ P < 0.05 as determined by paired t-test.
† GTP = guanosine triphosphate.
‡ TRAIL = tumor necrosis factor receptor-associated ligand.
Cell membrane receptors that were differentially expressed included cytokine receptors, adhesion receptors, and immune response receptors. Four of the five differentially expressed cell membrane receptors had lower expression in M. a. ptb–infected macrophages.
Seven cell-signaling genes were differentially expressed. STAT-induced STAT inhibitor 3, specific suppressor of cytokine signaling, had greater expression in M. a. ptb–infected macrophages.
Twelve genes related to cell metabolism were differentially expressed. Of these, three were involved in amino acid metabolism, two were involved in fatty acid metabolism, three were involved in purine/pyrimidine metabolism, three were involved in phospholipid metabolism, and one was involved in glycolysis.
Gene expression by M. a. ptb–infected compared with M. a. a–infected macrophages
Bovine macrophages exposed to M. a. a hybridized with 15.9–17.0% of the 12,600 probe sets on the U95Av2 chips. When M. a. ptb–infected macrophages were compared with M. a. a–infected macrophages, 27 genes were differentially expressed (Table 4). The differentially expressed genes were classified as inflammation/stress related (n = 3), cell membrane related (n = 2), cell signaling related (n = 8), apoptosis related (n = 5), transcription/translation related (n = 3), cell metabolism related (n = 2), and of unknown function (n = 4). Of the inflammation-related genes, TGF-β, matrix metalloproteinase 14, and thrombospondin-1 had greater expression in M. a. ptb-infected macrophages. Of the cell membrane receptors that were differentially expressed, MHC class-II DQ-β and β-adrenergic receptor had lesser expression.
Selected genes that were differentially expressed by bovine monocyte–derived macrophages incubated with Mycobacterium avium subsp. paratuberculosis compared with macrophages incubated with M. avium subsp. avium.∗.
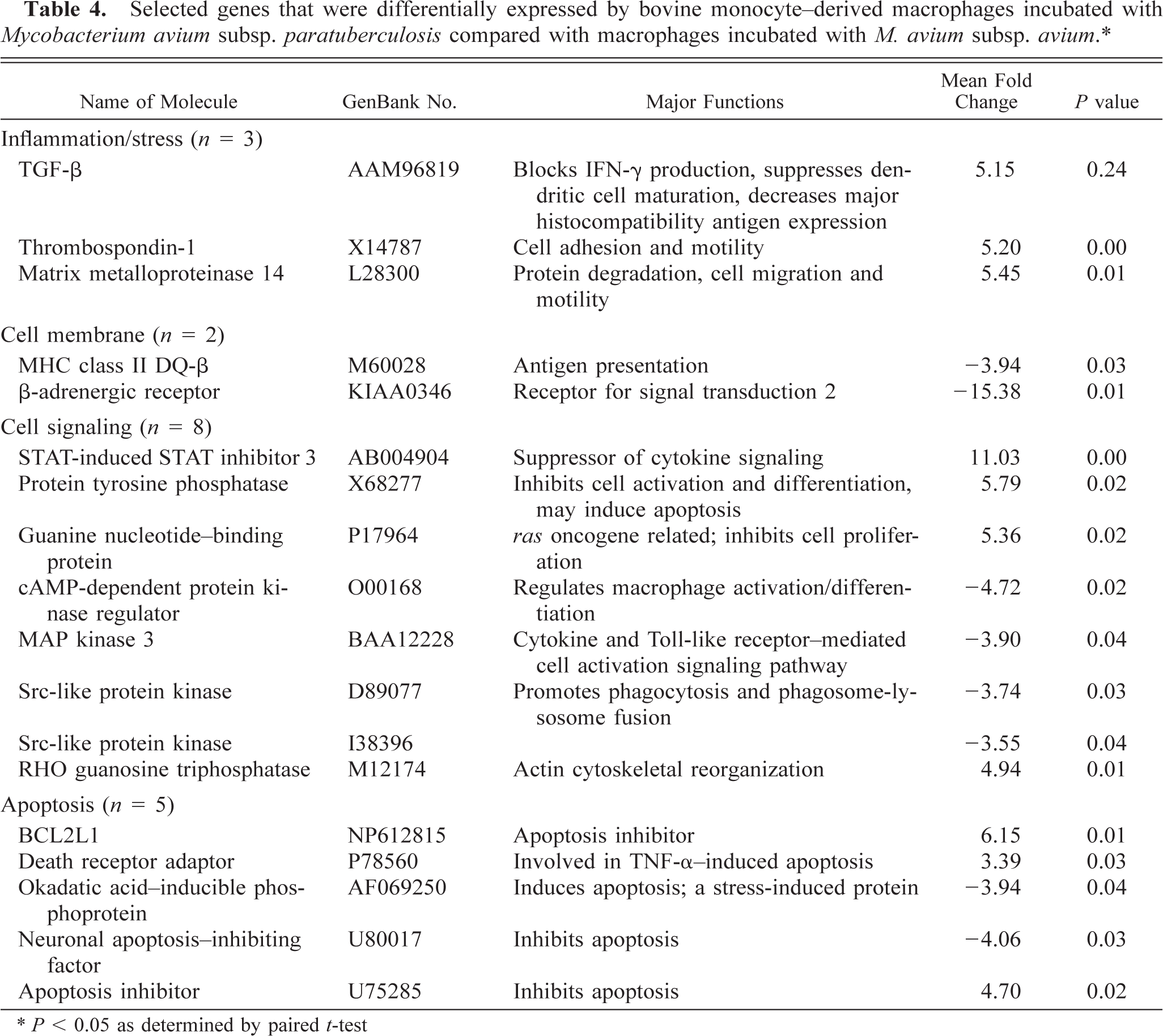
∗ P < 0.05 as determined by paired t-test
Of the cell-signaling genes that were differentially regulated, four protein kinases had lesser expression in M. a. ptb–infected cows, and one protein tyrosine phosphatase had greater expression. Additionally, STAT-induced STAT 3 inhibitor had greater expression in M. a. ptb–infected macrophages.
Five apoptosis-associated genes were differentially expressed. Two inhibitors of apoptosis had greater expression and one inhibitor of apoptosis had lesser expression in M. a. ptb–infected macrophages. One promoter of apoptosis had decreased expression in M. a. ptb–infected macrophages, and another promoter had increased expression.
Semiquantitative RT-PCR
Semiquantitative RT-PCR was done for selected genes of interest for macrophage inflammatory responses. These included TGF-β, STAT-induced STAT inhibitor 3, BCL2L1, thrombospondin-1, cAMP-dependent protein kinase regulator, β-adrenergic receptor, and two Src kinases. Of these, primers resulted in a single product of expected size for TGF-β, BCL2L1, thrombosponin-1, cAMP-dependent protein kinase regulator, β-adrenergic receptor, and one Src kinase (Fig. 1). Other reactions resulted in no detectable product, most probably because of poorly designed primers (i.e., primer sets were chosen based on the human sequence). When RT-PCR results were compared with microarray results, the direction of change was in agreement for all six genes, but the magnitude of change varied (Table 5). For all genes, RT-PCR gave lower fold changes; however, with the exception of β-adrenergic receptor, differences were relatively small.
Comparison of differential gene expression by bovine monocyte–derived macrophages infected with Mycobacterium avium subsp. paratubercuolsis or Mycobacterium avium subsp. avium as determined by DNA microarray and RT-PCR.

Gene expression by bovine macrophages 18 hours after addition of Mycobacterium avium subsp. paratuberculosis (M. a. ptb) or M. avium subsp. avium (M. a. a), as determined by semiquantitative RT-PCR. Data are representative of two separate experiments.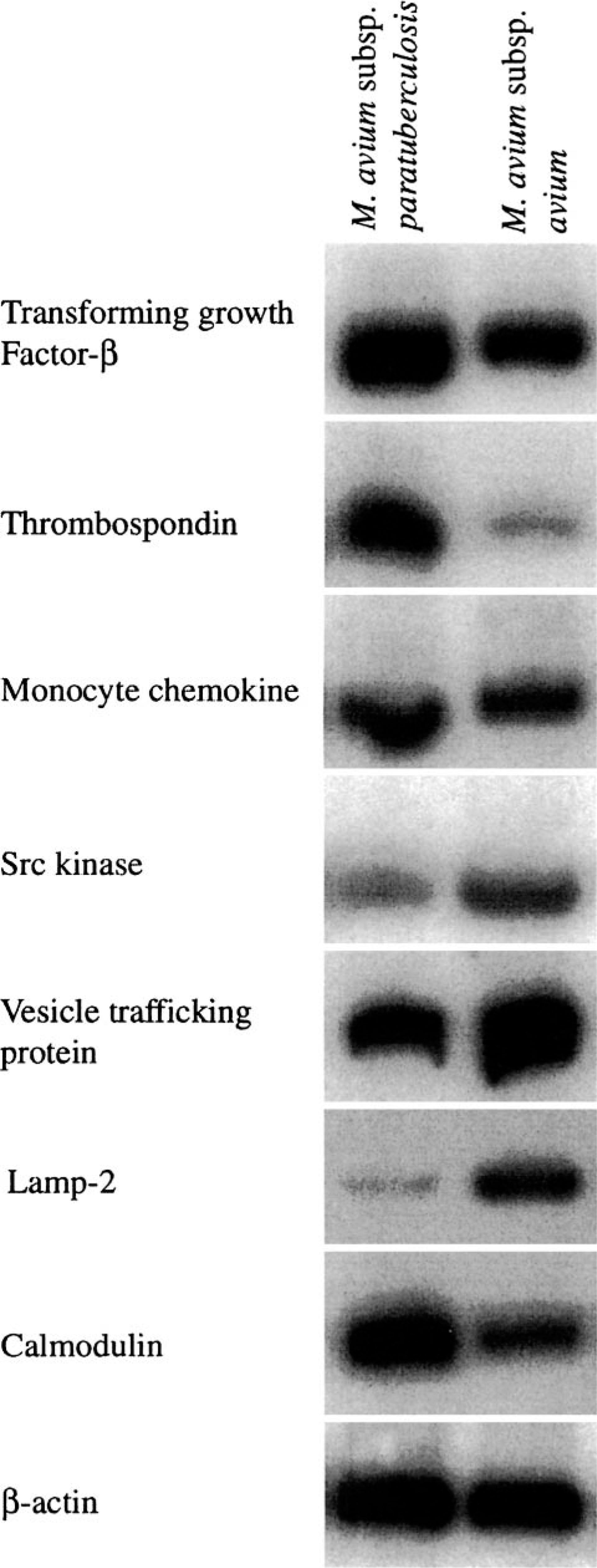
Killing of organisms
Killing of mycobacterial organisms was evaluated after 96 hours of coincubation, and the results were compared with the number of organisms present before incubation. The number of M. a. ptb organisms was similar to baseline values (Table 6). Alternatively, the number of M. a. a organisms was reduced by almost half.
Evaluation of mycobacterial ingestion and killing, phagosome acidification and apoptosis by bovine monocyte–derived macrophages incubated with Mycobacterium avium subsp. paratuberculosis (M. a. ptb) or Mycobacterium avium subsp. avium (M. a. a).
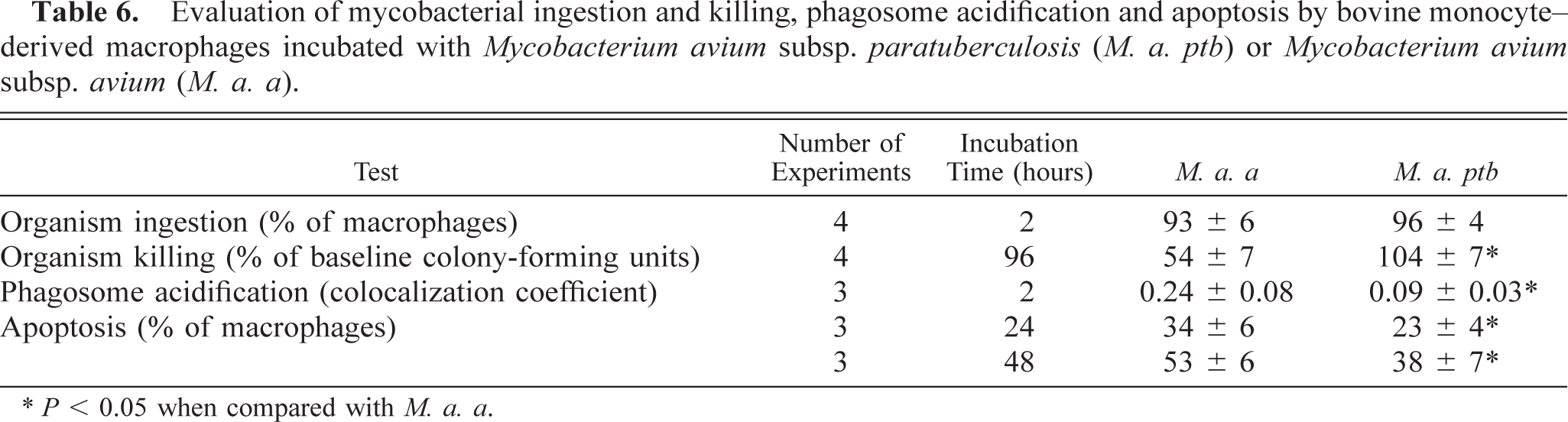
∗ P < 0.05 when compared with M. a. a.
Phagosome acidification
Phagosomes in macrophages incubated with M. a. ptb organisms were uniformly dark green in color. No yellow phagosomes were identified, and colocalization coefficients were consistently low (Table 6). Alternatively, macrophages incubated with M. a. a organisms had phagosomes that varied from green to yellow, indicating variable acidification of the phagosomes. Nine ± 4% of the phagosomes appeared distinctly yellow. Colocalization coefficients were variable, but in aggregate they were statistically different from those for macrophages incubated with M. a. ptb (P < 0.05).
Apoptosis
The percentage of apoptotic cells was evaluated for negative control macrophages and for macrophages incubated with M. a. ptb or M. a. a organisms. The percentage of apoptotic cells in control samples was 14 ± 7% and 25.2 ± 6% at 24 and 48 hours of incubation, respectively. Ingestion of either organism by macrophages resulted in a greater percentage of apoptotic cells (P < 0.05; Table 6). After 24 and 48 hours of incubation, the percentage of apoptotic cells was greater for M. a. a–infected macrophages than for M. a. ptb–infected macrophages (P < 0.05).
Discussion
In this study, we investigated gene expression and antimicrobial responses by bovine monocyte–derived macrophages after exposure to M. a. ptb organisms. Gene expression was evaluated by use of human oligonucleotide microarrays. The probe redundancy and match-mismatch analysis built into the U95Av2 microarrays provided a highly controlled means of detecting the presence or absence of hybridization. 15 However, we could study only genes that hybridized on the chip (i.e., failure to hybridize could have been due to lack of expression of the gene or failure of bovine RNA to hybridize with the human oligonucleotides).
We compared gene expression by macrophages phagocytizing M. a. ptb with that by nonactivated control macrophages to determine the extent to which baseline gene expression was altered by M. a. ptb ingestion. Several cytokines including TGF-β, IL-6, thrombospondin-1, and MIP-1β had greater expression in M. a. ptb–infected macrophages. TGF-β has been shown to inhibit inflammatory and immune responses by blocking IFN-γ production, suppressing dendritic cell maturation and differentiation, decreasing MHC class II expression, and suppressing macrophage activation. 8,19,35 Suppression of TGF-β production has been associated with increased killing of the bacillus Calmette Guerin by mouse macrophages. 23 Blastomyces dermatitidis has been reported to suppress macrophage proinflammatory responses in part through induction of TGF-β production. 11 IL-6 is a proinflammatory cytokine that is involved in many aspects of the acute inflammatory response. However, it has been reported to inhibit mycobactericidal activity of murine macrophages phagocytizing M. a. a. 3 Thrombospondin-1 is an extracellular matrix protein involved in cell adhesion and cell migration. 36
Interestingly, expression of vacuolar H+-adenosine triphosphatase (ATPase) was decreased in M. a. ptb–infected macrophages. A major function of vacuolar H+-ATPase is to acidify the phagosomes. 27,30 Virulent mycobacteria attenuate phagosome acidification by excluding the H+-ATPases from the phagosome membrane. 31 A previous study, in which a mouse macrophage cell line was infected with M. a. ptb organisms, reported that approximately 55% of phagosomes were acidified. 17 Our results indicate that primary cultures of bovine macrophages had minimal capacity to acidify M. a. ptb phagosomes during 2 hours of coincubation. Decreased expression of vacuolar H+ ATPase genes could contribute to this inability to acidify M. a. ptb-containing phagosomes.
Six molecules in the metallothionein family had greater expression in M. a. ptb–infected macrophages. Metallothioneins are a 10- to 12-member family of heavy-metal–binding proteins. 28,32 The importance of metallothioneins in the inflammatory response against intracellular microorganisms is poorly understood. Metallothionein I and II knockout mice had greater macrophage and T-lymphocyte infiltration and greater production of proinflammatory cytokines in an experimental model of autoimmune encephalomyelitis. 28 Alternatively, metallothioneins could sequester heavy-metal ions needed for survival and proliferation of microorganisms. 32
A variety of cell membrane receptors were differentially expressed by M. a. ptb–infected macrophages when compared with nonactivated macrophages. Of particular interest is MHC class II, which had lower expression in M. a. ptb–infected macrophages. Decreased cell surface MHC class I and class II molecules have previously been documented in macrophages phagocytizing M. a. ptb organisms. 33 These data indicate that decreased cell surface MHC class II may result from decreased transcription of the molecule. The combination of reduced expression of MHC class II, immunoglobulin superfamily member 4, and leptinlike receptor-1 indicates that M. a. ptb–infected macrophages may have reduced capacity to present antigens to T lymphocytes.
The results of the present study differ from those of a previous study. In that study gene expression by bovine peripheral blood mononuclear cells (presumably mostly lymphocytes) was compared with gene expression by inactivated cells. 6 A bovine leukocyte cDNA microarray, containing 721 unique sequence tags, was used to evaluate gene expression. Only six genes had greater expression (i.e., > 1.5-fold) by M. a. ptb–infected cells than by inactivated control macrophages. The differences between the two studies may have been due to the different microarray platforms used and the use of a mixture of lymphocytes and monocytes rather than monocyte-derived macrophages. However, these differences highlight the variability in gene expression that can occur under different experimental conditions and the need to evaluate the relevance of variable gene expression through functional assays or protein quantitation.
Gene expression by macrophages ingesting M. a. ptb was also compared with gene expression of macrophages activated by addition of IFN-γ and LPS. As with inactivated macrophages, M. a. ptb–infected macrophages had greater expression of thrombospondin-1, MIP-1β, several metallothioneins, and matrix metalloproteinase 12 and lesser expression of MHC class II DQ-β, immunoglobulin superfamily member 4, and integrin alpha 6. STAT-induced STAT inhibitor 3 had greater expression in M. a. ptb–infected macrophages. This molecule binds to the phosphorylated JH1 domain of Janus tyrosine kinase (JAK2) and inhibits signal transduction through the JAK-STAT pathway. 12,24,29 Greater expression of STAT-induced STAT inhibitor 3 was reported in a study in which mouse macrophages infected with M. tuberculosis were compared with macrophages activated by IFN-γ. 9
Gene expression by macrophages phagocytizing M. a. ptb was compared with that by macrophages phagocytizing M. a. a organisms to identify differences in gene expression that may explain the capacity of bovine macrophages to kill M. a. a but not M. a. ptb organisms. Differences in killing capacity were documented in this study as well as in a previous study. 34 Differentially expressed genes that might alter inflammatory or immune responses include TGF-β, MHC class II DQ-β, STAT-induced STAT inhibitor 3, Src kinases, and apoptosis-related genes. As noted above, TGF-β has anti-inflammatory and anti-immune properties. In a previous study bovine macrophage cell surface MHC class II molecules were shown to have decreased after phagocytosis of both M. a. ptb and M. a. a organisms. 33 However, unlike macrophages containing M. a. a organisms, macrophages containing M. a. ptb organisms failed to upregulate MHC class II molecules after treatment with IFN-γ. Persistent suppression of MHC class II on M. a. ptb–infected macrophages could abrogate the immune response to the organism.
Several cell-signaling genes were differentially regulated. Greater expression of STAT-induced STAT inhibitor 3 by M. a. ptb–infected macrophages suggests that cell activation signaling through the JAK-STAT pathway may be inhibited. A previous study reported that live and not dead M. tuberculosis organisms increased expression of STAT-induced STAT inhibitor 3 by murine macrophages. 9 Src kinases have been shown to associate with cell and phagosome membranes and to promote phagocytosis and phagosome-lysosome fusion. 20 Therefore, the decreased expression of Src genes might contribute to the failure of phagosome-lysosome fusion reported in M. a. ptb–infected macrophages. 4
Five apoptosis-related genes were differentially regulated by M. a. ptb–infected organisms when compared with M. a. a–infected organisms. However, the net effect of these changes on the rate of apoptosis could not be predicted from the data. In our functional studies phagocytosis of both organisms increased the degree of apoptosis; however, there was a greater increase in M. a. a–infected macrophages. Induction of apoptosis in infected macrophages is thought to be a protective mechanism for the host because it releases mycobacteria from their protected intracellular environment. 13,14 Previous studies reported a modest increase in apoptotic cells when a mouse macrophage cell line was incubated with M. a. ptb organisms. 4
In conclusion, human high-density oligonucleotide microarrays were used to compare gene expression profiles of macrophages incubated with M. a. ptb with those of of inactivated and activated control macrophages and macrophages incubated with M. a. a, an organism that is usually killed by bovine macrophages. Our results indicate that a variety of genes coding for molecules of diverse function were differentially expressed. It is probable that intracellular survival of organisms is dependent on a complex interaction between the organism and the macrophage that involves multiple host genes and metabolic pathways. Functional assays, including organism ingestion and killing, phagosome acidification, and apoptosis, indicate that some of the differentially expressed genes identified in this study were associated with functional alterations in the cells. Additionally, these results provide many other potentially interesting differences in gene expression for future study.
Footnotes
Acknowledgements
This research was supported in part by grants from the University of Minnesota Agricultural Experiment Station Rapid Response Fund and University of Minnesota, Academic Health Center.