
Abstract
Severe combined immunodeficiency (SCID) is an inherited disorder of humans, mice, horses, and dogs, in which affected individuals are incapable of generating antigen-specific immune responses. It occurs when lymphocyte precursors fail to differentiate into mature lymphocytes because of mutations within recombinase-activating genes 1 and 2 or within the genes encoding deoxyribonucleic acid (DNA)-dependent protein kinase (DNA-PK). It also occurs when differentiated lymphocytes are incapable of completing signal transduction pathways because of defects in cell surface receptors for interleukins (IL). A spontaneous mutation in DNA-PKcs of BALB/c mice results in SCID, as do experimentally induced mutations in RAG1 and RAG2. SCID in horses results from a spontaneous mutation in DNA-PKcs. Two molecular mechanisms account for SCID in dogs. Jack Russell Terriers have a mutation within the DNA-PKcs gene, whereas Cardigan Welsh Corgi and Basset Hound have different defects in the gene encoding the γ chain that is common to the receptors for IL-2, −4, −7, −9, −15, and −21. The location of the mutation within target genes influences the spectrum of diseases observed in affected animals.
Keywords
Severe combined immunodeficiency (SCID) is an inherited disorder of humans, mice, horses, and dogs, in which affected individuals are incapable of generating antigen-specific immune responses sufficient to protect them from infectious diseases. The disorder was first described in children during the 1960s and may result from molecular defects in at least 15 different enzymes or other proteins required for lymphocyte differentiation or signal transduction. 44 Inheritance may be autosomal recessive, X-linked, or sporadic, depending on the molecular defect in affected persons.
The first spontaneous animal counterpart of SCID was described in horses in 1973. 24 The disorder is limited to horses of the Arabian breed and is inherited as an autosomal recessive trait. 33 In 1983, SCID was described in C.B-17 BALB/c mice in which the disease is also inherited as an autosomal recessive disorder. 6 SCID was subsequently described in three breeds of dogs—Basset Hound (X-linked inheritance), Cardigan Welsh Corgi, and Jack Russell Terriers (autosomal recessive inheritance). 2,21,46 The spontaneously occurring molecular defects in mice, horses, and dogs have been defined and offer clues to the disease patterns observed in affected animals.
Molecular Requirements for Lymphocyte Differentiation
The differentiation of mature T- and B lymphocytes from lymphoid precursors requires the rearrangement and expression of genes encoding T-cell receptors and immunoglobulin receptors, respectively. Gene segments encoding Variable (V), Diversity (D), and Joining (J) proteins must be rearranged and brought into proximity for successful transcription and translation. Two groups of enzymes are critical to the success of these gene rearrangement events (Fig. 1). Recombinase-activating genes 1 and 2 (RAG1, and RAG2) are expressed only in lymphoid tissues, whereas the deoxyribonucleic acid (DNA)–dependent protein kinase (DNA-PK) gene family is expressed in virtually all tissues. The RAG and DNA-PK gene products act sequentially within T- and B-lymphocyte precursors to cut, rearrange, and anneal DNA that encodes T-lymphocyte antigen-specific receptors and B-lymphocyte surface immunoglobulin receptors, respectively. Failure to properly complete these gene rearrangement events results in the elimination of lymphocyte precursors and near absence of mature, functional T- and B lymphocytes. RAG-1 and RAG-2 make the initial cuts in DNA required for ultimate rearrangement of T-cell receptor and B-cell surface immunoglobulin receptors. Defects in RAG1 and RAG2 genes cause SCID in children. 38,39 Spontaneous mutations in RAG1 and RAG2 have not been described in species other than humans. However, mice engineered to express RAG1 and RAG2 mutant genes develop non–T, non–B SCID similar to that observed in children with RAG-1 or RAG-2 deficiencies. 28,43
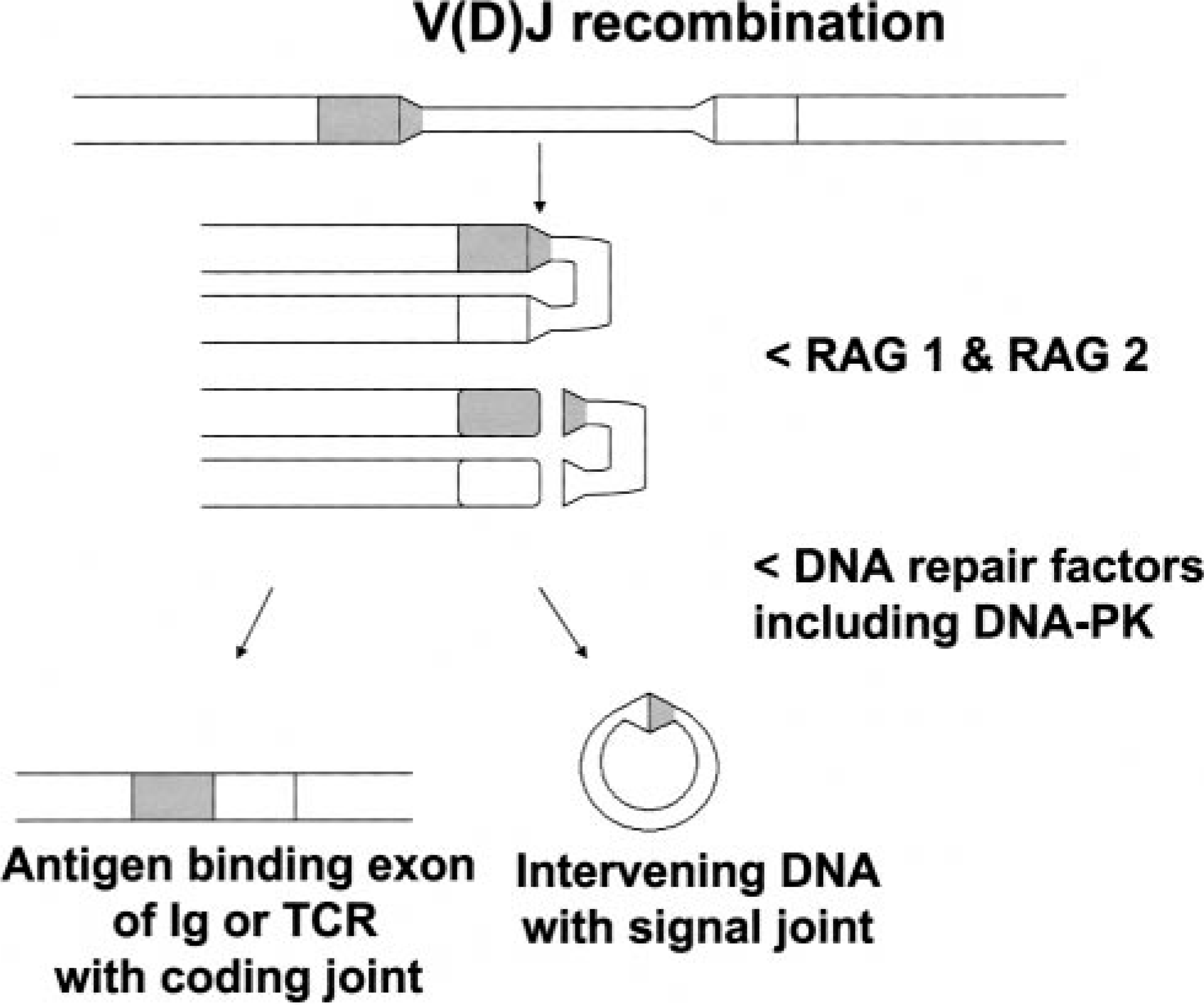
The sequential action of RAG 1, RAG 2, and DNA-PK completes V(D)J recombination required for transcription and translation of T-lymphocyte receptors and B-lymphocyte surface immunoglobulin receptors.
DNA-PK is believed to play an important role in DNA repair. Within lymphocyte precursors, DNA-PK acts after RAG-1 and RAG-2 and anneals severed DNA to produce genes that encode the antigen-specific T-cell receptors and surface immunoglobulin receptors characteristic of functionally mature lymphocytes. During the annealing process, coding joints and signal joints are formed. The signal joints are formed when intervening regions of DNA are excised and circularized during V(D)J recombination. The coding joints between V and (D) regions, as well as between (D) and J regions, are directly involved in antigen binding by B-lymphocyte surface immunoglobulin receptors and T-lymphocyte receptors.
DNA-PK has three subunits, the molecular mass of which varies among animal species (Fig. 2). The Ku portion of DNA-PK has two subunits with molecular masses of approximately 80 and 70 kd. The larger subunit (p80) binds to the free ends of double-stranded DNA in a sequence-independent manner. The DNA-PK catalytic subunit (DNA-PKcs) joins Ku after p80 has bound to double-stranded DNA. DNA-PK becomes a functional kinase in this configuration, adding phosphate groups to several acceptor proteins. As described below, mutations interfering with DNA-PK in C.B-17 mice, foals, and Jack Russell Terriers occur in DNA-PKcs, and proper function of DNA-PK is essential for the formation of coding joints during V(D)J gene rearrangement events. DNA-PK is essential for signal joint formation in horses but not in mice or dogs. 14
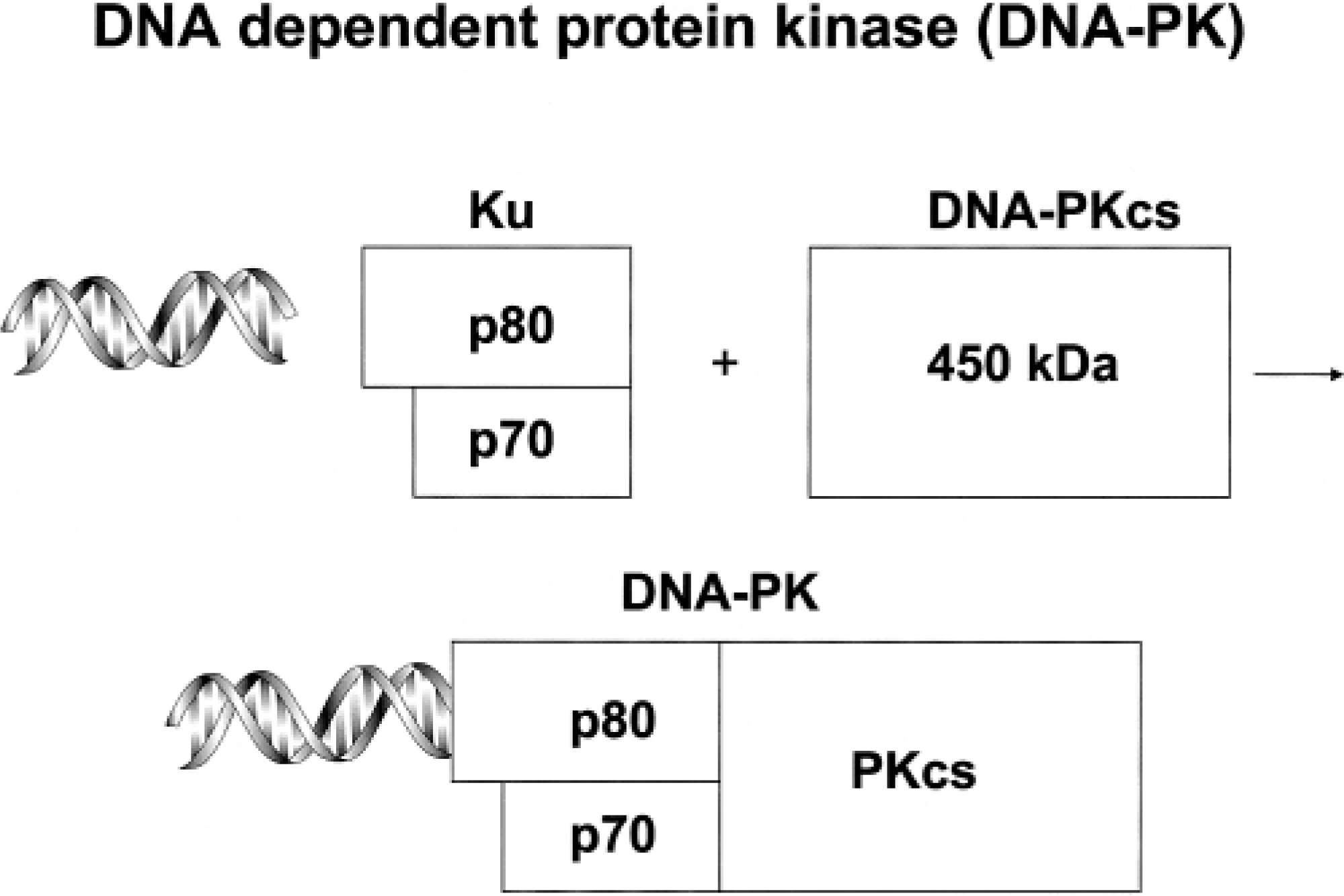
Subcomponents of DNA-dependent protein kinase.
T- and B-lymphocyte precursors that successfully complete V(D)J recombination and migrate to peripheral lymphoid tissues communicate through the release and binding of proteins collectively referred to as interleukins (IL). Interactions between IL and responsive cells occur only through specific IL receptors that are often composed of multiple proteins. Receptors for IL-2, −4, −7, −9, −15, and −21 are able to distinguish among and specifically bind their requisite ILs. However, the receptors for IL-2, −4, −7, −9, −15, and −21 contain a γ chain common to all six receptors. 35,47 The γc is essential for lymphokine-dependent signal transduction. Mutations in the gene encoding γc are responsible for most X-linked SCID in children, and numerous mutations impairing γc production and function have been documented. 35 Two breeds of dogs (described below) develop SCID because of mutations in γc.
SCID in Mice
SCID was described in C.B-17 mice on BALB/c background in 1983. 6 The disorder is inherited as an autosomal recessive trait and results in the near absence of mature T- and B lymphocytes (non–T, non–B SCID). 10 Affected mice are remarkably susceptible to bacterial, viral, fungal, and protozoal infections because of their inability to generate antigen-specific immune responses. However, the life span of immunodeficient mice can be extended beyond 1 year through proper management that minimizes exposure of these mice to infectious agents. Under such conditions, some SCID mice produce immunoglobulins beginning at 6 months of age, and virtually all are “leaky” by 10–14 months of age. 8–10,12 The term “leaky SCID” has been used to describe the phenomenon by which affected mice produce some immunoglobulins and lymphocytes with markers characteristic of T cells. T-cell lymphomas in the thymus were reported in 41 of 269 (15%) SCID mice 10–65 weeks of age. 13
Cells from SCID mice are highly susceptible to ionizing radiation, suggesting reduced ability to repair DNA defects. SCID mice also lack DNA-PK enzyme activity. 4,22 Further studies showed that the Ku component of DNA-PK was present, and that DNA-PK activity was restored by addition of functional DNA-PKcs. 4 The gene encoding DNA-PKcs, located on chromosome 16, was sequenced, and a single nucleotide substitution, resulting in a premature stop codon, was revealed. 5,7 The mutation prevents translation of the 83 C-terminal amino acids of DNA-PKcs and substantially reduces DNA-PK enzyme activity. 5 Coding joint formation occurs poorly, if at all, in mice deficient in DNA-PK activity, but signal joint formation proceeds normally. 29,37 This suggested that DNA-PKcs does not participate in signal joint formation, or that mice possess redundant pathways for completing this reaction. It now appears that the latter explanation is correct, and that murine RAG-1 and RAG-2 can catalyze signal joint formation in the absence of DNA-PK activity. 27,42
The molecular basis of leaky SCID is not known. The C-terminal sequence of DNA-PKcs shares remarkable homology with that of phosphatidylinositol-3-kinase. 19,34,40 By analogy to the known structure and function of phosphatidylinositol-3-kinase, truncation of DNA-PKcs 83 amino acids before the C terminus does not eliminate the full active site of the enzyme (Fig. 3). Reading through the premature ochre stop codon in SCID mice over 6 months of age may provide sufficient stable DNA-PKcs to catalyze coding joint formation, with subsequent immunoglobulin production and T-lymphocyte proliferation. 5

Comparison of the SCID mutations in DNA-PKcs from mice, dogs, and horses. The unmutated human DNA-PKcs contains 4,127 amino acids, of which the 325 C-terminal amino acids are homologous to phosphatidylinositol-3-kinase (PI3K). 19,34 . PI3K contains three subdomains, and the active site of the enzyme is within subdomain 2. DNA-PKcs gene mutations in SCID mice, dogs, and horses prevent translation of the C-terminal 83, 517, and 967 amino acids, respectively.
SCID in Horses
SCID was first described in two foals in 1973 and is known to be an autosomal recessive disorder limited to horses of the Arabian breed. 24,25,33 The paucity of mature T and B lymphocytes in affected Arabian foals is profound. These foals produce no antibodies after infection or immunization, and no evidence of leakiness has been detected. 18,23,30,31 SCID foals rarely survive up to 5 months of age before succumbing to infections caused by equine adenovirus, Pneumocystis carinii, Cryptosporidium parvum, or many species of bacteria. 3,32,45 Like SCID mice, the immunologic defects in SCID foals can be corrected through transplantation of normal histocompatible bone marrow cells. 11,31
Fibroblasts from SCID foals are more susceptible to the lethal effects of ionizing radiation, suggesting a defect in DNA repair systems. 48 Based on this observation, and the knowledge that SCID mice lack DNA-PK activity, we showed that SCID foals also lack this enzyme activity. 48 Equine DNA-PKcs is located on chromosome 9. 1 Cells from SCID foals contain Ku that is able to bind double-stranded DNA but lack detectable DNA-PKcs protein. 48 Sequence analysis of the equine DNA-PKcs revealed a five–base pair deletion, resulting in a frame-shift mutation and premature stop codon. 40 The mutation prevented translation of the 967 C-terminal amino acids, resulting in a nonstable protein. 40,48 By analogy to phosphatidylinositol-3-kinase, absence of the 967 C-terminal amino acids of DNA-PKcs eliminates the active site of DNA-PK (Fig. 3). SCID foals are incapable of V(D)J gene rearrangement for immunoglobulin heavy chains and T-cell receptors. 42 Neither coding joints nor signal joints are formed in SCID foals. 48 Therefore, in contrast to mice, DNA-PK is required for signal joint formation in horses. This suggests that horses lack redundant pathways for catalyzing signal joint reactions.
Neoplasia is uncommon in horses below 8 years of age, and SCID foals rarely survive up to 5 months of age. Therefore, it is not surprising that neoplasms have not been described in SCID foals. However, horses heterozygous for the SCID trait are at greater risk for developing sarcoids, suggesting that DNA-PK may function as a tumor suppressor through its ability to repair DNA. 14 The existence of a heterozygote detection assay makes it possible to reduce the frequency of the DNA-PKcs mutation through selective breeding. 41
SCID in Dogs
Two distinct molecular mechanisms result in SCID in dogs. Recently, SCID was reported in Jack Russell Terriers and shown to be due to near absence of DNA-PK activity. 2,26 A point mutation results in a premature stop codon at nucleotide 10,828 of DNA-PKcs and terminates translation of DNA-PKcs 517 amino acids before the C terminus. 14 This loss of C-terminal amino acids from DNA-PKcs is intermediate between that reported for SCID in mice and SCID in foals (Fig. 3). Interestingly, impairment of signal joint formation is also intermediate between that reported for mice with SCID and foals with SCID. 14 Jack Russell Terrier puppies with SCID succumb to infections at a few months of age. No evidence of neoplasia or leakiness has been reported.
Basset Hounds with SCID were first recognized in 1982, and the disorder was described as X-linked SCID in 1989. 16,18,21 X-linked SCID is characterized by moderate lymphopenia, with normal percentages of circulating B lymphocytes and low to normal percentages of phenotypically mature, but nonfunctional, T lymphocytes. Dogs with X-linked SCID are able to complete the gene rearrangement events depicted in Fig. 1. Therefore, these dogs possess lymphocytes with surface markers characteristic of mature T- and B lymphocytes. Affected puppies are remarkably susceptible to bacterial and viral infections and rarely survive past 3–4 months of age when raised in conventional environments. These puppies are able to synthesize IgM so that their serum concentrations are similar to those of normal littermates. 15 However, their serum IgG concentrations are significantly reduced, and IgA is typically not detectable. Affected puppies produce minimal quantities of specific antibodies after immunization with bacteriophage ΦX-174. Virtually all the antibody produced is of the IgM class, indicating inability to class-switch to IgG and IgA. 15 One case of acute monocytic leukemia was reported in a dog with X-linked SCID that was maintained in a gnotobiotic environment to 20 months of age. 17 After the characterization of this immunodeficiency disorder in the Basset Hound, a very similar manifestation of SCID was reported in puppies of the Cardigan Welsh Corgi. 36,46
SCID in Basset and Cardigan Welsh Corgi puppies involves a mutation in the gene encoding the γ chain of the IL-2 receptor (IL2RG). As mentioned, this γ chain (γc) is common to the receptors for IL-2, −4, −7, −9, −15, and −21, and mutations within it prevent lymphocytes in affected dogs from completing signal transduction pathways initiated by any of these ILs. 20 The canine IL2RG encodes a polypeptide 373 amino acids long. 20 The mutation in Bassets involves the deletion of four base pairs (CCTC at positions 30–33) in exon 1. This results in a premature stop codon and in translation of a predicted polypeptide that is 21 amino acids long and contains only 10 amino acids of the normal polypeptide sequence. Interestingly, the corresponding messenger ribonucleic acid is readily detectable on northern blots and appears to be stable. 20 In the Cardigan Welsh Corgi, insertion of a cytosine after nucleotide 582 in exon 4 results in a premature stop codon that also prevents full translation of γc. 46
It is not surprising that different mutations of IL2RG occur in two breeds of dogs. Mutation of IL2RG is the most common molecular mechanism for SCID in children. The human IL2RG coding region contains 1,124 nucleotides and eight exons. More than 145 distinct point, insertion, and deletion mutations, involving all eight exons, have been reported. 35
SCID in mice, horses, and dogs provides useful models for defining the molecular requirements for lymphocyte differentiation and function, the immunologic mechanisms of protection against infectious agents, and a venue for exploring treatments for immunodeficiency disorders in children. Investigation of other animals with apparent immunodeficiency disorders will likely reveal additional molecular mechanisms of comparative biomedical interest.
Footnotes
Acknowledgements
I thank Dr. Gerald Callahan for helpful comments on the manuscript.