
Abstract
A clinical, morphologic, ultrastructural, and genetic study was performed on five rough-coated dachshund semisiblings with osteogenesis imperfecta (OI). Clinical signs consisted of pain, spontaneous bone and teeth fractures, joint hyperlaxity, and reduced bone density on radiography. Primary teeth were extremely thin-walled and brittle. The hallmark of the disease was a severe osteopenia characterized by impairment of lamellar bone formation in the long bones, skull, and vertebral column. No deformity or dwarfism was present. The columns of chondrocytes and primary trabeculae in the epiphyses and metaphyses were histologically normal. An abrupt failure of secondary spongiosa and lamellar bone formation was evident in the medullary and cortical zones in all animals. The few existing trabeculae consisted of woven bone. There was no increase in the number and size of osteoclasts or lacunae. In the teeth, the dentine layers were thin and lacked a tubular pattern. Ultrastructurally, osteoid apposition on bone surfaces was reduced, and small numbers of large cytoplasmic vacuoles were present in a few osteoblasts. Molecular analyses of the collagen type I-encoding genes COL1A1 and COL1A2 revealed several nucleotide differences compared with the published canine sequences but were not significant for OI. Therefore, OI in these Dachshund litters was characterized by a severe, generalized osteopenia and dentinopenia. This pattern of reduced bone formation is suggestive of defective production of collagen type I.
Keywords
Introduction
Osteogenesis imperfecta (OI) is a well-documented inherited disorder in children but has rarely been described in animals. In humans, the clinical disease is characterized by a severe, nonmetabolic osteopenia with spontaneous fractures, sometimes combined with deformities, dwarfism, and disturbance of dentinogenesis. 27,28 The disease spectrum ranges from subclinical events to stillbirth. 27,30 Several studies have shown that the clinical outcome is difficult to classify by specific pathomorphologic features. 5,33,34 Nevertheless, most cases of OI have been characterized histologically by an immature pattern of bone formation, which is confirmed by a paucity of endochondral and intramembranous ossification and the absence of lamellar bone and the Haversian system. 9,33,34
The underlying causes of the disease are most often genetic defects of the COL1A1 or COL1A2 genes, which encode the procollagen molecules of collagen type I, resulting in quantitative and qualitative abnormalities of this collagen type. As part of the basic subunit of each procollagen chain, glycine is found in every third amino acid position, resulting in a (Gly-X-Y)n repeat structure. Any interruption of this structure leads to destabilization of the triple helix with subsequent intra- or extracellular degradation or expression of irregular collagen fibrils and ultimately to diminished collagen synthesis and bone formation. 3,4,12,24,25,38 Collagen type I represents 90% of the organic substance of bone, tendon, and tooth, and these are the major target organs of the disease. Other type I collagen–rich tissues, such as the skin, are rarely affected. 26,37
OI has been documented previously in cattle, sheep, domestic cats, mice, and tigers. 1,2,10,11,14,16,18,19,21,23,31 In dogs, OI has been reported in Golden Retrievers, Collies, Poodles, Beagles, Norwegian Elkhound, and Bedlington Terriers. 8,17,22,29 Young animals are mostly affected during the first few weeks of life. Defects in the COL1A1 or COL1A2 genes have thus far been found only in mice and Golden Retrievers. 7,8 Here, we describe the clinical, pathomorphologic, and genetic findings in five dogs showing signs of OI.
Material and Methods
Two of the four and three of the six puppies from two different litters (litters A and B) of rough-coated Dachshunds with a common 10-year-old sire had a history of pain, lameness, and multiple fractures starting at 3–4 weeks of age. All puppies were kept under the same environmental conditions and fed commercial diets. There was neither a history of similar diseases in descendants of this sire nor had the management been changed by the breeders.
A puppy from litter B (dog No. 5, female, 1.4 kg) was euthanatized at the age of 10 weeks by a local practitioner and was sent without further clinical or biochemical investigation for necropsy to the Institute of Pathology, School of Veterinary Medicine Hannover, Hannover, Germany.
Two affected puppies from each litter were presented to the Clinic for Small Domestic Animals of the School of Veterinary Medicine, Hannover. At presentation, these animals were aged 13 weeks (litter A, dog No. 1, female, 1.65 kg and dog No. 2, male, 1.55 kg) and 14 weeks (litter B, dog No. 3, female 1.7 kg and dog No. 4, female, 1.6 kg). The clinical investigation of these dogs (Nos. 1–4) included physical and orthopedic examinations, radiographs, complete blood cell counts (CBC), and determination of alkaline phosphatase (ALP) activity, serum concentrations of urea nitrogen, creatinine, total protein, albumin, calcium, potassium, and phosphorus, and levels of Vit-D3 (tested in dog Nos. 1–3) (Immundiagnostik AG, Bensheim, Germany), parathyroid hormone (tested in dog Nos. 3–4), and thyroxine/T4 (tested in dog Nos. 1–4). Because of the poor prognosis, the owners elected to have the dogs euthanatized, and necropsies were performed.
Samples of major tissues were fixed in 10% neutral buffered formalin for 48 hours. Bone specimens from epiphyses, metaphyses, and diaphyses of humerus, ulna, femur, tibia, ribs, skull, mandible, and maxilla with teeth were decalcified in a commercial solution (Ossa Fixona®, Waldeck GmbH, Münster-Roxel, Germany) for 6 hours (control group, 24 hours). Thereafter, tissues were embedded in paraffin, cut, and stained with hematoxylin and eosin (HE), toluidine blue, Masson-Goldner, and Picrosirius red. For undecalcified bone sections, bone samples were fixed in 4% paraformaldehyde (pH 7.4). After 48 hours at 4 C, the tissue was dehydrated in alcohol and embedded in glycolmethylacrylate (GMA; Heraeus, Wehrheim, Germany). These slides were stained with HE, toluidine blue (Toluidin Blau O®, Merck, Darmstadt, Germany), and Von Kossa.
For electron microscopy, samples were processed within 45 minutes after euthanasia. Tissue from ribs and femur, tibia, and humerus epiphyses and metaphyses of affected and control dogs were fixed in 5% glutaraldehyde in 0.1 M cacodylate buffer (pH 7.2) for 48 hours, postfixed and precontrasted in cacodylate-buffered osmium tetroxide (pH 7.2), and embedded in Epon 812 (Serva, Heidelberg, Germany) for ultrastructural examination. Ultrathin sections (0.2 µm) were cut with an ultramicrotome (Ultramikrotom OM U3, Reichert, Vienna, Austria), collected on 200-mesh 2 copper grids, stained with alcoholic uranyl acetate and lead citrate, and examined under a transmission electron microscope (EM 10, Zeiss, Oberkochen, Germany).
For molecular analysis of collagen genes, bone was collected immediately after euthanasia from metacarpal bones of each dog and stored in liquid nitrogen.
Total ribonucleic acid (RNA) was prepared with TRIZOL™ (Life Technologies, Karlsruhe, Germany). Forty picomoles of (T)24V primer was annealed to 4 µg of total RNA and reverse transcribed in a 40-µl reaction with 8 units of Omniscript reverse transcriptase (Qiagen, Hilden, Germany). After a 60-minute incubation at 37 C, the reaction was terminated by heating to 75 C for 10 minutes. Eleven reverse transcription–polymerase chain reaction (RT-PCR) primer pairs were designed that amplify overlapping 800- to 1,500-bp fragments of the canine COL1A1 and COL1A2 complementary deoxyribonucleic acid (cDNA) sequences (Table 1). For PCR, 2 µl of the synthesized cDNA was used as a template in 50 µl reactions containing 50 pmol of each primer, 200 µM deoxynucleotide triphosphates, and 2.5 units Taq DNA polymerase (Qiagen, Hilden, Germany). After an initial 5-minute denaturation step at 94 C, 35 cycles of 45 seconds at 94 C were performed, followed by 45 seconds at the annealing temperature of the specific primers and 60 seconds at 72 C in a PTC-200 thermocycler (Biozym, Hessisch-Oldendorf, Germany). RT-PCR products were checked for purity and yield on standard agarose gels.
PCR primers for the amplification of COL1A1 and COL1A2 cDNA fragments.
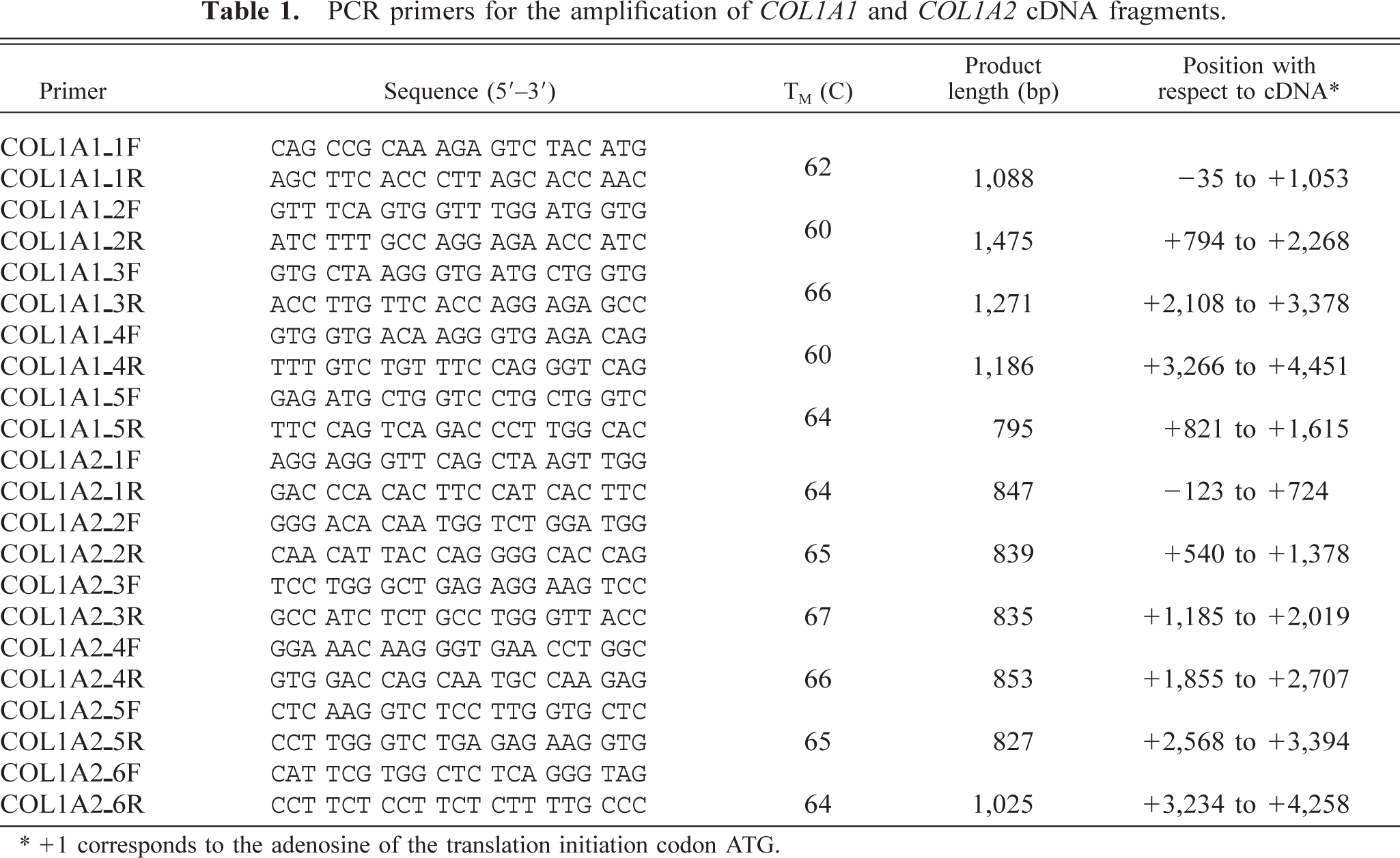
∗+1 corresponds to the adenosine of the translation initiation codon ATG.
RT-PCR products were directly sequenced using infrared dye–labeled internal primers (Table 2) and the thermosequenase kit (Amersham Bioscience, Freiburg, Germany). The sequencing reactions were separated on a LICOR 4200 L2/S2 automated sequencing system (MWG-Biotech, Ebersberg, Germany). Sequence data were analyzed with Sequencher 4.0 (GeneCodes, Ann Arbor, MI) and compared with the previously published canine and human COL1A1 and COL1A2 cDNA sequences (canine genes: GenBank accession Nos. AF153062, AF035120; human genes: GenBank accession Nos. Z74615, Z74616). Discrepancies between the determined COL1A1 and COL1A2 cDNA sequences and the database sequences (AF153062, AF035120) were subsequently verified at the genomic DNA level. The respective genomic regions were amplified in all four affected dogs and in eight unrelated control dogs with the primers given in Table 2. The genomic PCR products were sequenced as described above.
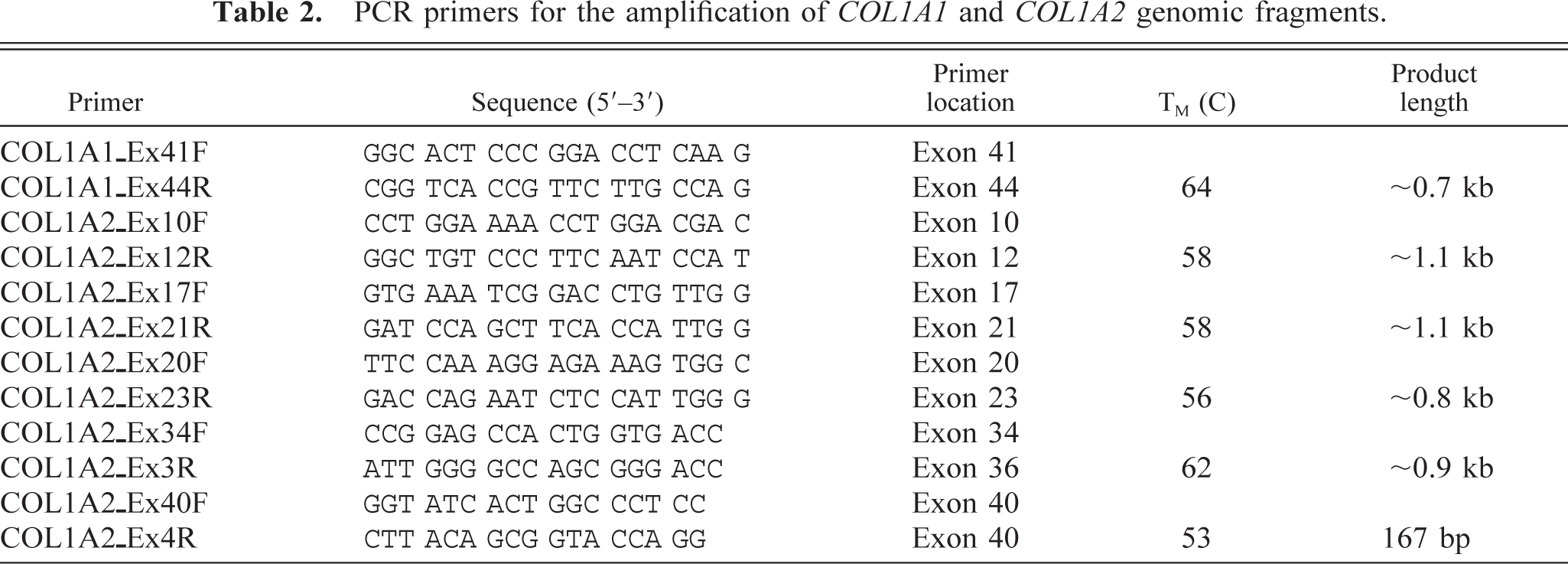
PCR primers for the amplification of COL1A1 and COL1A2 genomic fragments.
Results
All dogs showed reduced agility and were painful when picked up carefully, but otherwise showed normal behavior and good appetite. The animals were in appropriate body condition and well-proportioned, with no evidence of deformities or dwarfism. All teeth were brittle and translucent, and some were fractured (Fig. 1). There was moderate to severe joint hyperlaxity, notably with hyperextension in the tibiotarsal joint. Routine blood analyses, including parameters reflecting mineral homeostasis, were within normal ranges in all puppies. A reduced radioopacity of bones, a thinning of cortices in long bones (Fig. 2), and several serial fractures of ribs were found on radiographs.
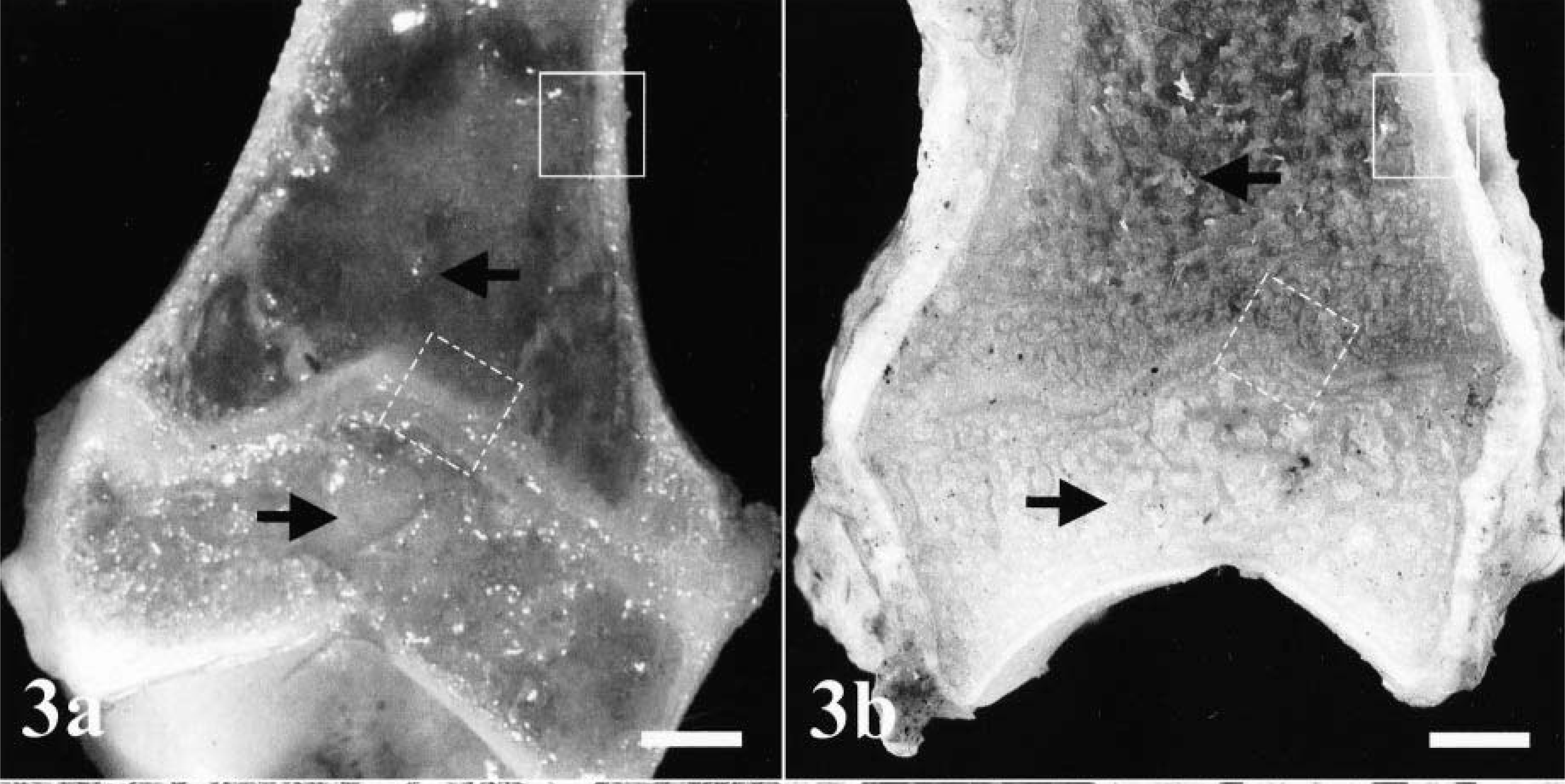
At necropsy, a general osteopenia was found in all dogs, as indicated by reduced cancellous bone mass in diaphyses and metaphyses (Fig. 3). The bodies of the vertebrae also were affected and showed a thinning of the cortical bone. Several serial fractures of ribs were found in all dogs. Articular cartilages and tendons, skin, parenchymatous organs, central nervous system, and endocrine glands were normal. Dog Nos. 3 and 4 were found to have mild endoparasitosis consisting of a few Toxocara canis larvae in the small intestine.
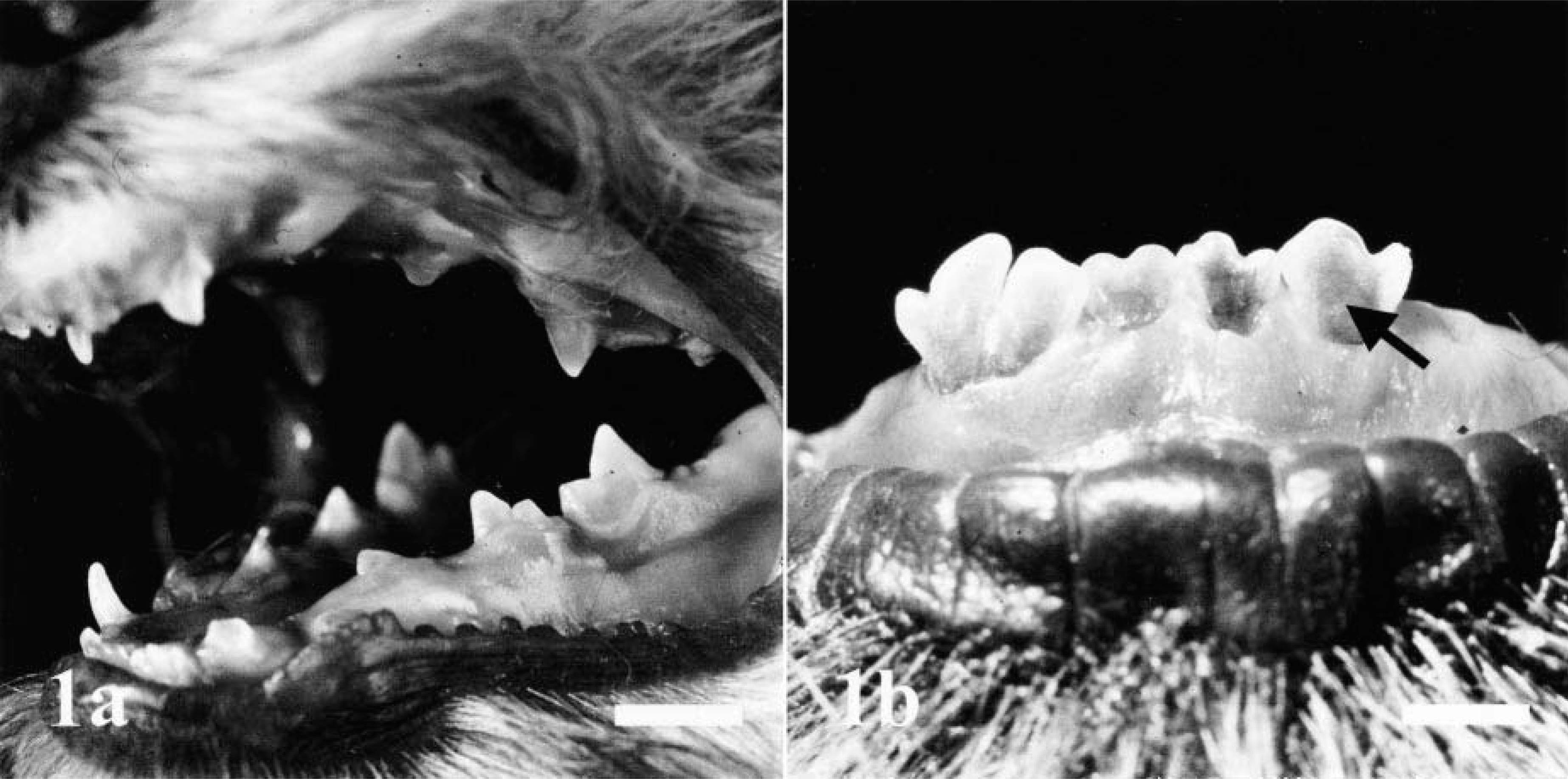
Oral cavity; dog No. 2.
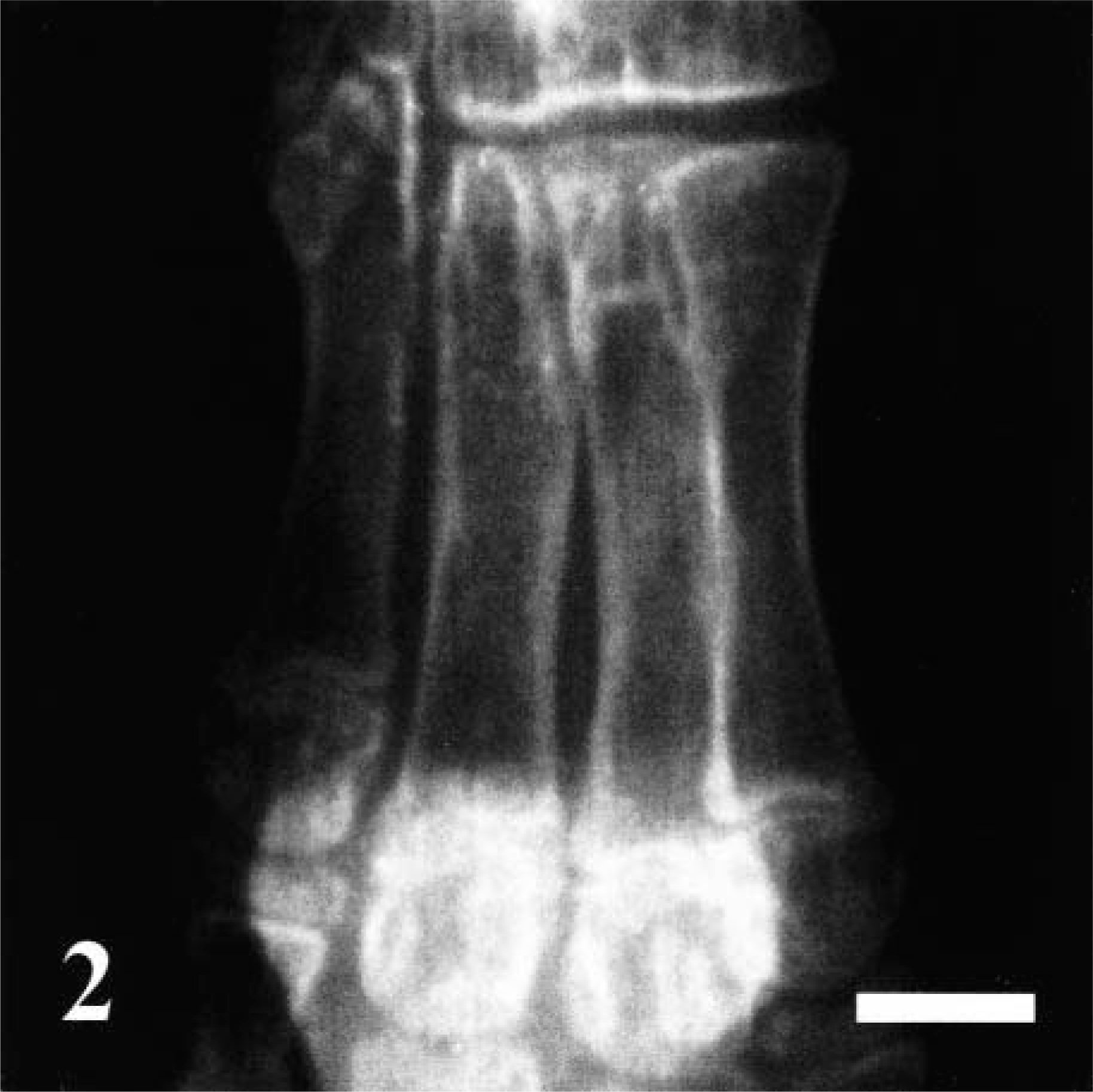
Radiograph of metatarsal bones; dog. No. 3. The radiodensity of long bones was extremely reduced. Note the thinning of the cortices. Bar = 10 mm.
Tibia, distal epiphysis:
Histologically, all bones were affected, but alterations were most prominent in long bones, ribs, mandibles, and teeth. In the epiphyseal and physeal cartilage, chondrocytes were arranged in regular columns, resulting in normal primary spongiosa, but there was a marked absence of secondary spongiosa in the metaphyses (Fig. 4). There were only a few trabecular islands, consisting of woven bone, present in the medullary cavity of some metaphyses and diaphyses. The surfaces of these trabeculae contained a discontinuous seam of osteoid and were lined by a single layer of small osteoblasts. Some of the osteoblasts contained single cytoplasmic vacuoles. In the long bones, the flat bones of the skull, and the vertebral bodies, the cortices consisted of irregularly arranged, thin trabeculae of woven bone with few osteocytes. No Haversian system was observed, and there was a loose fibrovascular stroma within the wide spaces between the trabeculae (Fig. 5). No extensive osteoclastic activity was observed in any bone. Mild focal subperiosteal new bone formation was evident in the proximal humerus and in the femoral diaphysis of dog No. 2 and in the proximal femoral diaphysis and metaphysis of dog No. 4. There was callus formation at the sites of rib fractures in all dogs. The mandibular bone also was composed of small, poorly mineralized trabeculae and thin cortices (Fig. 6). Small foci of proliferating fibrous tissue were present within the medullar cavity.
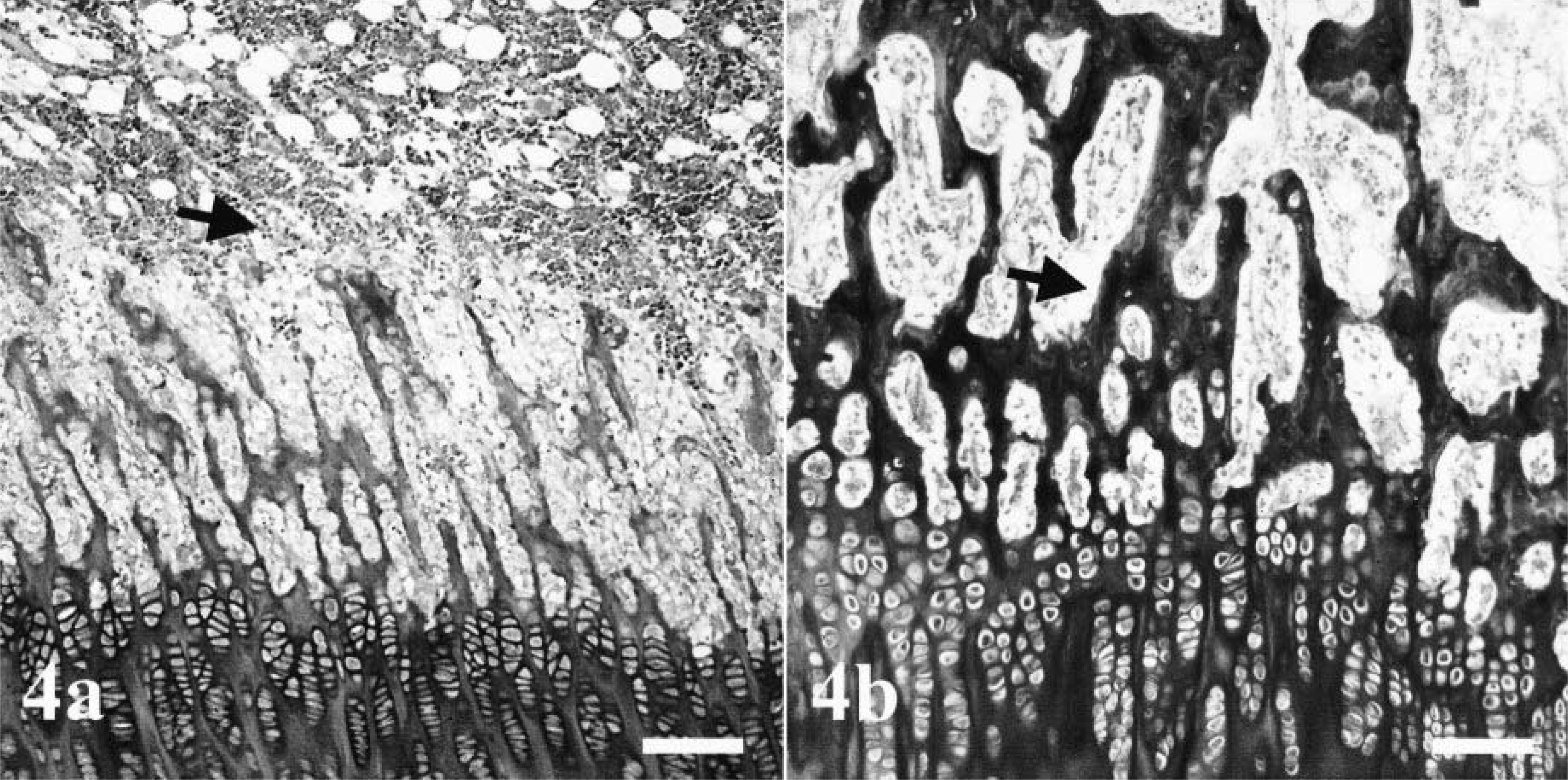
Tibia, distal growth plate:
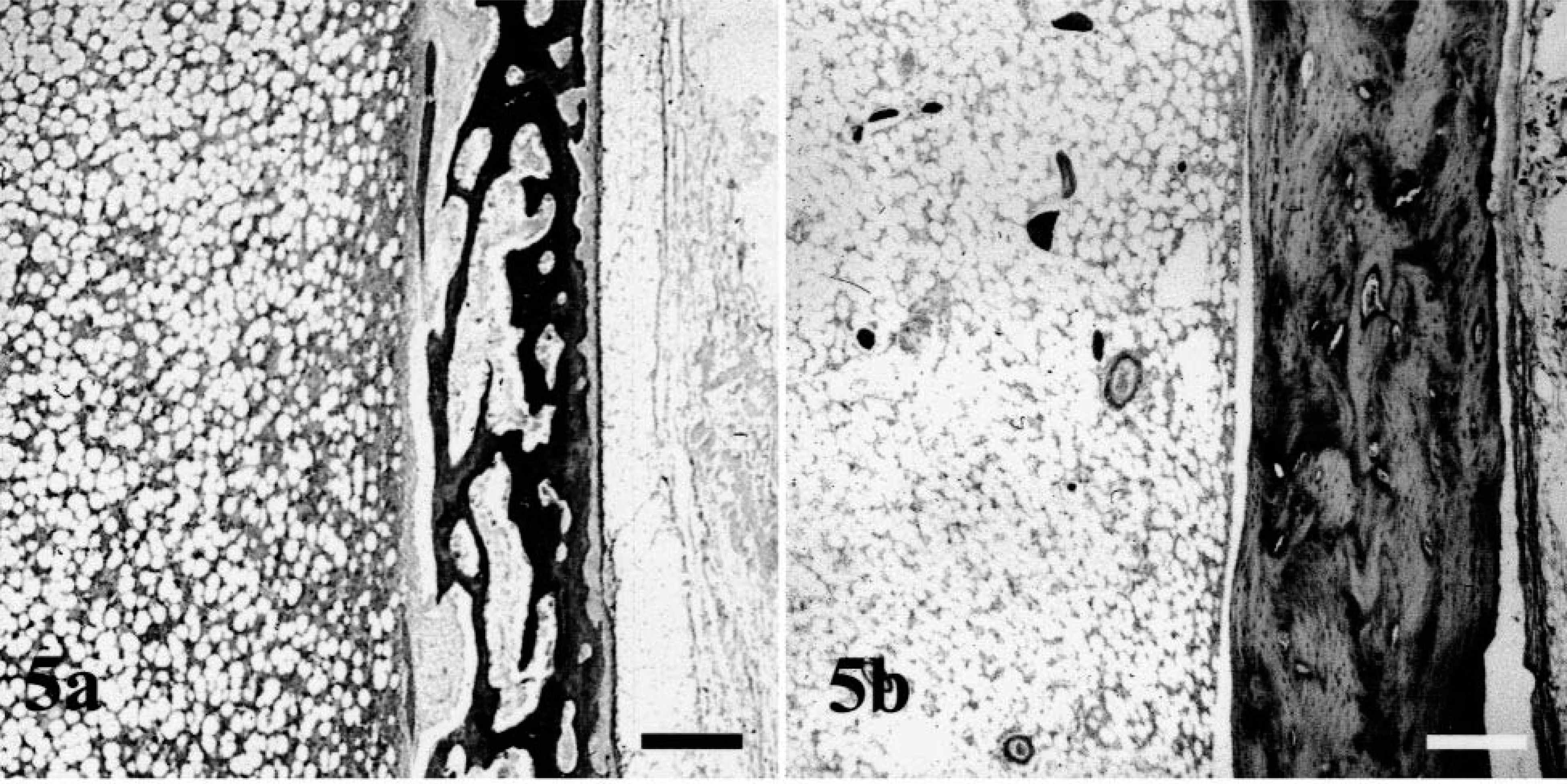
Femoral cortex:
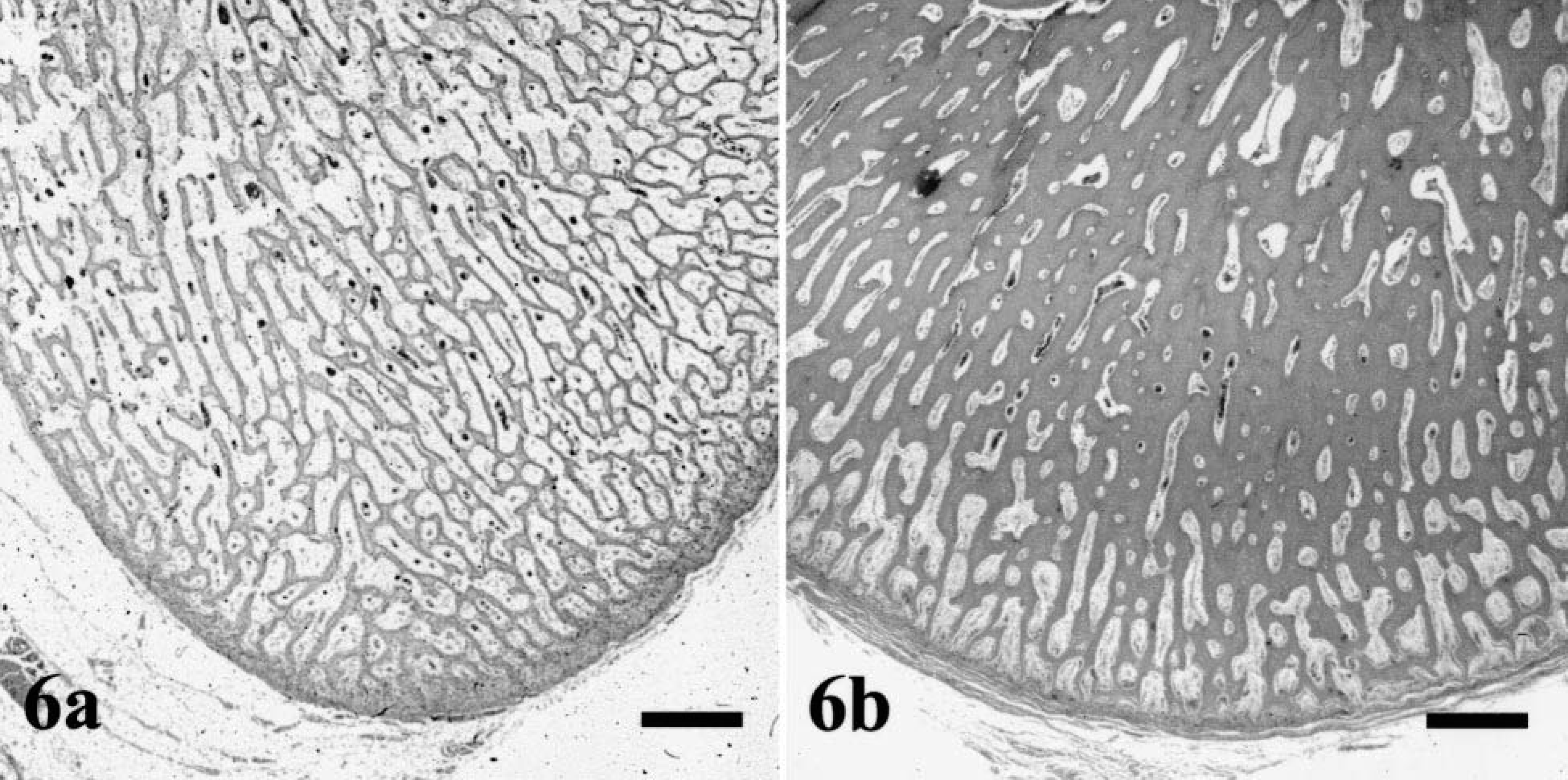
Mandibular bone:
The teeth were of normal size, but the dentine layer was only one-quarter normal thickness and had an atubular pattern (Fig. 7). The width of the predentine layer varied and was lined by a few, spindle-shaped odontoblasts in the outer part of the pulp (Fig. 8). Some roots of primary teeth showed a few lacunae-like lesions containing single osteoclasts. The density and structure of collagen fibers in the skin and ligaments of dog Nos. 1, 2, and 4 were normal.
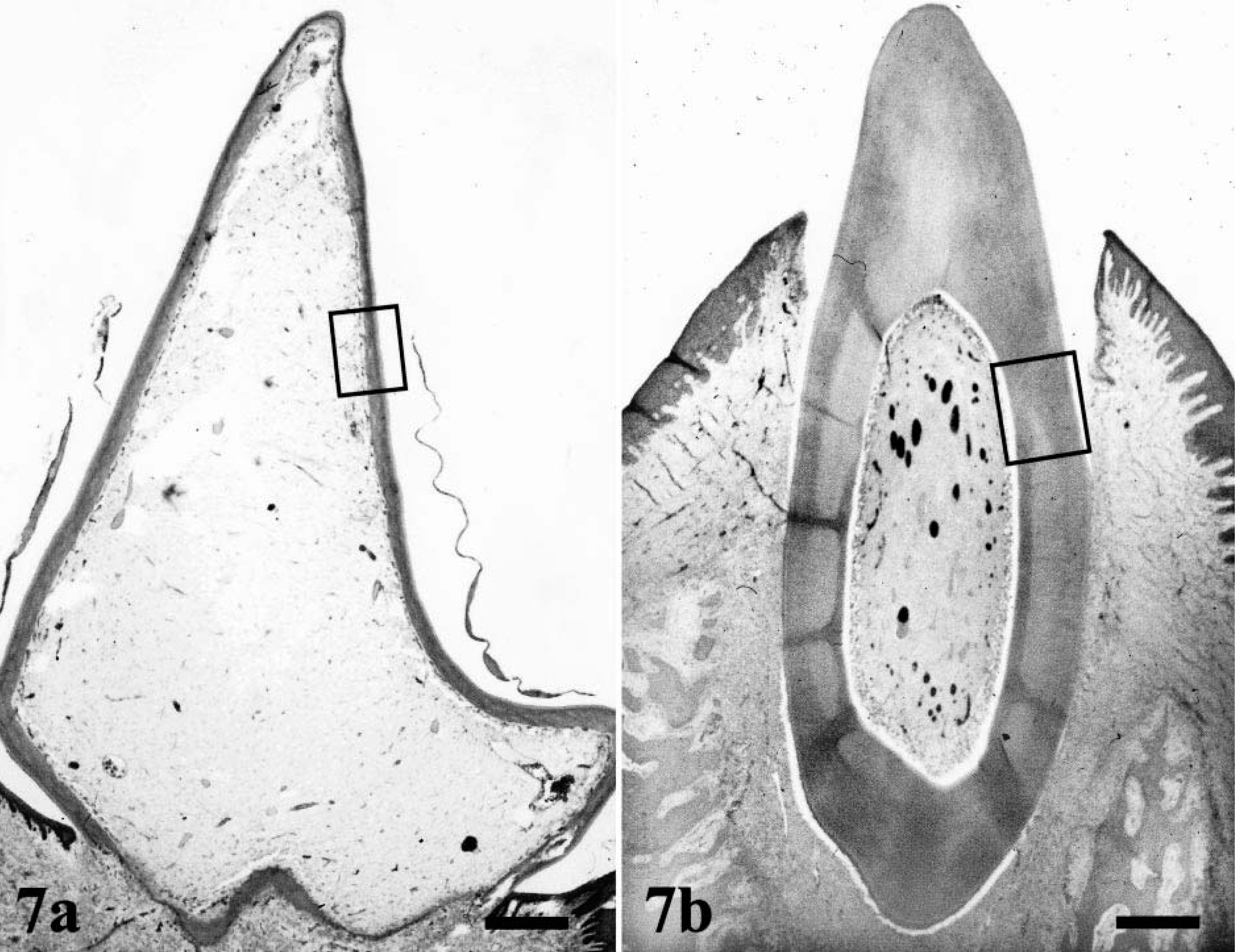
Left second incisor, mandible; enamel layers are absent after demineralization procedure:
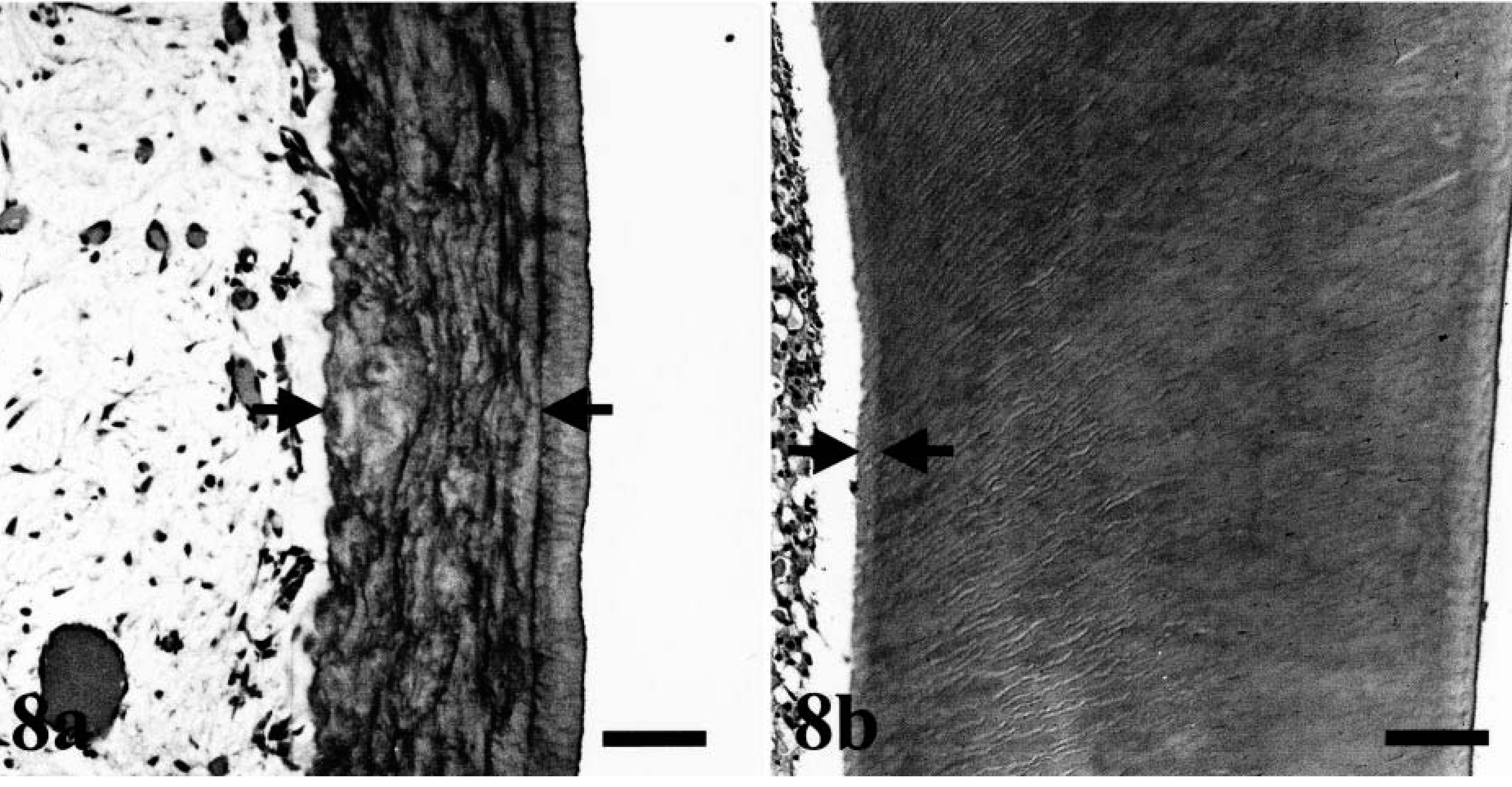
Dentine layer:
A slightly increased bone marrow density of all cell lines was observed in the affected dogs compared with control dogs.
Ultrastructurally, thin layers of osteoid were present on all trabecular surfaces of affected bones. The collagen fibers were reduced in number but showed a regular, banded pattern. The mineralization front was thin, and there were cartilage cores in large, poorly mineralized areas. Some osteoblasts contained cytoplasmic vacuoles and had a moderate dilatation of the rough endoplasmic reticulum (Fig. 9).
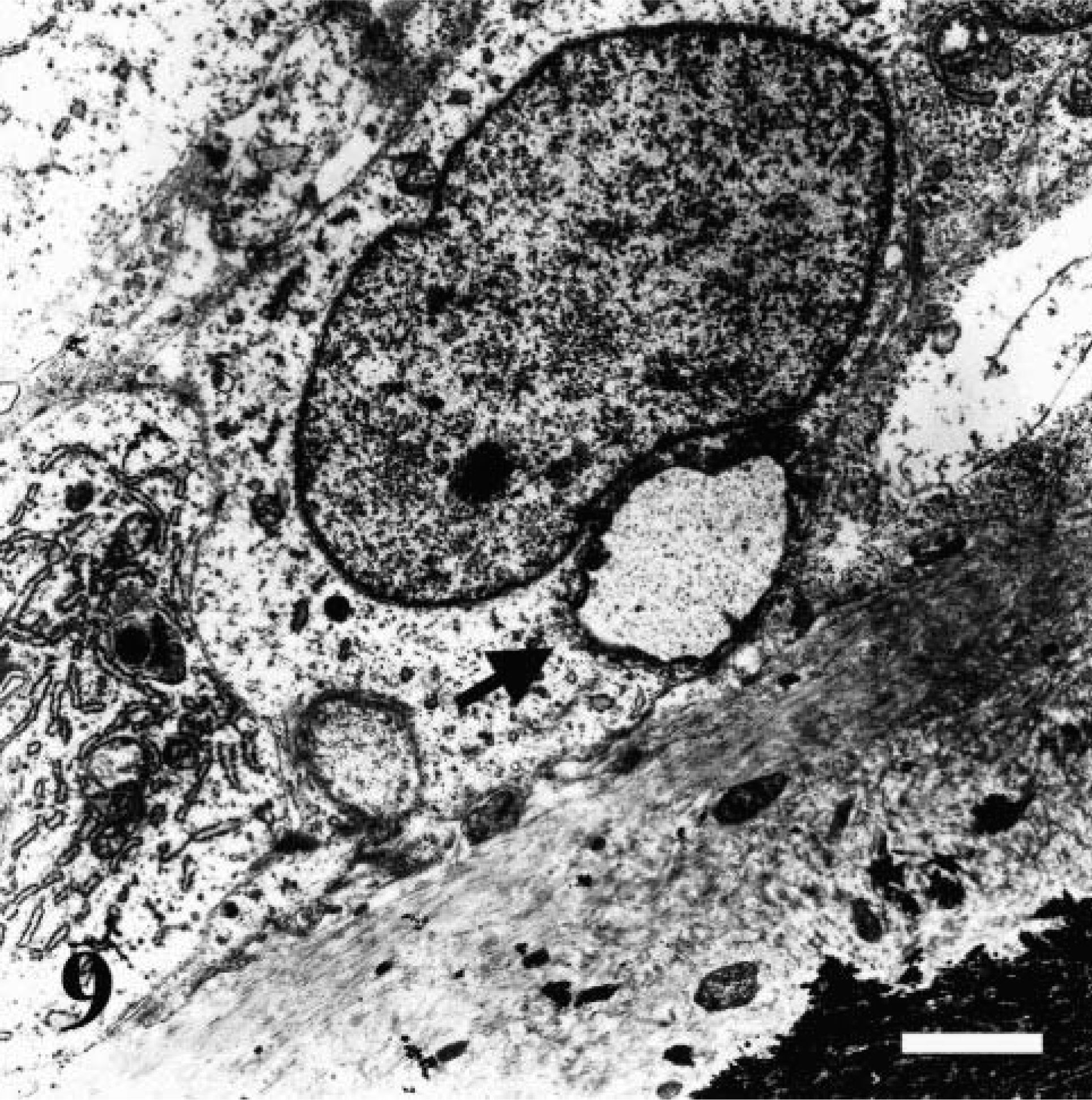
Transmission electron micrograph. Femoral physis; dog No. 4. Thin layers of regular osteoid are present on the mineralization front. Some osteoblasts contain large vacuoles (arrow). Uranyl acetate counterstain. Bar = 1.5 µm.
The molecular analysis of the collagen genes COL1A1 and COL1A2 revealed one nucleotide substitution within the COL1A1 cDNA and six nucleotide substitutions in the COL1A2 cDNA compared with the sequences in the database. These nucleotide differences were present in the affected dogs and in their unaffected littermates (Table 3). Four of the seven nucleotide changes lead to amino acid exchanges, whereas the other three were silent mutations.
Variations of the COL1A1 and COL1A2 genes.
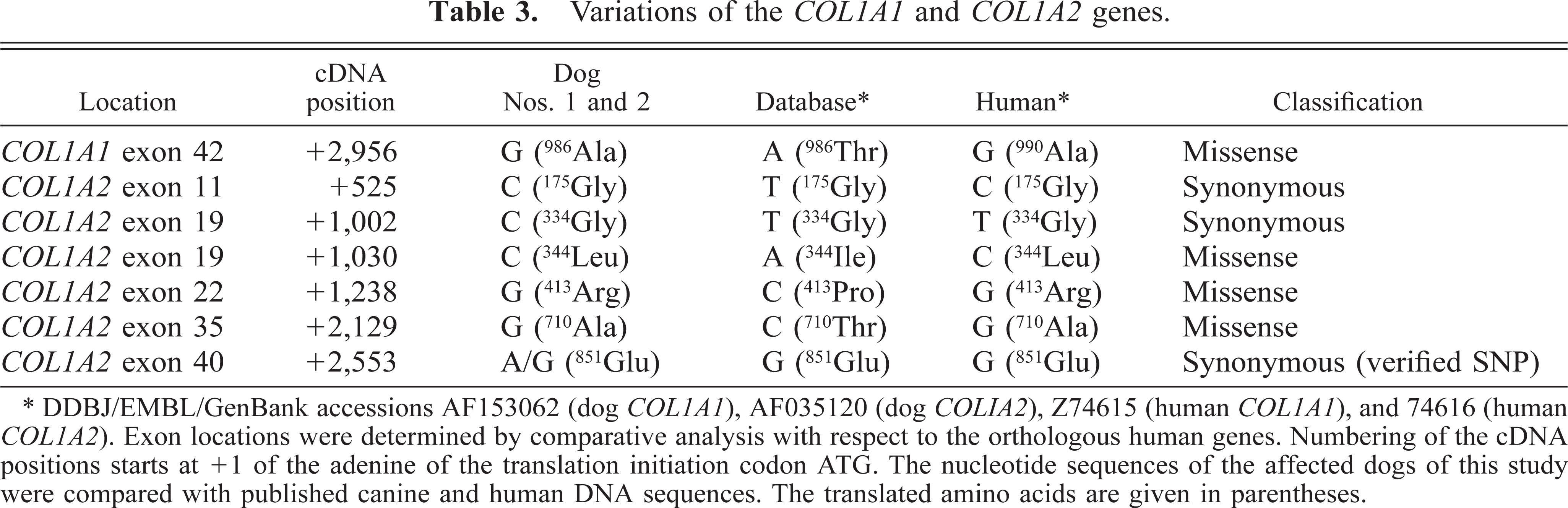
∗DDBJ/EMBL/GenBank accessions AF153062 (dog COL1A1), AF035120 (dog COL1A2), Z74615 (human COL1A1), and 74616 (human COL1A2). Exon locations were determined by comparative analysis with respect to the orthologous human genes. Numbering of the cDNA positions starts at +1 of the adenine of the translation initiation codon ATG. The nucleotide sequences of the affected dogs of this study were compared with published canine and human DNA sequences. The translated amino acids are given in parentheses.
Discussion
The clinical signs of OI in these five rough-coated Dachshund puppies started within the first 3 months of life, as has been reported for Golden Retrievers, Collies, Beagles, and Bedlington Terriers. 6,13,22,29 The early onset of the clinical disease, the involvement of male and female puppies from different litters with a common sire, and the number and sex of affected animals suggested a hereditary, most likely autosomally recessive, condition. The affected dogs exhibited symptoms reflecting severe osteopenia and dentinopenia but showed no significant changes in hematology, serum chemistry, and hormonal levels, including ionized calcium and parathyroid hormone, as in the previous descriptions of OI in dogs and humans. 6,13,22,27,28,35 Therefore, metabolic diseases such as primary and secondary hyperparathyroidism and hypothyroidism or nutritional osteoporosis could be ruled out. 15,27,28 The main pathomorphologic features were extensive, generalized bone fragility due to a paucity of cancellous and cortical lamellar bone in the absence of malformation, deformities, or dwarfism. In contrast, Bedlington Terriers have been reported with severe malformation of the pelvis, long bones, and vertebral column. 22 In young Collies, moderate deformities of extremities have been interpreted to be the result of multiple microfractures. 29 Brachygnathia inferior, skin fragility, and malformation in long bones also were common features in six cases of lethal OI in New Zealand Romney lambs. In that report, an increase in the diaphyseal diameter of long bones in combination with an excessive formation of spongiosa, resulting in a decreased space of the medullary cavity, also was described. 2
The histopathologic changes observed here in these Dachshunds are strongly suggestive of decreased bone formation. A common feature of OI is the normal appearance of growth plates and primary spongiosa, whereas lamellar bone formation is deficient. 6,22,27,28,33–35 The present cases of OI are similarly characterized by the failure of development of secondary spongiosa and by defective membranous ossification. The degree of histologic change can be highly variable in OI. 5,9,28,32,34 Although some humans showed an approximately normal content and architecture of cancellous bone, there have been reports of severe, nonlethal cases of OI, similar to those described here. 27 Additionally, the loss of bone due to hyperparathyroidism would lead to an increased osteoclastic activity, with increasing number and size of osteoclasts and lacunae, increasing serum calcium and parathyroid hormone levels, and in later stages fibrous osteodystrophy. 15
Similar to the present cases of OI, decreased secondary spongiosa has been reported in Golden Retrievers with OI. 6 However, that report gave no additional information about the quality and size of the residual spongiosa. In sheep, trabeculae have been reported to be increased in number but were found to be very narrow and covered by a thin layer of basophilic bone tissue. 2 The small amount of fibrous tissue with the moderate new bone formation observed in the mandible of dog No. 3 and the periostal new bone formation in dog Nos. 2 and 4 was interpreted as healing microfractures.
Because the organic matrix of dentine is composed primarily of collagen type I, alterations of collagen metabolism also should influence dentine development in teeth. For this reason, dentinogenesis imperfecta has been consistently observed in humans and also has been described in calves and Bedlington Terriers. 1,2,6,13,22,28
Ultrastructural changes were moderate. The cytoplasmic vacuolation of osteoblasts could be the result of an intracytoplasmic accumulation of procollagen caused by a secretion defect, as has been described in humans. 33 Furthermore, there are only a few reports that correlate specific types of OI with ultrastructural changes. 28
Most cases of human OI are due to an autosomal dominant inheritance caused by point mutations, leading to single-base changes that cause substitutions of glycine, or exon deletions within one copy of the COL1A1 or COL1A2 gene. 24,25 A recently reported mutation in the canine COL1A1 gene also led to a substitution of glycine with alanine at position 208 and was responsible for a severe case of OI. 6 However, autosomal recessive–inherited humans also are known. 27 In some of these forms and in several dominantly inherited forms of OI, the collagen type I genes have been excluded as disease loci. 20 It has been suggested that the best candidates for the causative gene(s) in these COL1A1 and COL1A2 unlinked cases are the 15 genes involved in the posttranslational modification steps required for the maturation of the nascent procollagen monomers. 20,24,36 In the affected Dachshunds studied here, seven nucleotide changes in the COL1A1 and COL1A2 cDNAs were detected, including four missense mutations, which were, however, also present in the healthy control dogs. At these four positions, the sequences were identical to the human COL1A1 and COL1A2 sequences. Therefore, it is unlikely that the encoded proteins were defective. A possible explanation for the observed nucleotide discrepancies with respect to the previously published canine sequences could be a natural variation between different dog breeds or sequencing or PCR errors. At least one of the silent base substitutions represented a single-nucleotide polymorphism because both alleles were found in several control animals.
In summary, OI is a rare disease in a group of inherited disorders of the connective tissue. It is characterized by severe, generalized osteopenia and dentinopenia. Pathomorphologic features strongly indicate a pathogenesis of reduced bone and dentine formation due to defective production of collagen type I.
Footnotes
Acknowledgements
We thank Professor Steve Weisbrode (Department of Veterinary Biosciences, Ohio State University, Columbus, OH) for his comments on the histopathology of these OI cases.