
Abstract
Apoptosis can be defined as a carefully regulated process, characterized by specific morphologic and biochemical features. It is initiated by both physiologic and pathologic stimuli, and its full expression requires a signaling cascade in which caspase activation plays a central role. Knockout mice lacking key genes encoding proteins constituting the core apoptotic cascade have helped us to establish the functional hierarchy of the mechanisms controlling apoptosis in animal development and, to a lesser extent, in disease. Induced mutant mice have also revealed the intimate crosstalk between apoptotic and other homeostatic pathways and have defined distinct temporal and tissue-specific roles of individual apoptotic effectors. Eliminating genes controlling caspase-dependent apoptosis can convert an apoptotic phenotype to a necrotic one, both in vitro and in vivo. This suggests that necrosis and apoptosis represent morphologic expressions of a shared biochemical network through both caspase-dependent mechanisms as well as non-caspase-dependent effectors such as cathepsin B and apoptosis-inducing factor. The cell death program, whether by apoptosis or necrosis, is mediated through an integrated cascade, which can be accessed at multiple sites, and propagated through numerous branch points. An understanding of the physiologic conditions that influence these decisions is required to adequately prevent, or induce, cell death.
Although the morphologic features of apoptotic cell death were noted as early as 1885 by Fleming, 22,62 the term apoptosis was not used until 1971, when it was introduced by Kerr. 32 Historically, apoptosis defined a type of cell death that was not only morphologically distinct from necrosis but also possessed distinct biochemical and molecular features. 33 The impact of this distinction was far-reaching, in basic science and clinically, because it implied the existence of an orderly genetic program, which could potentially be manipulated to prevent or to induce cell death. The genes encoding major portions of the apoptotic network have since been cloned, and their in vivo function has been explored using genetically altered mice. This has been enormously helpful in establishing the functional hierarchy of genes controlling apoptosis in animal development. Additional complexities, which are rooted in the intimate crosstalk between the cell death machinery and other homeostatic mechanisms, have emerged. Whether a cell dies and whether death occurs by apoptosis or necrosis is dependent on physiologic milieu, developmental stage, tissue type, and the nature of the cell death signal.
Developing tissues walk an exquisitely fine line between proliferation and death. For a tissue to grow, it must resist apoptosis. However, cell subpopulations of cells at specified times and locations must succumb to apoptosis for the tissue to assume its normal form and function. This dichotomy underscores the intimate relationship between mechanisms of development and neoplasia and reflects the intersection between the apoptotic pathway and cell-cycle machinery. 35 Knockout (KO) animals for components of the central apoptotic cascade primarily reflect the impact of apoptosis on development. 29,88 Embryonal lethality and discrete developmental defects of central nervous and immune systems predominate. 84 These experiments reveal distinct temporal and tissue-specific roles of individual apoptotic effectors in animal development. In addition, they confirm that eliminating key components of the apoptotic cascade will not prevent cell death but may change its form from primarily apoptotic to primarily necrotic. 59,61 This finding suggests that differing morphologies can result from alternate expressions of a shared biochemical network.
Both necrotic and apoptotic morphologies occur in the hepatic ischemia model originally used by Kerr to introduce the term apoptosis. 32 Given the intensity of the debate regarding the precise definitions of apoptosis and necrosis that these phenomena can coexist within the same lesion prompts consideration of whether drawing such distinctions is essential. Although facets of the apoptotic machinery are activated in many pathologic conditions, a host of physiologic and environmental factors can influence the segregation of downstream cellular events so that apoptotic or necrotic morphology prevails. Heterogenous toxic, 65 ischemic, 70 degenerative, 64 and immunologic 41 stimuli that are typically associated with necrosis can induce the apoptotic phenotype. Stimuli that may under some circumstances result in apoptosis, can under other circumstances (particularly energy depletion or reduced caspase activation) induce a necrotic phenotype. 59,61 In addition, lethal stimuli may use mechanisms that are caspase independent, such noncaspase proteases 58 and apoptosis-inducing factor (AIF). 7
The diversity of roles in which apoptosis is central resist simplification. It is clear that apoptotic mechanisms are part of a biochemical continuum, which are reflected by a similar diversity of morphologic features. This review begins with a brief discussion of morphologic and biochemical features of apoptosis and necrosis. The phenotypes of mice that lack one or more apoptotic components are used to illustrate the fundamental processes controlling developmental cell death, as well as how these mechanisms are highly context specific. Finally, means by which the core apoptotic pathway can be subverted to induce a necrotic phenotype are described.
Apoptosis as a Morphologic Descriptor
At the light microscopic level, the morphologic features of irreversible cell death are easily detectable and include any of the following: in the nucleus—karyolysis, pycnosis, and karyorrhexis; and in the cytoplasm—condensation, swelling, loss of cytoplasmic detail, and fragmentation. 46 Of these changes, apoptosis is characterized by the sequence of chromatin margination and fragmentation, cellular shrinkage, budding, fragmentation, and engulfment by neighboring cells. 33 This morphology predominates in developing tissue, as well as in adult tissue responding to certain physiologic stimuli. The dying cells whose morphology deviates from this definition are labeled as succumbing to necrosis. 46 Terms such as oncosis, 46 cytoplasmic cell death, 12 autophagic cell death, 6 or paraptosis 78 describe specific variants of necrotic cell death. The confusion surrounding these terms has arisen largely through attempts to ascribe a specific mode of death to a particular morphology. This approach fails when confronted by processes in which both necrotic and apoptotic phenotypes result from the same stimulus. Coexistence of necrotic and apoptotic morphologies characterizes both developmental 8 and many acquired disease processes. 41,65,70 There is an emerging concept that characteristic apoptotic morphology is the result of a caspase-dependent cascade and that deviation from this pathway will result in cell death with a spectrum of necrotic morphologic features. 3,61 Ascribing a specific mode of death to a given morphologic appearance inevitably leads to confusion; however, it is not without value. As evidence accumulates linking specific components of the cell death machinery to particular variants of necrotic morphology, we can begin to understand the morphology of cell death in the context of a biochemical continuum, one end of which is occupied by classic apoptosis.
Apoptosis as a Biochemical Process
The morphologic features that characterize apoptosis are the result of a number of detectable biochemical features, all of which are induced by caspase-induced proteolytic destruction of cytoskeletal and metabolic proteins. 26,72 These include poly(adenosine 5′ diphosphate-[ADP]ribose) polymerase (PARP), deoxyribonucleic acid (DNA)–dependent protein kinase, lamin, protein kinase C, and actin. Apoptotic cells are reported to have a relatively low ratio of adenosine triphosphate (ATP) to ADP, apparently indicating decreased synthesis of ATP in the mitochondria. 17,42 Lipid membranes sustain progressive peroxidation, and the cell membrane composition alters. For example, in normal viable cells, phosphatidylserine is located on the cytoplasmic surface of the cell membrane. In apoptotic cells, phosphatidylserine is translocated from the inner to the outer leaflet of the plasma membrane, where it is recognized by macrophages, mediating phagocytic removal of the apoptotic cell. 16 Externalization of phosphatidylserine is an indicator of intermediate stages of apoptosis and can be detected by annexin V conjugates. 63
Immunohistochemical and biochemical detection of a growing number of apoptotic effectors can be performed. Caspase-3 is a key effector in the apoptosis pathway, amplifying the signal from initiator caspases (such as caspase-8) and signifying full commitment to cellular disassembly. 79 Caspase-3 activation can be detected immunohistochemically using an antibody to the cleaved portion or biochemically using caspase substrates. 1 Nuclear fragmentation, mitochondrial membrane potential flux, and caspase-3 activation apparently precede phosphatidylserine externalization during apoptosis, whereas permeability to propidium iodide (a marker of lost membrane integrity) and cytoskeletal collapse occur later. 72 Of the available tissue-based methods to detect apoptosis, terminal deoxyuridine triphosphate nick end labeling (TUNEL) and immunohistochemical detection of activated apoptotic effectors (e.g., caspase-3) are most suited for formaldehyde-fixed, paraffin-embedded material. Examples of reagents useful in mouse tissues include the In Situ Cell Death Detection Kit (a TUNEL kit from Roche Diagnostics Corporation, Indianapolis, IN) and antibodies against cleaved caspase-3, lamin A, and PARP (Cell Signaling Technology, Inc., Beverly, MA). TUNEL assays are widely used for detecting DNA fragmentation that occurs in apoptotic cells. 72 Once the cells are fixed, DNA strand breaks can be detected in situ using mammalian terminal deoxynucleotidyl transferase, which covalently adds labeled nucleotides to the 3′-hydroxyl ends of these DNA fragments in a template-independent fashion.
Recently, in vitro evidence suggests that the overlap between apoptosis and necrosis is not limited to morphology. 11,37 Necrosis from freezing can produce an orderly pattern of DNA fragmentation despite pharmacologic pan-caspase suppression. 37 Therefore, TUNEL staining, which is an indication of chromatin fragmentation, should not be considered proof of an exclusively apoptotic process. Phagocytic removal of nuclear and cytoplasmic fragments has long been considered a hallmark of apoptosis. Again, evidence in the nematode C. elegans suggests that phagocytosis is not an exclusive process and is used to remove cells killed by both apoptotic and necrotic processes. 11 Consequently, it would be unwise to rely exclusively on any single diagnostic method to differentiate between the morphologic variants of cell death.
Mouse Models of the Core Apoptotic Cascade
Two distinct but interconnected apoptotic pathways have been characterized—the cell surface death receptor pathway (extrinsic) and the mitochondria-initiated (intrinsic) pathway (see Fig. 1 and Tables 1, 2). Both pathways terminate in activation of effector caspases, which mediate the proteolytic events characterizing apoptosis. 5,26 Intracellular signals are primarily transduced through the intrinsic program, whereas extracellular ligands use the cell surface death receptor pathway. Downstream events initiated by the latter pathway result either in direct caspase activation or in caspase activation indirectly through mitochondria. An additional, less well-characterized intrinsic cell death pathway has also been described. 66 Accumulation of misfolded proteins and alterations in Ca2+ homeostasis in the endoplasmic reticulum (ER) cause ER stress and lead to cell death.
Abbreviations and current terminology for proteins and mechanisms involved in apoptosis.

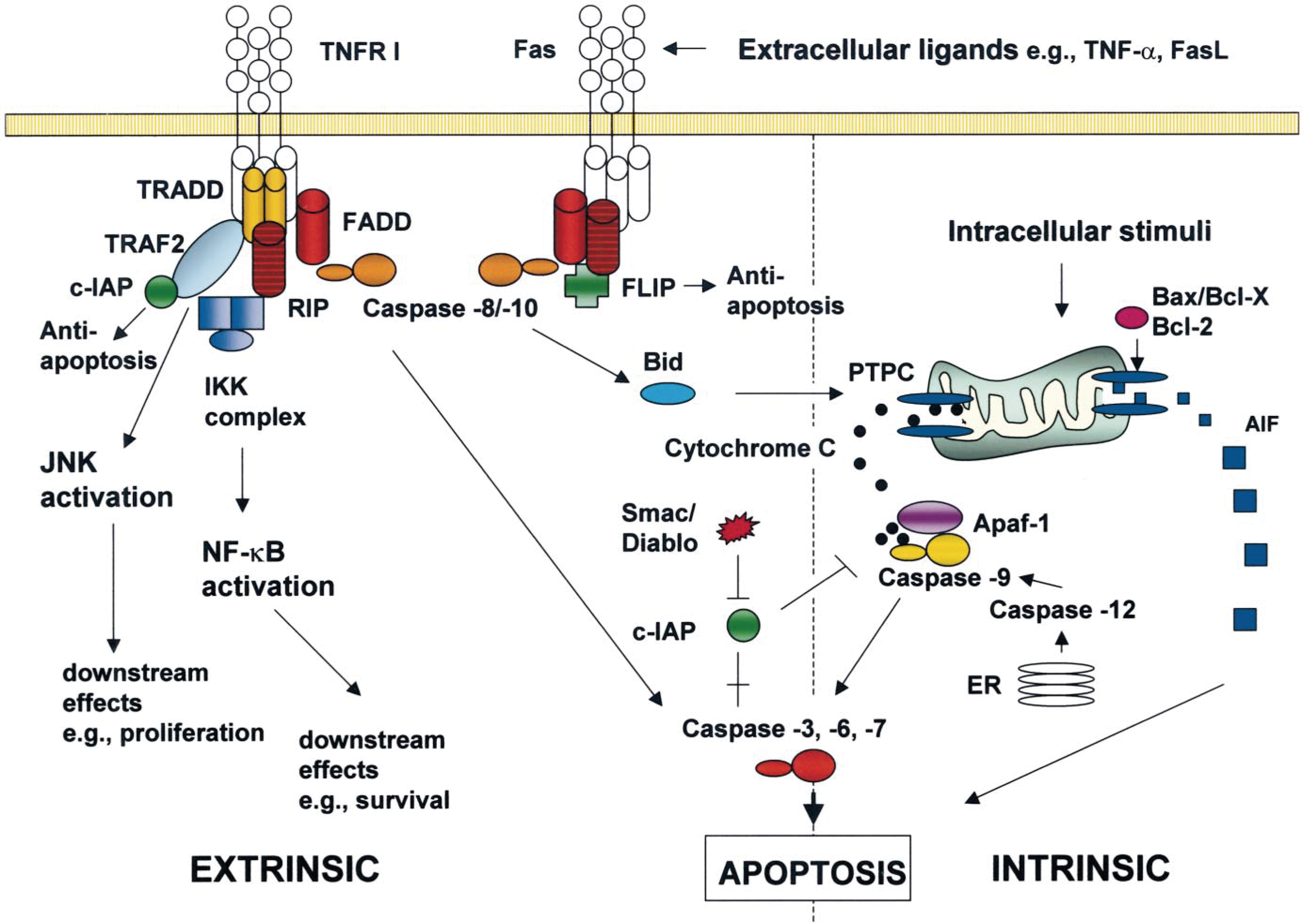
Major components of the core apoptotic cascade. The two major apoptotic pathways—the cell surface death receptor pathway (extrinsic) and the mitochondria-initiated (intrinsic) pathway are depicted. The extrinsic pathway emanates from extracellular stimuli, which are transduced through membrane-associated receptors of the TNFR superfamily. Intracellular stimuli induce apoptosis primarily through the mitochondria and, to a lesser extent, through the ER. Only those proteins or processes for which in vivo roles have been identified using genetically modified mice (see Table 2) are included in the figure.
Phenotypes of knockout mice lacking components of the core apoptotic pathways. ∗
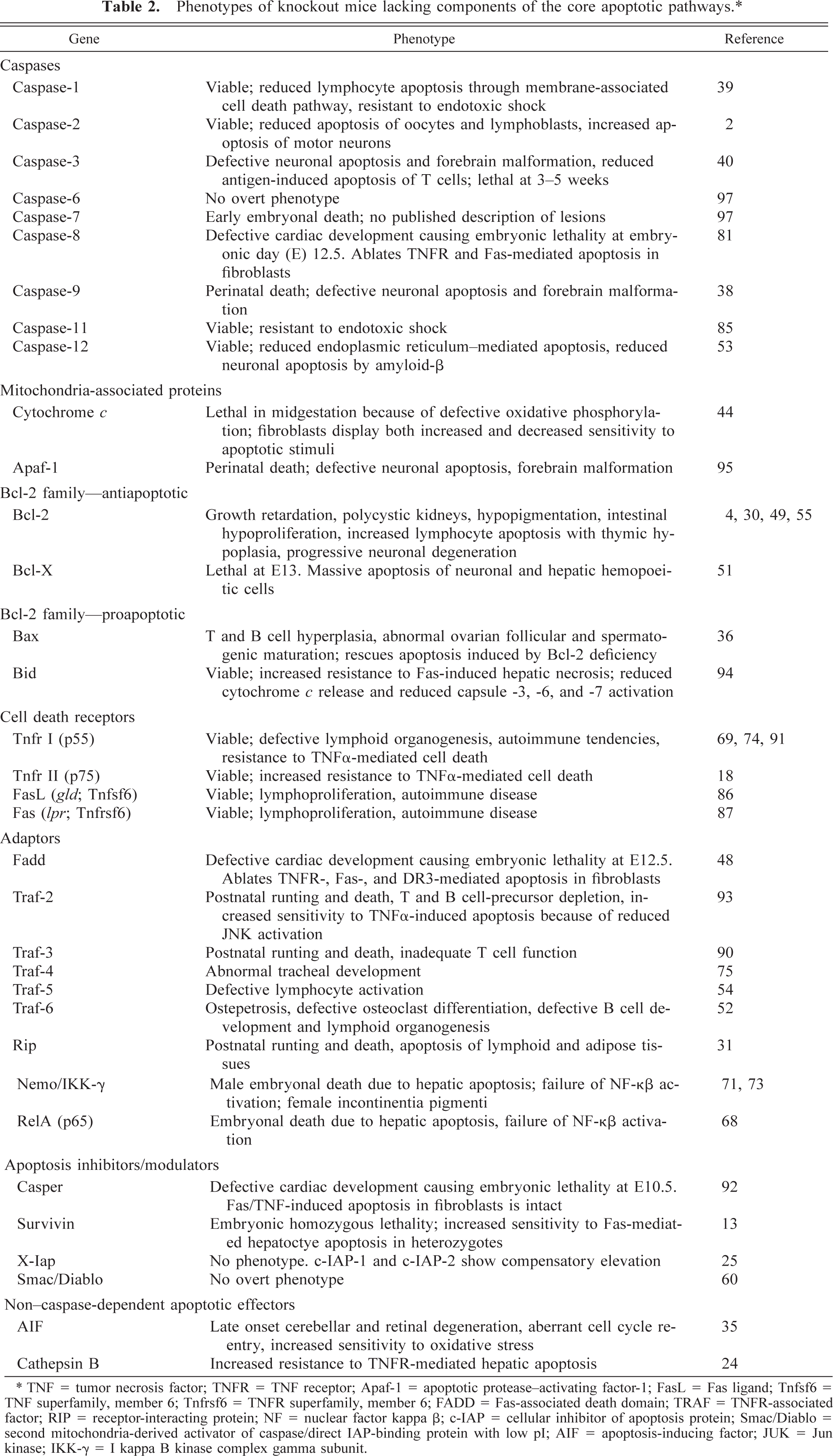
∗ TNF = tumor necrosis factor; TNFR = TNF receptor; Apaf-1 = apoptotic protease-activating factor-1; FasL = Fas ligand; Tnfsf6 = TNF superfamily, member 6; Tnfrsf6 = TNFR superfamily, member 6; FADD = Fas-associated death domain; TRAF = TNFR-associated factor; RIP = receptor-interacting protein; NF = nuclear factor kappa β; c-IAP = cellular inhibitor of apoptosis protein; Smac/Diablo = second mitochondria-derived activator of caspase/direct IAP-binding protein with low pI; AIF = apoptosis-inducing factor; JUK = Jun kinase; IKK-γ = I kappa B kinase complex gamma subunit.
Caspases
Caspases (the family of cysteine proteases related to mammalian interleukin-1β–converting enzyme) exist as dormant proenzymes, which when activated by proteolysis cleave a host of cellular substrates at specific aspartate residues. 26,57 Their activation requires proteolytic cleavage between their three domains: N-terminal prodomain, large P20, and small P10 domains. They constitute a hierarchical cascade in which initiator caspases (caspase-8, -9, and -10) are activated in response to apoptotic stimuli and in turn, activate the effector caspase-3, -6, and -7. Caspase-8 and -10 share similar structural features (a death effector domain, DED), which allows them to interact with DED-containing adaptor molecules and thus transduce apoptotic signals from cell surface death receptors to the mitochondria 9 or to effector caspases. Caspase-9 is a major component of the apoptosome (see Table 1 and below) and is activated in response to increased mitochondrial permeability. Effector caspases mediate the biochemical and morphologic features constituting apoptosis. 79 Apoptotic stimuli emanating from the ER require activation of caspase-9 by caspase-12 66 and appear to result in cell death in a mitochondria-independent fashion.
Knockout mice. The results of KO mouse studies indicate that caspase functions can be divided into two broad phenotypic classes—those which primarily affect development and those which mediate immune system function. 84,98 The limitation of developmental abnormalities to specific organ systems or anatomic regions in mice deficient for apoptosis-associated molecules suggests that individual cell populations depend on specific apoptotic mechanisms during specific points of their development. Ablation of caspase-1, -11, and -12, 39,53,85 thought to primarily mediate the inflammatory response, has no overt consequences on mouse development but does display defects of immune system structure and function. Mice that lack caspase-12 are resistant to ER stress-induced apoptosis, but their cells undergo apoptosis in response to other death stimuli. 53 Caspase-2 KO mice develop without an overt phenotype but exhibit dichotomous, tissue-specific apoptotic defects. 2 For example, they develop excessive, apoptosis-resistant oocytes but experience accelerated apoptosis of facial motor neurons during development. Caspase-8 KO mice die in utero at embryonic day (E) 11.5 and exhibit impaired formation of cardiac muscles and marked abdominal congestion. 81 Retarded developmental apoptosis in caspase-9 KO mice results in perinatal death accompanied by severe cortical malformation associated with supernumerary cells. 38 A similar but more variable and less severe outcome is induced by caspase-3 ablation. 40 These mice may live for approximately 1 month after birth before succumbing with similar cerebral malformations.
Elimination of the other two effector caspases, caspase-6 and -7, results in vastly divergent phenotypes. Caspase-6 KO mice develop normally; however, eliminating caspase-7 results in embryonal lethality. 97 Taken together, these data suggest that the requirements for caspase-3, -6, and -7 during murine development differ drastically. Of the three effector caspases, only caspase-3 has been found to be an essential mediator of the morphologic and biochemical hallmarks of apoptosis. 97 Cells from caspase-3 KO mice display reduced cytoplasmic bleb formation, DNA fragmentation, and reduced cleavage of known caspase substrates such as lamins, DFF45, and fodrin-alpha. Nevertheless, caspase-3–deficient cells undergo apoptosis as readily as wild-type cells, implying that caspase-6 and -7 play distinct roles in the execution of apoptosis.
Background strain has a tremendous impact on expression of the induced genetic defect. For example, caspase-3 KO mice on a B6;129X1/SvJ background die during the perinatal period and exhibit marked neural precursor cell expansion and exencephaly. 43 In contrast, caspase-3 KO mice on a C57BL/6J background reach adulthood, are fertile, and show minimal brain pathology. 43 Intercrosses of C57BL/6J and B6;129X1/SvJ mutants reveal that the vast majority of caspase-3 KO F1 mice display the severe B6;29X1/SvJ-like phenotype. 43 These findings are consistent with incompletely penetrant strain-dependent genetic modifier(s), which alters the neurodevelopmental consequences of caspase-3 ablation.
The role of mitochondria
The acquisition of the apoptotic phenotype, including characteristic changes in nuclear morphology and biochemistry (chromatin condensation and DNA fragmentation), depends on the action of apoptogenic proteins released from the mitochondrial intermembrane space. 96 The release of cytochrome c from the intermembrane space is the commitment step in both the intrinsic and extrinsic pathways. In the cytosol, cytochrome c interacts with apoptotic protease–activating factor-1 (Apaf-1) to form the apoptosome. Apaf-1 normally resides in an inert state in the cytosol; however, on its interaction with cytochrome c, it recruits and activates procaspase-9. Activated caspase-9 in turn cleaves the effector caspase-3, -6, and -7 to execute apoptosis. 77
The precise mechanisms through which cytochrome c is released remain elusive. Increased mitochondrial permeability can occur by activation of the permeability transition pore complex (PTPC or “megachannel”). 26 The PTPC regulates matrix Ca2+, pH, and transmembrane potential. It comprises the adenine nucleotide translocator (inner membrane), the voltage-dependent anion channel (outer membrane), and cyclophilin D (matrix). Activation of the PTPC by a host of proapoptotic stimuli (elevated Ca2+, reactive oxygen species, ceramide metabolites, and p53-mediated changes in redox potential) creates a pore spanning both inner and outer mitochondrial membranes. This results in release of cytochrome c into the cytoplasm and loss of the transmembrane potential. However, studies describing retention of mitochondrial transmembrane potential during initial stages of apoptosis suggest an alternative route of cytochrome c release via specific permeabilization of the outer mitochondrial membrane, possibly by BH3-only (see Table 1 for definition) members of the Bcl-2 family. 26
Increased mitochondrial permeability through the PTPC or through selective outer membrane permeabilization is controlled by the Bcl-2 family of proteins. 5,26 The Bcl-2 family comprises both pro- and antiapoptotic members that function by promoting or retarding cytochrome c release, respectively. Bcl-2 and Bcl-X are well-characterized inhibitors of apoptosis and retard cytochrome c release by preventing opening of the megachannel. 5,26 Their cytoprotective effects can be counteracted by the BH3-only members of the Bcl-2 family, which, in mammals, include Bid, Bad, Bik, Bim, Blk, Hrk, and Noxa. 5,26 The similarity of these proteins to the Bcl-2 family resides only in their 18–amino acid BH3 domain. They promote cytochrome c release by oligomerizing within the mitochondrial membrane. Bid is an important bridge between the cell surface death receptor pathway and the mitochondria. 19 When cleaved by caspase-8, Bid will translocate to the mitochondrial membrane, where it induces cytochrome c release.
Knockout mice. Apaf-1 KO mice display perinatal mortality with cerebral malformation similar to that seen in caspase-3 and -9 KO mice. 95 Embryos defective in cytochrome c die by midgestation. 44 Cells derived from these embryos display reduced caspase-3 activation and are resistant to some apoptotic stimuli. However, they display enhanced sensitivity to apoptosis induced by tumor necrosis factor–alpha (TNFα), indicating that apoptosis by the cell surface death receptor pathway is not cytochrome c dependent.
Bcl-2 KO mice exhibit pleiotropic abnormalities, which include accelerated apoptosis of thymus and spleen, polycystic renal disease, hypopigmentation attributable to melanocyte loss, intestinal dysplasia, and insufficient long-bone growth due to osteoblast abnormalities. 4,30,55 In the postnatal nervous system, substantial degeneration of motor, sensory, and sympathetic neurons occurs after the physiologic cell death period, suggesting that Bcl-2 is crucial for the maintenance of specific neuronal populations during the early postnatal period. 49 Elimination of a related gene, Bcl-X, results in an accelerated and more severe phenotype. 51 Bcl-X KO mice die around E13, with extensive apoptotic cell death in postmitotic immature neurons of the brain, spinal cord, and dorsal-root ganglia as well as in hematopoietic cells in the liver.
Bax KO mice are viable but display lineage-specific aberrations in cell death. 36 Thymocytes and B cells are hyperplastic, and Bax-deficient ovaries contain atretic follicles with excess granulosa cells. Bax KO males are infertile as a result of disordered seminiferous tubules with an accumulation of atypical premeiotic germ cells but with no mature haploid sperm. In the nervous system, Bax KO mice display reduced developmental cell death of Purkinje and retinal precursors. 21,50 Bid KO mice are viable and are resistant to apoptotic stimuli emanating from cell surface death receptors. 94
The cell surface death receptor pathway
The intrinsic pathway may be modulated at several levels. Extracellular stimuli may use molecules constituting the cell surface death receptor pathway. The majority of these receptors are members of the TNF receptor (TNFR) superfamily. 9 The intracellular domains of these receptors associate with adaptor molecules, which then initiate a complex of downstream signaling events controlling proliferation, apoptosis, immune regulation, inflammation, and stress responses, as well as apoptosis. The majority of the intracellular events after TNFα binding are mediated by TNFR type I (TNFR I; p55), with the role of TNFR type II (TNFR II; p75) being less clearly understood. Both p55 and p75 are coexpressed on cells, and both bind to TNFα. 45 After TNFα binding, aggregation of intracellular domains of TNFR I is recognized by the adaptor protein Fas-associated death domain (FADD), which recruits procaspase-2, -8, and -10 to the complex and results in their activation. Activated caspase-8 is a central upstream mediator of the death receptor apoptotic pathway. It can directly activate the effector caspase-3, -6, and -7 and can indirectly activate the mitochondria-initiated pathway by cleavage and translocation of Bid to the mitochondrial membrane. The membrane-associated receptors possess additional cytoplasmic domains, which recruit additional adaptors such as TNFR-associated death domain (TRADD). TRADD in turn recruits additional adaptor proteins, receptor-interacting protein (RIP), and TNFR-associated factor 2 (TRAF-2), ultimately resulting in activation of two major transcription factors—nuclear factor kappa β (NF-κβ) and c-Jun. 9 These mediate a number of downstream signaling pathways controlling proliferation and survival and provide a good example of how intimately apoptotic mechanisms are associated with other facets of cellular homeostasis. For example, in the absence of NF-κβ activity, cellular susceptibility to TNFR I–mediated apoptosis increases, 71 and mice deficient in the NF-κβ–transactivating gene Rel A (p65) die at E14–15 with massive liver apoptosis. 68 NF-κβ exerts antiapoptotic effects through an endogenous caspase inhibitory system mediated by cellular inhibitor of apoptosis protein 2 (c-IAP-2), which subsequently inhibits caspase-3 activation. 34
Fas is a death domain–containing member of the TNFR superfamily and plays a central role in the regulation of apoptosis, particularly in the immune system. The Fas ligand (FasL) gene is transcriptionally silent in most cells. 83 Once induced, FasL interacts with Fas, resulting in formation of a death-inducing signalng complex (DISC), which includes FADD and caspase-8 and -10. Autoproteolytic activation of these caspases initiates events similar to those induced by activation of TNFR I. The autoproteolytic activation of caspase-8 in DISC is regulated by FADD-like interleukin converting enzyme–like inhibitory protein (FLIP). 83 FLIP can be incorporated into the DISC of death receptors where it exists in isoforms, which are structurally similar to caspase-8 but lack enzymatic activity. 83
Knockout mice. Mice deficient for upstream components of the cell surface death receptor pathway display defects primarily in the immune system. 91 Mice that lack Tnfr I have defective organogenesis of splenic white pulp and Peyers patches, 69 are susceptible to certain infectious agents, and display accelerated atherosclerosis. 74 Tnfr II KO mice show normal T-cell development and activity but have increased resistance to TNFα-induced death. 18 Mice homozygous for lpr (originally for lymphoproliferation, due to a mutation in Fas; now TNFR superfamily, member 6 [Tnfrsf6]) or gld (originally for generalized lymphoproliferative disease, due to a mutation in FasL; now TNF (ligand) superfamily, member 6 [Tnfsf6]) develop lymphadenopathy and splenomegaly and suffer from autoimmune disease. 87 Northern hybridization with a FasL complementary DNA probe indicates that the lymphocytes accumulating in lpr and gld mice abundantly and constitutively express the FasL messenger ribonucleic acid. By means of in situ hybridization and immunohistochemistry, the cells from both lpr and gld mice can be identified as CD4−/−/CD8−/− double-negative T cells. 86
Mice that lack Fadd, the adaptor molecule bridging cell surface death receptors and caspase-8, display a phenotype similar to that displayed by caspase-8–deficient mice, 48 suggesting that these molecules occupy sequential positions in a hierarchical pathway. Mice with null mutations of Traf-2 appear normal at birth but become progressively runted and die prematurely. 93 Atrophy of the thymus and spleen and depletion of B-cell precursors also are observed. Thymocytes and other hematopoietic progenitors are highly sensitive to TNFα-induced cell death, and serum TNFα levels are elevated in these Traf-2 KO animals. 93 Mice deficient for other Trafs display phenotypes that primarily affect lymphocyte function. Traf-1 KO mice are viable and have normal lymphocyte development. 80 Traf-1–deficient T cells exhibit stronger proliferation to anti-CD3 mAb than wild-type T cells. Traf-3 KO mice appear normal at birth but become progressively runted. 90 Their T cells are functionally defective. Traf-5 deficiency results in defective lymphocyte activation. 54 Traf-6 KO mice exhibit severe osteopetrosis. 52 Myeloid-derived precursor cells from Traf-6 KO mice are unable to differentiate into functional osteoclasts. In addition, Traf-6 KO mice are defective in B-cell development, lymph node organogenesis, and interleukin-1 signaling in thymocytes. Lack of Traf-4 expression results in a localized developmental defect of the upper respiratory tract. 75 Traf-4 KO mice are born with a constricted upper trachea at the site of the tracheal junction with the larynx.
Rip KO mice appear normal at birth but fail to thrive, displaying extensive apoptosis in both the lymphoid and adipose tissues and dying at 1–3 days of age. 31 In contrast to a normal thymic anti-Fas response, Rip-deficient cells are highly sensitive to TNFα-induced cell death. Rip stimulates apoptosis, jun-kinase, and NF-κβ activation in vitro. Therefore, it appears that sensitivity to TNFα-mediated cell death in Rip KO cells is the result of a failure to activate the transcription factor NF-κβ. 31
Disruption of the X-linked gene encoding NF-κβ essential modulator/inhibitor of kappaB kinase gamma (Nemo/Ikk γ) produces male embryonic lethality attributable to hepatic apoptosis and completely blocks NF-κβ activation. 71 Heterozygous female mice develop patchy skin lesions with massive granulocyte infiltration and hyperproliferation as well as increased apoptosis of keratinocytes. Affected animals present with severe growth retardation and early mortality. Surviving mice recover almost completely, presumably through clearing the skin of Nemo/Ikk γ KO keratinocytes. Male lethality and strikingly similar skin lesions in heterozygous females are hallmarks of the human genetic disorder incontinentia pigmenti. 73
Apoptosis inhibitors/modulators
Apart from the Bcl-2 family members Bcl-2 and Bcl-X, which function by regulating mitochondrial cytochrome c release, at least two additional antiapoptotic mechanisms have been described. Casper (c-FLIP) associates with FADD and caspase-8 in signaling complexes downstream of death receptors like Fas. It appears to exert both proapoptotic effects (through caspase-8 activation) and antiapoptotic effects (through maintenance of cellular NF-κβ activity). 92 Members of the inhibitor of apoptosis (IAP) family, comprising c-IAP-1 and -2, X-linked IAP, and Survivin, regulate apoptosis downstream of apoptosome assembly by binding to and selectively inhibiting initiator and effector caspases. 23 Inhibition of apoptosome assembly by the IAPs can be counteracted by a proapoptotic molecule called second mitochondria-derived activator of caspase/direct IAP-binding protein with low pI (Smac/Diablo), which is released from mitochondria during apoptosis. 67 Smac activity, can in turn, be inhibited by its binding to survivin.
Knockout mice. Casper KO embryos do not survive past E10.5 and exhibit impaired heart development. 92 This phenotype is reminiscent of that reported for Fadd KO and caspase-8 KO embryos. However, unlike Fadd-deficient and caspase-8–deficient cells, Casper-deficient embryonic fibroblasts are highly sensitive to FasL- or TNFα-induced apoptosis. These results suggest that Casper has two distinct roles: to cooperate with FADD and caspase-8 during embryonic development and to mediate cytoprotection against death factor–induced apoptosis through maintenance of NF-κβ activity. 92 X-Iap KO mice are viable, and their cells do not demonstrate any defects in induction of caspase-dependent or -independent apoptosis in vitro. 25 The levels of c-Iap-1 and c-Iap-2 protein are increased, suggesting that a compensatory mechanism leads to upregulation of other family members when X-Iap expression is lost. Survivin KO mice fail to complete gestation. 13 In heterozygotes, the liver is more sensitive to Fas-induced apoptosis via the caspase-8-mitochondrial axis. Smac KO mice have a normal phenotype, suggesting the existence of redundant molecules capable of compensating for a loss of Smac function. 60
Caspase-independent apoptotic effectors
AIF can induce apoptotic cellular changes and mediates a caspase-independent pathway. 7 Under normal circumstances, AIF is secluded behind the outer mitochondrial membrane. However, on induction of apoptosis, AIF translocates to the cytosol and the nucleus. The Bcl-2 family regulates the release of AIF from the mitochondria. 14 An expanded role for AIF has been described in the Harlequin (Hq) mutant mouse. 35 Aged Hq mutants, express less AIF with increased susceptibility of cerebellar and retinal neurons to free-radical damage and resultant unscheduled cell cycle reentry and subsequent apoptosis. 35
Evidence for the role of apoptogenic proteases residing in the endosomal–lysosomal system in apoptosis is accumulating. Lysosomal cysteine peptidases cathepsin B and cathepsin L are ubiquitously expressed members of the papain family and contribute to the terminal degradation of proteins in the lysosome. Cathepsin B KO mice demonstrate increased resistance to apoptosis through the Tnfr pathway. 24 The reasons for this are unclear, although cathepsin B is thought to promote release of cytochrome c from the mitochondria. Cathepsin B substrates that are specifically cleaved during apoptosis are not known. 24
Apoptosis is a Cell Type– and Stimulus-Specific Process
This phenomenon is demonstrated clearly in many single-gene mutants (i.e., mice lacking individual caspase genes have distinct tissue-specific phenotypes) and can be more closely examined using double-mutant animals. These studies are of two general types. The first design examines apoptotic events in animals lacking two or more genes known to mediate functionally opposing components of the apoptotic process. For example, Bax ablation will not prevent embryonal death in Bcl-X KO mice but will prevent the premature apoptosis of neuronal precursors, 76 suggesting that Bcl-2 interacts with Bax in the nervous system but with another proapoptotic protein in hemopoeitic cells of the liver. In the absence of Rel A (an NF-κβ transactivating enzyme) the fetal liver is sensitized to Tnfr I–mediated apoptosis. 68 Mice that lack both Rel A and Tnfr I survive embryonic development and are born with normal livers without evidence of increased hepatocyte apoptosis. 68 These animals survive an average of 10 days, dying from acute hepatitis with an extensive hepatic infiltration of immature neutrophils.
The second scheme examines the effect of altered expression of pro- or antiapoptotic genes on the progression of apoptotic cell death in animals carrying a known genetic defect. Experiments of this nature are often used to evaluate the therapeutic potential of apoptosis inhibitors in treating the corresponding human diseases and to deepen the understanding of the specific apoptotic pathways at work. A good example is seen with mouse models of retinitis pigmentosa (RP), a genetic disorder of rod photoreceptors, resulting in their death by apoptosis. Overexpression of both Bcl-2 and Bag (both antiapoptotic mediators) in mouse models of RP will provide synergistic protection of photoreceptors, 10,20 indicating that the intrinsic pathway functions in the pathogenesis of the disease. On the contrary, elimination of Fas in tested models of RP fails to protect photoreceptors, 56 suggesting that the extrinsic pathway through Fas is not a critical mediator of apoptosis in this model.
The Apoptosis-Necrosis Continuum
The phenomenon of coexistent apoptotic and necrotic morphologies resulting from the same stimulus finds some explanation in the results of in vitro and in vivo studies. Two clearly identifiable factors that will convert an ongoing apoptotic process to a necrotic process are the availability of intracellular ATP 42 and the availability of caspases. 15 Uncoupling of mitochondrial oxidative phosphorylation is an inevitable consequence of loss of mitochondrial membrane potential. Release of cytochrome c disrupts electron transport, resulting in a drop in ATP production and generation of reactive oxygen species, both of which can contribute to cell death in the absence of caspases. In addition, because the apoptosome is dependent on ATP for its proapoptotic activity, 17,42 progressive energy depletion would result in a greater proportion of cells being committed to necrotic, rather than apoptotic, death. 59
Extensive in vitro evidence supports the dependency of apoptotic morphology on an intact caspase signaling pathway. These experiments use cells that are either deficient in a particular caspase or are subjected to pharmacologic caspase inhibition. For example, Fas-induced cell death in the absence of caspase-8 occurs with a necrotic morphology and without release of mitochondrial cytochrome c. In this case, an alternate pathway using Fadd and Rip results in caspase-independent cell death. 28,47 Similarly, caspase-independent, cell type–dependent necrosis can be induced by Bax, 89 TNF α, 82 or pharmacologic agents that typically produce apoptosis. 27 In vitro findings are borne out by in vivo results in genetically altered mice. Cells undergoing developmental cell death express both apoptotic and necrotic morphologies, although apoptotic morphology typically prevails. For example, limb buds from wild-type mice will display dying cells of both morphologies in interdigital regions. 8 In Apaf-1 KO mice, which fail to undergo caspase-3 activation, digit separation still occurs but is delayed. 8 In addition, the number of cells displaying nonapoptotic morphology increases dramatically. These experiments indicate that typical apoptotic morphology is caspase dependent and that elimination or inhibition of caspases does not prevent cell death but results in an increased prevalence of cells with nonapoptotic morphology. 8 Similar findings are noted in neuronal populations of caspase-3 and caspase-9 K mice. 61 As with limb development, cell death in the nervous system is delayed but when completed still results in normal organ morphology. Dying cells display a morphology that differs from that of typical apoptosis in that nuclear margination fails to occur and cytoplasmic vacuolation prevails. Reduced chromatin fragmentation is reflected by reduced TUNEL staining. 61
Conclusions
Apoptosis can be readily defined as a carefully regulated process, characterized by specific morphologic and biochemical features. It is initiated by both physiologic and pathologic stimuli, and its full expression requires a signaling cascade in which caspase activation plays a central role. Necrosis is less easily summarized. However, it would be a mistake to view necrosis as less orderly or less regulated. It is becoming increasingly clear that necrosis and apoptosis represent morphologic expressions of a shared biochemical network. The final form of a cell's death is highly dependent on its physiologic context at the time when the death signal is received. The signaling cascade that culminates in typical apoptosis is affected by a host of homeostatic mechanisms that not only create the tissue-specific pathology peculiar to mice lacking individual apoptotic proteins but also result in a spectrum of necrotic morphologies. Consequently, from a practical viewpoint, any single biochemical marker of apoptosis cannot be relied on to distinguish between the variants of cell death. From a therapeutic viewpoint, the issue of whether a cell dies by apoptosis or necrosis is arguably less relevant than it used to be. Attempts to prevent cell death reveal that if a cell is destined to die, it will find a way to do so, despite interference with key apoptotic mechanisms. Cell death, whether it is expressed by apoptosis or necrosis, is mediated through an integrated cascade, which can be accessed at multiple points and which contains numerous branch points through which the death program can be propagated. An understanding of how the physiologic milieu of the cell influences these decisions is required to adequately prevent, or induce, cell death.