
Abstract
This investigation found that genetic inactivation of mouse Atg4b, 1 of the 4 mammalian homologs of the autophagy-related gene Atg4, resulted in amorphous globular bodies in the neuropil of the deep cerebellar nuclei and adjacent vestibular nuclei but nowhere else in the brain or other tissues. The spheroid-like bodies in the deep cerebellar and vestibular nuclei showed heterogeneous composition, reactivity with anti-ubiquitin antibody, and staining characteristics of proteinaceous material. Atg4b-deficient (Atg4b –/–) mice also showed a mild but measurable impairment of motor performance on the Rotarod. Atg4b –/– mice produced by breeding heterozygous parents were produced at a slightly lower than expected ratio to heterozygous and wild-type siblings but showed no other clear abnormalities in a battery of screening tests. These findings appear to be different than those reported for inactivation of other Atg4 homologs, suggesting that these homologs have tissue-specific functions beyond redundancy.
Autophagy (literally “eating of self”) refers to a group of processes for degradation of cytoplasmic contents. Autophagy is a cellular process by which the cell forms an internal double membrane-bound vesicle (autophagosome) that somewhat randomly sequesters a portion of the cytoplasm, which then fuses with the lysosome, forming the autophagolysosome, to digest the contents. This process both recycles the constituent building blocks and removes unneeded or unwanted components from the cytoplasm such as aggregates of denatured protein, effete organelles (eg, mitochondria), or other structures that are not or cannot be degraded through other means. Additional forms of autophagy (microautophagy, chaperone-mediated autophagy, mitophagy) have been described (see reviews 16,31 ).
Considerable interest in autophagy is apparent in the recent medical research literature, as autophagy has been demonstrated to be an important pathophysiological process involved in several neurodegenerative disorders, 22 myopathies, innate resistance to infection with bacteria 13 and viruses, 2,20 aging (life span extension), 22,31 neoplasia, autoimmunity, 15 development, and some forms of programmed cell death. 3,15,16
Autophagy-related (Atg) 8 genes and proteins have been shown to be evolutionarily conserved from yeast to higher eukaryotes, although multicellular organisms have evolved more complex systems. Several autophagy-related genes were originally identified in yeast as being critical for survival in nutrient-deprived conditions. 30 At least 31 ATG genes have been identified in the human genome so far, 31 largely associated with formation of the autophagosome membrane. The presence of 4 mammalian homologs for both Atg4 (A, B, C, and D) 18 and Atg8 (GATE-16, GABARAP, LC3, and Atg8L) 6 in humans and animals (some with multiple orthologs and distinct expression patterns in different tissues 5 ) suggests that autophagy has evolved to have a greater diversity of purpose and control.
Atg4 (also known as autophagin) is a cysteine protease that activates Atg8 by proteolytic clipping, allowing Atg8 to become esterified with phospholipid during formation of the nascent autophagosome membrane. As well, Atg4 has esterase activity that hydrolyzes the Atg8-phospholipid link, apparently recycling Atg8. 24 Marino and coworkers 18 identified a family of 4 human cysteine proteinases homologous to yeast Atg4. Of these 4, human ATG4B (also known as autophagin-1) was found to be most active in proteolytic activation of the 4 human Atg8 homologs (GATE-16, GABARAP, LC3, and Apg8L), 6,28,29 although ATG4C was most widely distributed in human tissues. Both ATG4B and ATG4C were capable of rescuing deficient yeast. 18 The proteolytic active site of ATG4B has been shown to involve Cys74 by mutagenesis 28 and crystal structure. 12,27 Recently, reactive oxygen species generated during starvation or stress have been shown to act on nearby Cys 78 in the regulation of ATG4b activity. 25,26
A few Atg genes have been studied in knockout mice. Disruption of Beclin-1, one of the Atg8 homologs in mammals, is homozygous lethal early during early embryogenesis, 33 and halpoinsufficiency of this gene is associated with increased tumorigenesis. 21,33 Atg5 and Atg7 knockout mice appear nearly normal but die shortly after birth, probably because of an inability to upregulate autophagy during the early neonatal starvation period between the end of placental and beginning of enteral nutrition. 10,11 Both Atg5 and Atg7 conditional knockouts targeted to neurons showed severe neurodegeneration, but the Atg7 neuron-conditional knockout was more severely affected and died by 28 weeks of age. 4,9,19 Atg4c knockout mice were reported to be viable, fertile, and generally normal into adulthood to external appearances but had decreased locomotion and autophagic activity in the diaphragm muscle under starvation conditions and increased susceptibility to chemical carciongenesis. 17
As part of a large-scale gene-trapping effort, we report findings in mice homozygous (hom) for inactivation of Atg4b (Atg4b –/–) compared with their wild-type (wt, Atg4b +/+) littermates.
Methods
Mouse Husbandry
Mice were housed in a barrier facility at 22°C on a fixed 12-hour light and 12-hour dark cycle and were fed rodent chow #5001 (Purina, St. Louis, MO) ad libitum. Procedures involving animals were conducted in conformity with the Institutional Animal Care and Use Committee guidelines, which are in compliance with the state and federal laws and the standards outlined in the Guide for the Care and Use of Laboratory Animals. For phenotypic comparison, male and female hom and wt cohort littermates were bred from heterozygous (het) F1 parents with a mixed (C57BL/6J-Tyr c-Brd × 129S5/SvEvBrd) genetic background at Lexicon Pharmaceuticals (The Woodlands, TX).
The loss of function mutation of Atg4b was performed using a gene-trap method in embryonic stem (ES) cells that generates mutations by randomly inserting a promoterless construct within expressed genes to capture transcription and prematurely terminate translation. 34 In this method, mutagenized ES cell clones are initially isolated and screened by transcriptional tagging in high-throughput fashion to identify the mutated gene in each clone. ES cell clone OST264114 (OmniBank, Lexicon Pharmaceuticals) was selected for microinjection based on sequence similarity to the mouse Atg4b gene (NCBI nucleotide accession NM_174874). The exact genomic insertion site of the gene trap vector in OST264114 was determined by inverse polymerase chain reaction (PCR). The insertion was localized to the first intron of the gene, shortly after the initiation codon, thus preventing translation of almost all of the structural coding sequence (Fig. 1 ), and absence of gene expression was confirmed by reverse transcriptase–PCR of hom mouse tissues.
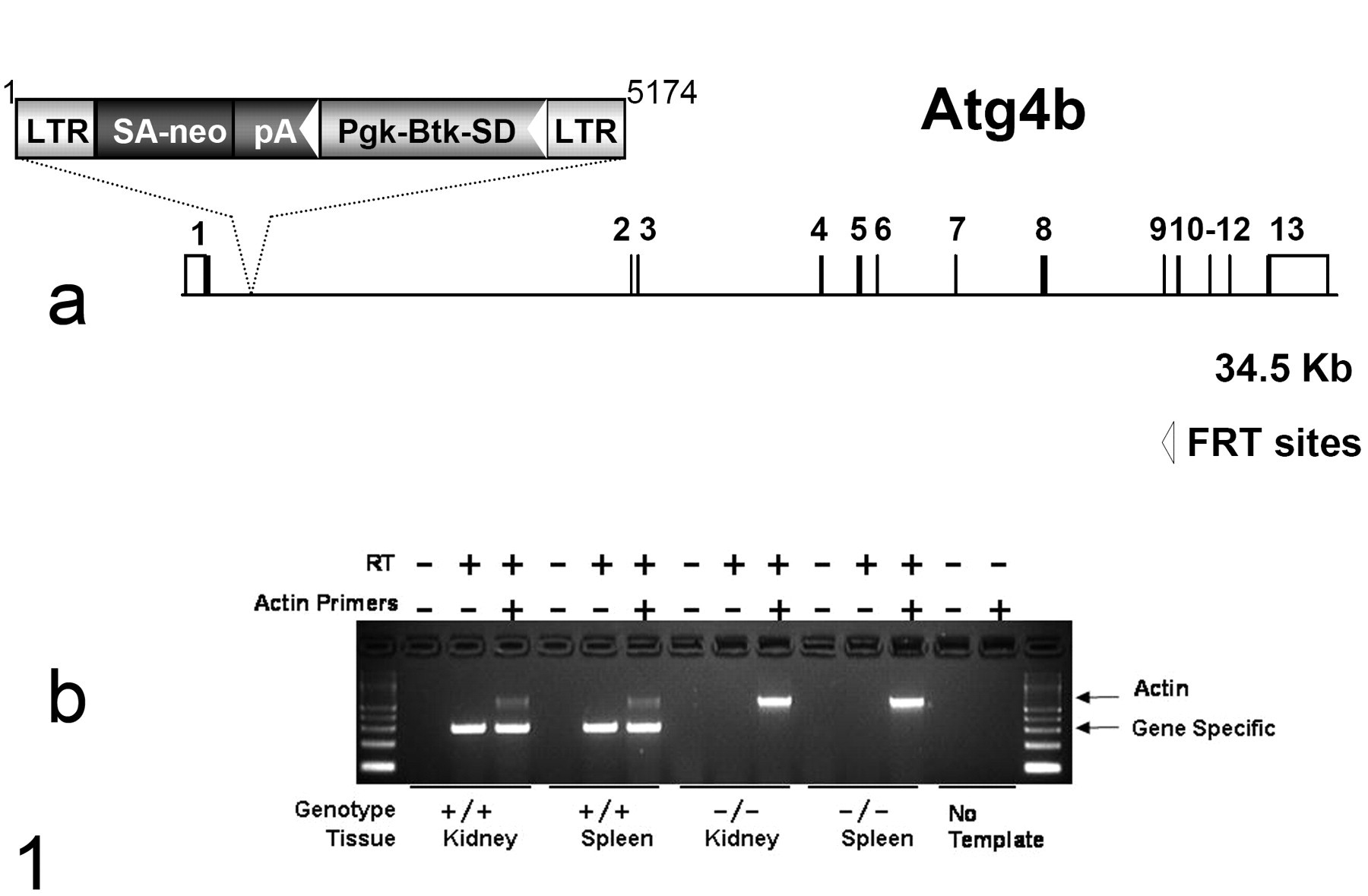
Disruption of the Atg4b gene. (a) Atg4b-deficient mice were generated from OmniBank ES cell clone OST264114, which contains a gene trap vector insertion within the first intron of Atg4b (accession NM_174874). Filled boxes represent coding exons; open boxes represent noncoding exons. LTR, long terminal repeat; neo, neomycin phosphotransferase gene; SA, splice acceptor sequence; pA polyadenylation sequence; Pgk, phosphoglycerate kinase-1 promoter; Btk-SD, Bruton tyrosine kinase splice donor sequence. (b) Gel showing polymerase chain reaction (PCR) product bands generated using gene-specific primers and with the presence and absence of reverse transcriptase (RT) and primers for actin. Messenger RNA for Atg4b is present in the wild-type (+/+) but not the homozygous knockout (–/–) tissues.
Histopathology
Tissues were collected and immersed in 10% neutral buffered formalin for 48 hours, except for the eyes, which were removed and fixed by immersion in Davidson’s fixative (Poly Scientific, Bay Shore, NY) overnight at room temperature. All tissues were embedded in paraffin, sectioned at 4 μm except as otherwise noted, mounted on positively charged glass slides (Superfrost Plus, Fisher Scientific, Pittsburgh, PA), and stained with hematoxylin and eosin (HE) for histopathologic examination.
Histopathology was performed on full necropsy tissues from a total of 10 hom mice (2 males and 2 females at 12–14 weeks, 2 males and 1 female at 45–54 weeks, 1 female at 74 weeks, and 2 females at 84 weeks of age), with 5 wt littermates (1 male and 1 female at 12 weeks, 2 females at 45 weeks, and 1 female at 84 weeks of age) serving as controls. The full necropsy tissue checklist included heart, skeletal muscle, aorta, lung, kidney, renal lymph node, trachea, thyroid, parathyroid gland, adrenal gland, pituitary gland, thymus, salivary gland, cervical lymph node, esophagus, stomach, pancreas, duodenum, jejunum, ileum, cecum, colon, rectum, mesenteric lymph node, liver, gallbladder, spleen, brain, spinal cord, eyes, harderian gland, urinary bladder, uterus, ovaries, fallopian tube, skin, mammary gland, bone, bone marrow, adipose tissue, blood, ear, tooth, turbinate, mediastinal lymph node, prostate, testes, epididymis, seminal vesicle, vas deferens, urethral glands, and mammary lymph node. An additional 4 hom and 6 wt mice (all female) were used to evaluate only the central nervous system (CNS) at 59–86 weeks of age. Brain histopathology was performed on male hom mice aged 12, 14, 54, and 54 weeks and female hom mice aged 12, 14, 45, 74, 84, 84, 84, 86, 86, and 86 weeks.
Bielschowsky stain for neurofibrils was performed using a kit (#k080, Poly Scientific) as was periodic acid–Schiff (#k047, Poly Scientific), according to the manufacturer’s instructions. Immunohistochemistry for ubiquitin was performed using rabbit anti-ubiquitin (#Z-0458, Dako, Carpinteria, CA) at 1:400 dilution, biotinylated goat anti-rabbit IgG (#BA-1000, Vector Laboratories, Burlingame, CA) at 1:500 dilution and detection using horseradish peroxidase–streptavidin (#SA-5004, Vector Laboratories), and diaminobenzidine chromogen with light hematoxylin counterstain.
For fluorescent staining, formalin-fixed paraffin-embedded brain sections were cut at 10 μm and dried onto positively charged slides (Superfrost, Fisher Scientific); deparaffinized with xylene; washed with 3 changes of absolute ethanol; stained with 0.01% Fast DiI (D7756, Invitrogen, Carlsbad, CA) in absolute ethanol for 10 minutes; differentiated with three 1-second dips in each of 100%, 95%, and 80% ethanol; washed with dH2O (3 times for 1 minute); and covered with aqueous mounting medium containing DAPI (Vectashield, #H1200, Vector Laboratories). Images were obtained with a confocal laser scanning microscope (Leica SP5), using excitation wavelengths of 543 nm (DiI) and 405 nm (DAPI). Autofluorescence was evaluated on deparaffinized, hydrated, 10-μm brain sections covered with aqueous mounting medium.
Phenotypic Testing
Starting at 9 weeks of age, animals were analyzed using a standard phenotypic analysis battery used at Lexicon Pharmaceuticals (as an example, the phenotypic screen of VGLUT1 mice is accessible at http://www.informatics.jax.org/external/ko/lexicon/2383.html). Briefly, wt and hom mice were subjected to a comprehensive battery of phenotype screening exams as previously described. 1,34 Screening assays included behavioral tests (such as circadian rhythm, open field, functional observation battery based on a subset of behavioral tests derived from the Irwin screen, 7 inverted screen, hot plate, prepulse inhibition of the acoustic startle response, tail suspension, marble burying, formalin paw assay, and context trace conditioning), fundoscopy and retinal angiography exams, blood pressure and heart rate measurements, serum chemistries, insulin levels, glucose tolerance testing, hematology, peripheral blood fluorescence activated cell sorting analysis, urinalysis, dual-energy x-ray absorptiometry scans, computed tomography scans, microcomputed tomography scans, fertility testing, skin fibroblast proliferation assays, and pathology.
The phenotypic analysis revealed no notable abnormalities in either sex across a wide range of behaviors; however, in view of histopathological findings, additional testing of the motor skills was performed on a separate cohort of female hom and wt mice using the Rotarod apparatus and the Smartrod software from AccuScan Instruments Inc. (Columbus, OH). The Rotarod consisted of 5 semi-enclosed chambers containing a rod 35 cm above the floor (diameter 3 cm, width 5 cm) made of ribbed plastic and flanked by round plates on either side to prevent escape (Accuscan Instruments, Columbus, OH). The speed profile was set to gradually increase Rotarod speed from 0 to 20 rpm during a 60-second trial: 10-second duration to accelerate from 0 to 10 rpm; 10-second duration at 10 rpm; 3-second duration to accelerate from 10 rpm to 15 rpm; 10-second duration at 15 rpm; 3-second duration to accelerate from 15 rpm to 20 rpm; and 24-second duration at 20 rpm. All animals were given a total of 12 trials divided in sets of 4 trials every 30 minutes until the completion of 12 trials. There was a 30-minute intertrial interval between every block of 4 trials (eg, trials 1–4 were run consecutively, but there was a 30-minute rest period between the end of trial 4 and the beginning of trial 5). The latency to fall and speed of the Rotarod when the animal fell were automatically recorded by the software. Repeated-measures analysis of variance statistical analysis was used to illustrate differences between hom and wt groups or group by trial differences using the Statistica software package (Statsoft Inc., Tulsa, OK). Where significant differences were detected, they were followed up by Tukey’s HSD post hoc analysis. The differences between genotypes during the first trial for all ages were determined using Student’s unpaired 2-tailed t-test.
Results
Homozygous knockout mice appeared normal to gross examination and in behavior through adulthood but were born in slightly less than the expected Mendelian proportions. Mating of mice heterozygous for the gene inactivation produced wt/het/hom = 75:139:48 rather than the expected 1:2:1 ratio (chi-square, P = .038). Both male and female hom were found to be fertile when mated to wt mice, and no difference was seen in growth rates between wt and hom up to 12 weeks of age. As well, no clearly significant abnormalities were initially identified by an extensive battery of phenotyping exams (described in Methods) with the exception of histopathology and later with extended Rotarod testing. These and all statements of results should not be interpreted to suggest that other or subtle differences were not present but only that screening examinations of the small populations used for analysis failed to show clear or statistically significant differences.
Histopathology
Initial histological examination of mice at 12 and 14 weeks of age (4 hom, 2 wt) identified abnormal spheroid-like structures or bodies in the neuropil of the deep cerebellar nuclei (DCN) and the vestibular nuclei of all hom brains (Fig. 2 ) but failed to identify any lesions related to the gene knockout in nonneural tissues. Similar bodies were present in brains of 14 hom mice, examined at 12, 14, 45, and at 84 weeks of age, but were absent in the brains of 10 wt littermates from the het × het crosses, indicating that lesions were associated with the gene trap inactivation of Atg4b. These bodies consisted of ovoid to globular structures composed of eosinophilic amorphous pale granular material that were scattered in the affected white matter tracts and less commonly adjacent to neurons or other cells. These bodies often had a dense center and pale periphery, but a few smaller ones were round, densely eosinophilic (hyaline), and more sharply circumscribed, as if formed by condensation of a larger body. The spheroid-like bodies varied from tiny granules up to 20 μm or more across, and many, particularly in older mice, had a heterogeneous asymmetric appearance with a hyaline side, core, or small hyaline inclusions within an asymmetric larger pale structure (Fig. 3 ).
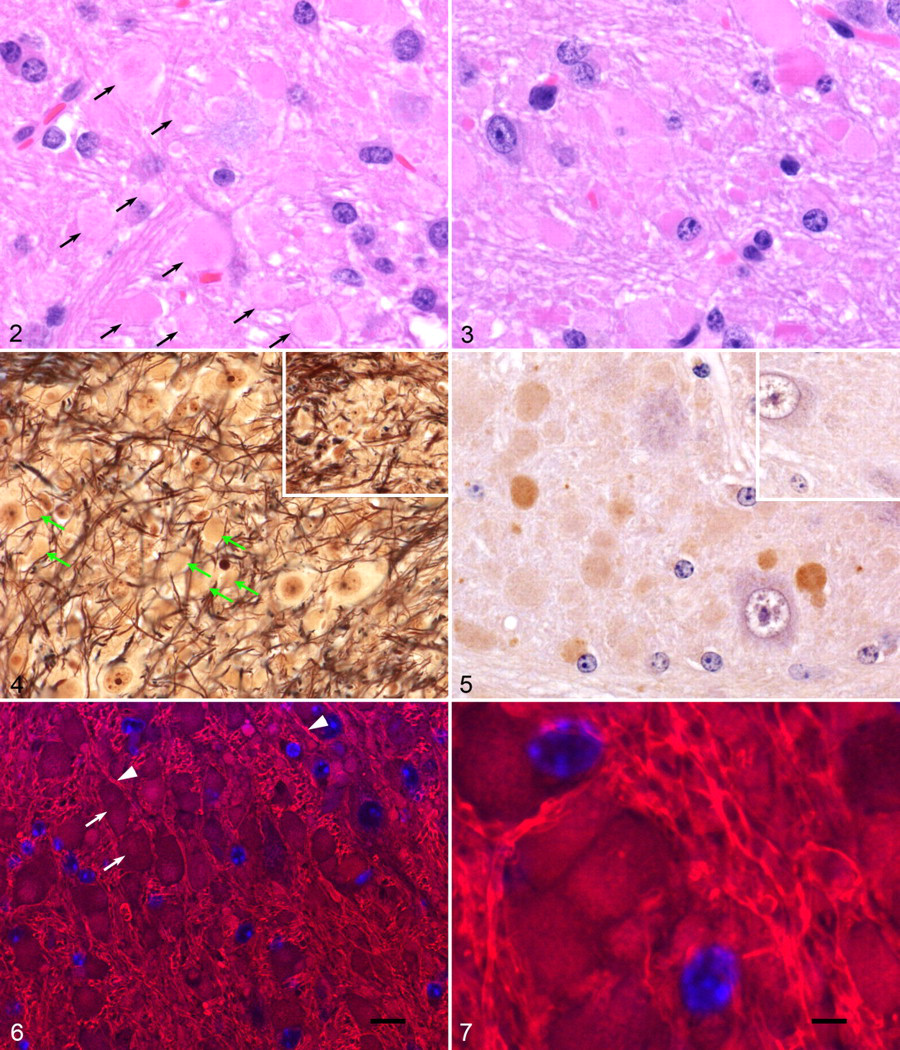
Bielschowsky’s silver impregnation method for neurofibers failed to stain these bodies (Fig. 4 ), except for minor scattered punctate staining within structures, and did not otherwise show swelling of axons, indicating that these were not formed from swollen axons. The neurofiber stain also showed the displacement of normally closely packed neurofibers, giving a tangled appearance. Periodic acid-Schiff stain similarly produced only weak, focal, or punctate staining of these bodies (not shown), which further indicates a heterogeneous and proteinaceous composition. Immunohistochemistry with anti-ubiquitin antibody showed distinct specific staining of lesion bodies with variable intensity and heterogeneity (Fig. 5 ); however, we did not find the distinct ubiquitin-positive inclusions in neuron cell bodies as reported for other Atg knockouts. 4,9
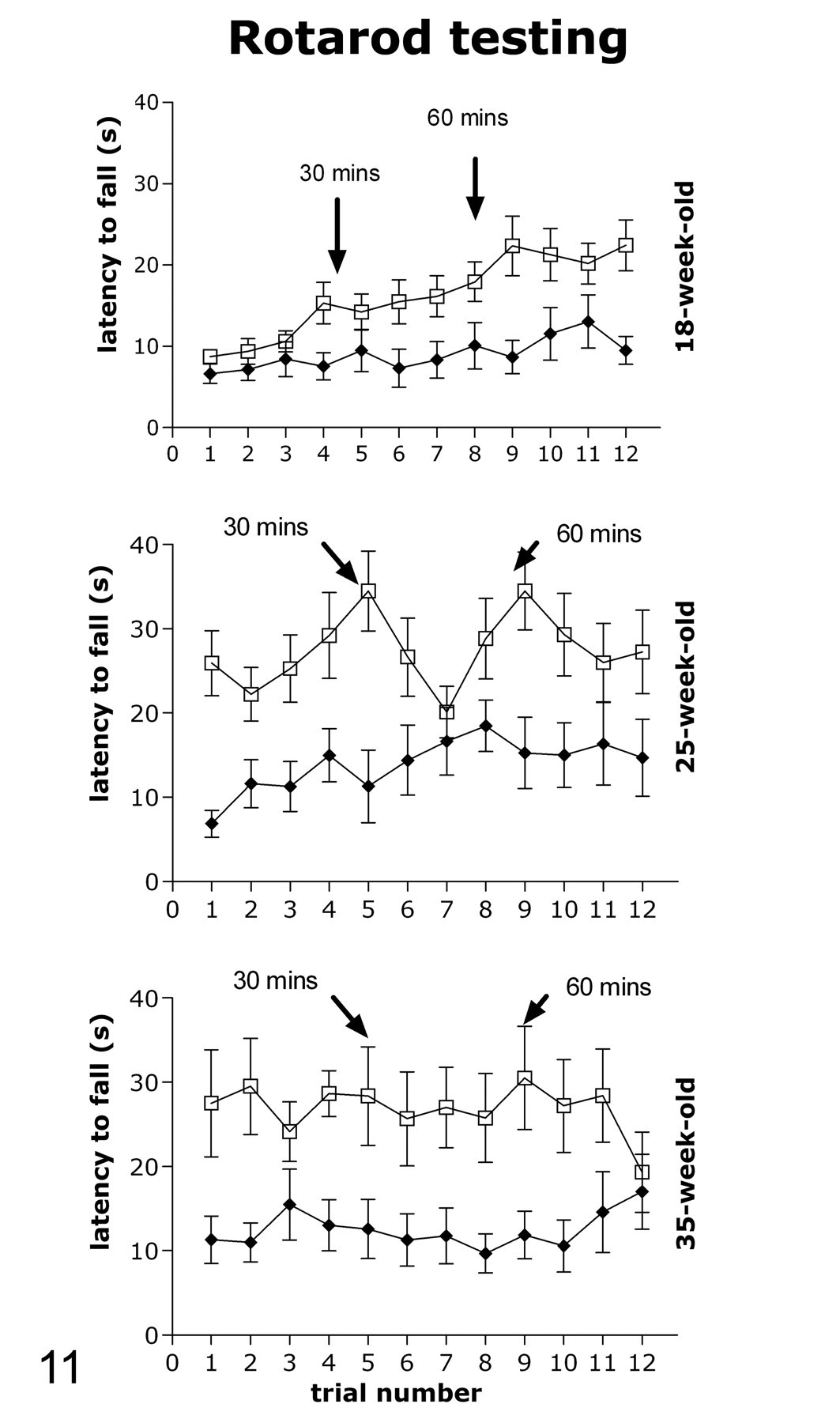
Rotarod testing at 18, 25, and 35 weeks of age. Atg4b hom knockout mice (filled circles) show impaired performance and learning with the Rotarod task compared with their wild-type littermates (open squares). Testing consisted of 3 blocks of 4 trials each, separated by a 30-minute rest period (see Methods for protocol details).
Fluorescent dyes were used to stain sections for high-resolution confocal images. The lipophilic tracer dye fast-DiI produced bright fluorescence of axon sheaths with lesion bodies and cell bodies showing much lower affinity for the stain (Figs. 6 and 7 ). These confocal images showed that the larger heterogeneous bodies observed with other stains were most commonly composed of smaller coalescing masses (Fig. 7). Finally, the bodies in unstained sections did not autofluoresce, indicating that they are not lipofuscin, which shows a broad spectrum autofluorescence. HE-stained sections showed weak fluorescence of these bodies in the fluorescein isothiocyanate channel and when capturing wavelengths above 488 nm laser stimulation on confocal microscopy; this was attributed to eosin fluorescence.
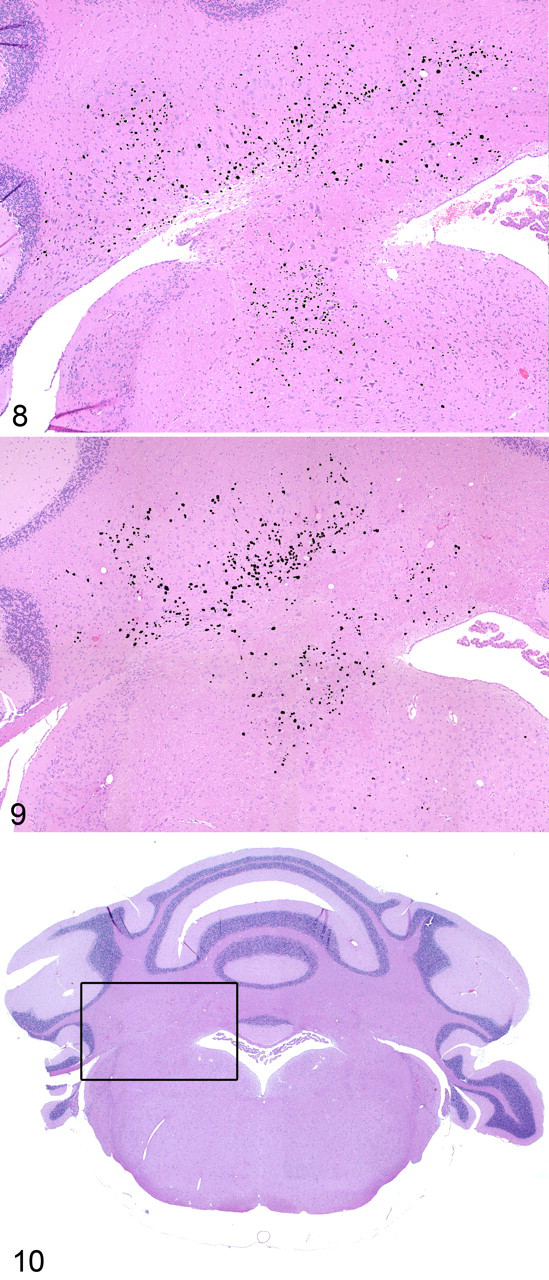
Comparison of serially spaced sections of DCN from mice of different ages showed considerable interindividual and section-to-section variation but no clear evidence of age-related increase in severity. The concentration of lesion bodies was greatest in the white matter tracts along the ventral border of the fastigial and interposed DCN (Figs. 8–10). As well, no clear alteration of neuron or glial cell density or evidence of neuron necrosis or degeneration was ever detected in the affected nuclei of the hom brain. These observations, based on a limited number of mice, certainly do not eliminate the possibility of degenerative and reactive changes under all conditions, especially in light of the neurological findings that suggest a functional deficit.
Neurological Evaluation
Initially we evaluated a cohort of male and female Atg4b hom and wt mice in a standardized phenotyping battery. Those mice were tested at 12 and 16 weeks of age, and no differences were found between genotypes in locomotor activity, acoustic startle response and sensorimotor gating, depressive-like behavior, learning and memory, and tonic inflammatory pain sensitivity as assessed by open field, prepulse inhibition, tail suspension, trace fear conditioning, and formalin paw assays, respectively. As well, no differences in motor strength and coordination were detected using the inverted screen assay.
In view of the potentially adverse lesion in the DCN, the agility of a cohort of aging female mice was tested with the Rotarod assay using the protocol developed specifically to better assess potential differences between genotypes. Mice were tested for the first time when they were 18 weeks old (Fig. 11 ). There was a significant effect of genotype on latency to fall (F 1,16 = 5.1, P < .05) and a significant genotype by trial interaction (F 11,176 = 3.3, P < .001). These data indicated that hom mice had significantly shorter time (latency) to fall off starting at trial 8 (least significant difference test post hoc analysis of trial × genotype interaction), suggesting impaired motor coordination or reduced stamina in hom mice. There was no difference between genotypes in latency to fall during the first trial (mean ± standard error of the mean [SEM], 6.6 ± 1.2 rpm in hom, and 8.8 ± 0.8 rpm in wt). Analysis of the genotype by trial interaction revealed that wt mice improved their performance over trials (significant difference from trial 1 was present starting at trial 4 for the wt mice), but hom mice did not show the same proficiency in learning the Rotarod task, possibly indicating impaired motor learning in addition to impaired motor coordination. Impairment was also evident by the average speed at which mice fell off: during trial 12, this speed was 7.5 ± 2.5 rpm (mean ± standard deviation) in hom and 13.1 ± 3.5 rpm in the wt mice (the speed data over trials is not shown in Fig. 11). The second Rotarod testing occurred when mice were 25 weeks old (Fig. 11). At that time the effect of genotype was not statistically significant, and genotype by trial interaction only approached significance (F 11,143 = 1.79, P = .06). However, analysis of only the first trial revealed a significant difference between genotypes in latency to fall (mean ± SEM, 6.9 ± 1.6 rpm in hom and 25.9 ± 3.7 rpm in wt, P < .001). On average, all the mice showed improved performance during test 2 compared with test 1, indicative of memory consolidation and motor learning. The average speed at which mice fell off during trial 1 was 5.8 ± 2.5 rpm in hom and 14.2 ± 5.6 rpm in wt. The average speed at which mice fell off during trial 12 was 10.1 ± 4.6 sec in hom and 14.2 ± 4.5 sec in wt. The final Rotarod test was applied to mice when they were 35 weeks old (Fig. 11). It was evident that wt mice further improved their performance from test 2 to test 3 whereas the hom mice did not. There was a difference between genotypes in latency to fall during the first trial (mean ± SEM, 11.3 ± 2.8 rpm in hom and 27.5 ± 6.4 rpm in wt, P < .05). There was also a significant overall effect of genotype on latency to fall (F 1,16 = 4.7, P < .05) and no genotype by trial interaction, indicating overall impaired performance in the hom mice.
Discussion
Compared with the inactivation of some of the other autophagy-related genes, such as Atg5, Atg7, and Atg8, inactivation of Atg4b is clearly of less importance to the development and survival of mice. This may be partially attributable to redundancy of the other 3 Atg4 homologs. From the ratio of genotypes produced from het × het breeding, it appears that about one third of the number of expected hom mice were not present for genotyping (approximately 2 weeks of age) before weaning, although experienced staff reported no problems with neonatal viability. Unfortunately, we were unable to further investigate this possible effect. Despite this, hom mice appeared completely normal to external appearances and none of the hom mice were found to suffer the neurodegeneration or hepatopathy associated with the inactivation of Atg7 or Atg5. The presence of these bodies in the CNS of Atg4b hom mice by 12 weeks of age and the lack of accumulation or significant alteration with age suggest to us that these masses are in some sort of steady state: either being continually formed and resorbed over time or, the alternative hypothesis, forming before 12 weeks of age and remaining static. The latter hypothesis seems unlikely given that virtually all internal biological structures are continually being degraded and replaced, and age-related lesions such as amyloid and lipofuscin pigments accumulate with time. Furthermore, the absence of a clear cellular reaction (gliosis, neuronal degeneration) suggests that these structures are fairly innocuous and that functional impairment caused by the gene inactivation was comparatively mild. These observations based on histology of a limited number of mice should not be interpreted as a guarantee that subtle neuron loss or gliosis does not occur. In future, carefully controlled studies, it may well be found that stressful conditions such as starvation (which we did not do), age, sex, and other factors can influence the severity, distribution, and quality of these lesions.
Although no clear evidence of neurodegeneration was identified histologically in Atg4b hom mice through 84 weeks of age, Rotarod testing clearly showed a functional deficit. By way of comparison, mice having a nestin-conditional knockout of Atg5 in neural cells showed prominent growth retardation, behavioral abnormalities, and tremor by 12 weeks of age, 4 none of which were observed in our Atg4 hom. As well, the Atg5 nestin-conditional knockout mice showed intracytoplasmic ubiquitin-positive inclusions in neurons that were not seen in our mice although “eosinophilic spheroids” that are reported to be widespread in this knockout appear to be similar to the spheroid-like bodies we found with a much more restricted distribution in Atg4b hom mice. Nestin-conditional inactivation of Atg7 in the CNS of mice also produced increased ubiquitin staining in the cytoplasm of neural cells. 9
The mechanism by which these bodies are formed is unknown. From the inferred gene function, a homolog of Atg4, and the accumulation of ubiquitinated material, it seems reasonable to assume that defective autophagy is involved. The ubiquitin staining of these bodies suggests that denatured proteins tagged for destruction comprise a significant component of these extracellular structures. We hypothesize that impairment of normal autophagy leads to accumulation of denatured protein and that this material is exocytosed from neuronal cell bodies or by an unknown mechanism.
Using Northern blotting, Atg4b appeared to be at least as strongly or more strongly expressed in many tissues of the mouse, including heart, liver, kidney, and skeletal muscle, compared with brain, with 2 major transcripts of approximately 2 Kb and approximately 3 Kb. 17,18 In the rat, immunoblotting and PCR showed that both Atg4b protein and messenger RNA were widely expressed in the brain and most other tissues, with in situ hybridization showing strong expression in the cerebellum, in Purkinje cell bodies and processes, 32 but precise localization of cellular expression for all 4 Atg4 homologs has not been reported in any species. The absence of lesions in other locations suggests that cells of other tissues and other areas of the brain might have functional redundancy by virtue of expression of other Atg4 homologs or by requiring less Atg4B-specific activity. As well, anatomic features of the DCN may render cell processes in this area more dependent on Atg4B-driven autophagy to clear aggregated denatured protein or less able to clear exocytosed material.
Interestingly, targeted disruption of Atg4C, the most widely expressed Atg4 homolog in mouse and human tissues, appeared to be well tolerated and no histological lesions were reported from the Atg4C knockout. As well, inactivation of Atg4C did not greatly alter expression of Atg4A, Atg4B, or Atg4D in the liver, where all 4 genes appear to be most strongly expressed. 17 The presence of 4 Atg4 homologs allows for redundancy of some functions; however, not all homologs appear equal. For example, ATG4B and ATG4C were the only human homologs capable of substituting for Atg4 in deficient yeast. 18 Studies in cultured cells from rats showed little if any effect of 90% knockdown of Atg4B expression on levels of and the extent of lipidation of LC3; however, in a cell free system, delipidation of the Atg8 homolog LC3 (a known function of Atg4) was greatly impaired, 32 suggesting that Atg4B is most important in the delipidation and recycling of LC3, and possibly other Atg8 homologs.
The fact that Atg4B knockout mice survive well through 84 weeks of age, with no gross abnormalities and minimal but reproducible CNS lesions, seems to offer a unique opportunity to study the function of this gene in autophagy homeostasis, particularly in the CNS, over a prolonged period that is not afforded by other currently identified autophagy knockout models. Viable autophagy-deficient models are clearly desirable to assist in the evaluation of autophagy in the pathogenesis of many human conditions including aging and in neurodegenerative diseases such as Huntington’s disease 23 and Niemann-Pick disease. 14 We hope these preliminary data will provide a starting point for future studies.
Footnotes
Acknowledgements
The authors express their appreciation to Kathy Henze, Mary Thiel, and Ryan Vance for expert technical assistance in these studies as well as to the staff and administration of Lexicon Pharmaceuticals for making this work possible.
The authors declared that they had no conflicts of interest with respect to their authorship or the publication of this article.
The authors declared that they received no financial support for their research and/or authorship of this article.