
Abstract
Mast cell tumors are among the most commonly seen tumors of the skin in dogs and are more highly aggressive than mast cell tumors of other species. Some breeds display a markedly higher incidence of mast cell tumor development than others and appear to have some genetic predisposition. Recently, mutations have been found in canine mast cell tumor tissues and cell lines within the juxtamembrane domain of the protooncogene c-KIT. In previous studies utilizing a small number of cases, no association between the presence of a mutation and the breed of dog or grade of the tumor could be identified. An expanded study with a larger sample set was performed to explore this possibility. The juxtamembrane domain of c-KIT was amplified using the polymerase chain reaction from genomic DNA preparations of 88 paraffin-embedded mast cell tumors from selected breeds. Mutations, consisting of duplications and deletions, were found in 12 of the tumors. A significant association was found between the presence of a mutation and a higher grade of tumor but not between breed and grade or between breed and the presence of a mutation.
Keywords
Mast cell tumors are one of the most frequently seen skin neoplasms in dogs, accounting for up to 21% of cases.5,12,18 Most of these tumors are benign, developing slowly and persisting for years without increasing in size or metastasizing. However, a large number are highly aggressive and present a significant threat to canine health.18 In contrast, mast cell tumors in humans are rare and usually benign.18 Some dog breeds appear to be at relatively little risk, whereas others such as the Boxer have been reported to have a high incidence of mast cell tumor development.5 This difference among breeds indicates a possible genetic influence on both susceptibility to tumors and tumor aggressiveness.
Mast cell tumors are usually graded on a histologic scale.11 Grade I tumors are highly differentiated, with regular nuclei, rare or no mitotic figures, and a large number of metachromatic granules. They are generally considered to be benign. Grade III tumors are highly undifferentiated, with large misshapen nuclei, many mitotic figures, and few cytoplasmic granules. This is the most aggressive of the three grades. Grade II tumors are intermediate between the other two types. Mast cell tumors are often also identified by clinical stage based on the extent of their growth. Incompletely excised tumors are designated stage 0, single tumors without and with lymph node involvement are designated stage I and stage II, respectively, multiple tumors comprise stage III, and recurrent or metastatic tumors make up stage IV.18 Both histologic grading and clinical staging are good predictive factors for the final outcome.18
The genes involved in the development of mast cell tumors are currently under study. Analyses of human7 and rodent15–17 mast cell tumor cell lines have revealed a number of mutations in the proto-oncogene c-KIT. The c-KIT gene encodes for a cell surface receptor, which upon binding of its cognate ligand induces a signal transduction cascade responsible for the development, maturation, and survival of many cell lineages, including hematopoietic stem cells, melanocytes, and mast cells.3,14,19 The c-KIT receptor consists of an extracellular domain of five immunoglobulin-like loops and an intracellular tyrosine kinase domain.13,20 Mutations in the kinase domain or the neighboring juxtamembrane domain have been shown to cause constitutive activation of c-KIT in the absence of ligand binding. Constitutive activation of c-KIT in certain murine cell lines leads to the uncontrolled cell growth and aggressive behavior typical of tumor development.8 The mutations identified to date in humans and rodents are confined to the kinase and juxtamembrane domains of c-KIT and consist of point mutations and small deletions. Many of these mutations cause constitutive activation of c-KIT in vitro and some have been identified in situ.
Mutations in canine c-KIT have only recently been identified, and all have been found exclusively in the juxtamembrane domain. Ma et al.10 discovered point mutations and small deletions in three of seven tumors and a duplication in two of three cell lines. London et al.9 found duplications in 5 of 11 tumors; however, they did not see any of the other types of mutations. Although the duplications encompassed approximately the same area of c-KIT, no two were identical. Both groups found mutations in approximately 50% of the animals studied. In a recent study, 2 of 15 mast cell tumors tested contained juxtamembrane domain duplications, and in one of these dogs, a recurrence of a tumor with the exact same duplication allowed molecular confirmation of the tumor type when the diagnosis based on pathology was questionable.21 Because duplications in the juxtamembrane domain of c-KIT as seen in canine mast cell tumor have not been identified in any other species to date, these duplications may be related in some way to the increased aggressiveness of these tumors in dogs. In the previous studies, the limited number of animals involved made it impossible to accurately determine the percentage of canine mast cell tumors with detectable mutations or to identify any correlation between the type of mutation seen and the breed of dog or the aggressiveness of the tumor. The present study of c-KIT duplications was conducted on a much larger scale to determine the significance of these mutations.
Materials and Methods
Sample selection
Mast cell tumor cases were selected from tissue samples submitted to the Michigan State University Animal Health and Diagnostic Laboratory (AHDL) during the period of 1998–1999. Cases were selected from breeds with the highest number of cases so that differences between breeds could be examined. The Boxer and Boston Terrier were chosen based on their suspected predisposition for mast cell tumors, whereas the Labrador Retriever, Golden Retriever, and mixed-breed dogs were not suspected to be predisposed and were chosen for comparison. The 15 dogs from the preliminary study were included; this group consisted of a Pit Bull in addition to the breeds listed above. For each dog, a single block of formalin-fixed, paraffin-embedded tumor tissue was retrieved from the AHDL archives.
Paraffin block DNA isolation
Sections of each block were cut and stained with hematoxylin and eosin. Stained sections were examined under a microscope by a certified veterinary pathologist (B. Yamini) to determine the borders of the tumor, which were marked on the block. A small piece of tissue approximately 2 mm in diameter was excised from each block within the boundaries of the tumor. DNA was isolated by a modified version of a previously described method.1 The tissue was placed into 400 µl of digestion buffer (50 mM Tris, pH 8.5, 1 mM ethylenediaminetetraacetic acid (EDTA), 0.5% Tween). The paraffin in the samples was melted by heating at 95 C for 10 minutes and heating for 30 seconds twice in a microwave at full power, with thorough vortexing after each heating step. The samples were allowed to cool, and 5 µl of 15 mg/ml proteinase K was added to each. The samples were then incubated at 42 C overnight or until the piece of tissue was completely digested. The proteinase K was inactivated by heating at 95 C for 10 minutes, and the samples were centrifuged at 12,000 rpm in a microcentrifuge for 10 minutes. An aliquot of 200 µl was then transferred to a clean tube, avoiding the transfer of paraffin.
Amplification of the juxtamembrane region of c-KIT
For each sample, the undiluted DNA preparation and dilutions of 1:10, 1:25, and 1:50 were used as templates for polymerase chain reaction (PCR) amplification of the juxtamembrane domain of c-KIT. PCRs with a total volume of 25 µl were set up using 5 µl of template, 20 pmol of each primer, 0.625 U Taq polymerase (Gibco BRL), and final concentrations of 100 μm dNTPs, 1.5 mM MgCl2, 50 mM Tris Cl (pH 8.3), and 10 mM KCl. Amplification was performed using primers JuxtF and JuxtR (Fig. 1). PCR conditions consisted of an initial denaturation step of 4 minutes at 94 C, followed by 35 cycles of 1 minute at 94 C, 2 minutes at 66 C, and 3 minutes at 72 C, and a final extension step of 8 minutes at 72 C.
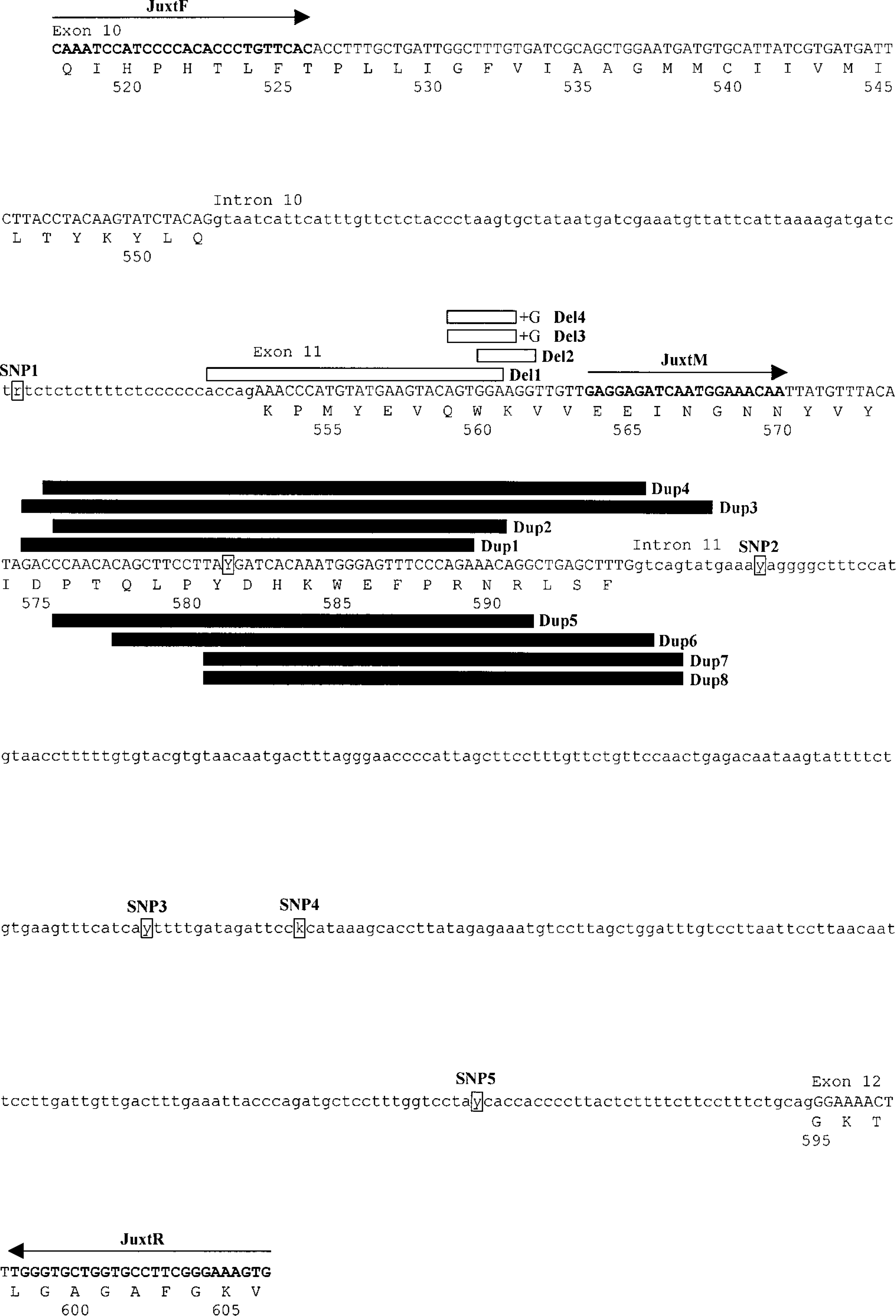
Genomic sequence of the juxtamembrane domain of canine c-KIT and flanking regions (amino acids 518–606). The amino acid sequence of the exons is given beneath the DNA sequence. Single nucleotide polymorphisms are shown in boxed letters (R = A or G; Y = C or T; K = G or T). Primers used for amplification and sequencing are shown in bold type, with arrows indicating the direction of the primer. Exon sequences are in uppercase letters; intron sequences are in lowercase letters. Deletions are denoted by open bars; duplications are denoted by solid bars.
Purification of amplification products
The PCR amplification products were separated on 2% agarose gels in Tris–acetate–EDTA buffer. Individual bands were stabbed with a pipette tip and transferred to 15 µl of water, 5 µl of which was used as a template in an additional PCR with the same conditions as the initial amplification. The secondary amplification products were separated on 2% agarose gels, and the resulting bands were excised and purified using the QIAEX II kit (Qiagen) following the manufacturer's protocol, with each sample resuspended in a final volume of 30 µl of 10 mM Tris, pH 8.
Sequencing of purified bands
Manual sequencing was performed using the Thermo Sequenase radiolabeled terminator cycle sequencing kit (USB). Samples were sequenced from both the JuxtF and JuxtR primers used for PCR amplification plus an internal primer JuxtM (Fig. 1). Sequencing followed the manufacturer's standard protocol for the dGTP termination mix except for an annealing temperature of 50 C, using 5 µl of template DNA and 0.5 pmol of primer per sample. The reaction products were separated on 6% denaturing polyacrylamide gels and visualized by exposure to x-ray film.
Automated sequencing was performed on an ABI Prism 377 DNA Sequencer using Big Dye terminators. Samples were sequenced from JuxtF and, when readable length produced was insufficient, JuxtM. Cycle sequencing was performed using 10 µl of template DNA and 3.2 pmol of primer per sample, following the manufacturer's protocol. Unincorporated terminators were removed by isopropanol precipitation following the manufacturer's protocol, except for the addition of 1 µl of 20 mg/ml glycogen as a carrier. Samples were resuspended in 4 µl of loading dye, 2 µl of which was loaded onto the gel.
Results
The juxtamembrane domain of c-KIT was successfully amplified from a total of 88 canine mast cell tumor cases. The number of dogs of each breed and the grade distribution of tumors within those breeds is shown in Table 1. PCR amplification revealed that 8 of the 88 tumors contained two larger bands in addition to the expected band when separated on agarose gels, indicating possible duplications. An additional tumor also contained two extra bands, one below and one above the expected band, indicating a possible deletion. All bands from these tumors were purified and sequenced manually. For each sample, the three bands represented the normal sequence, either a duplication or deletion, and a mixture of the two sequences. Figure 2 shows an example of a duplication and a deletion as seen on an agarose gel. The remaining samples were sequenced either manually or with an automated sequencer to screen for mutations too small to be seen as separate bands. Three additional deletions were found during the course of this sequencing.
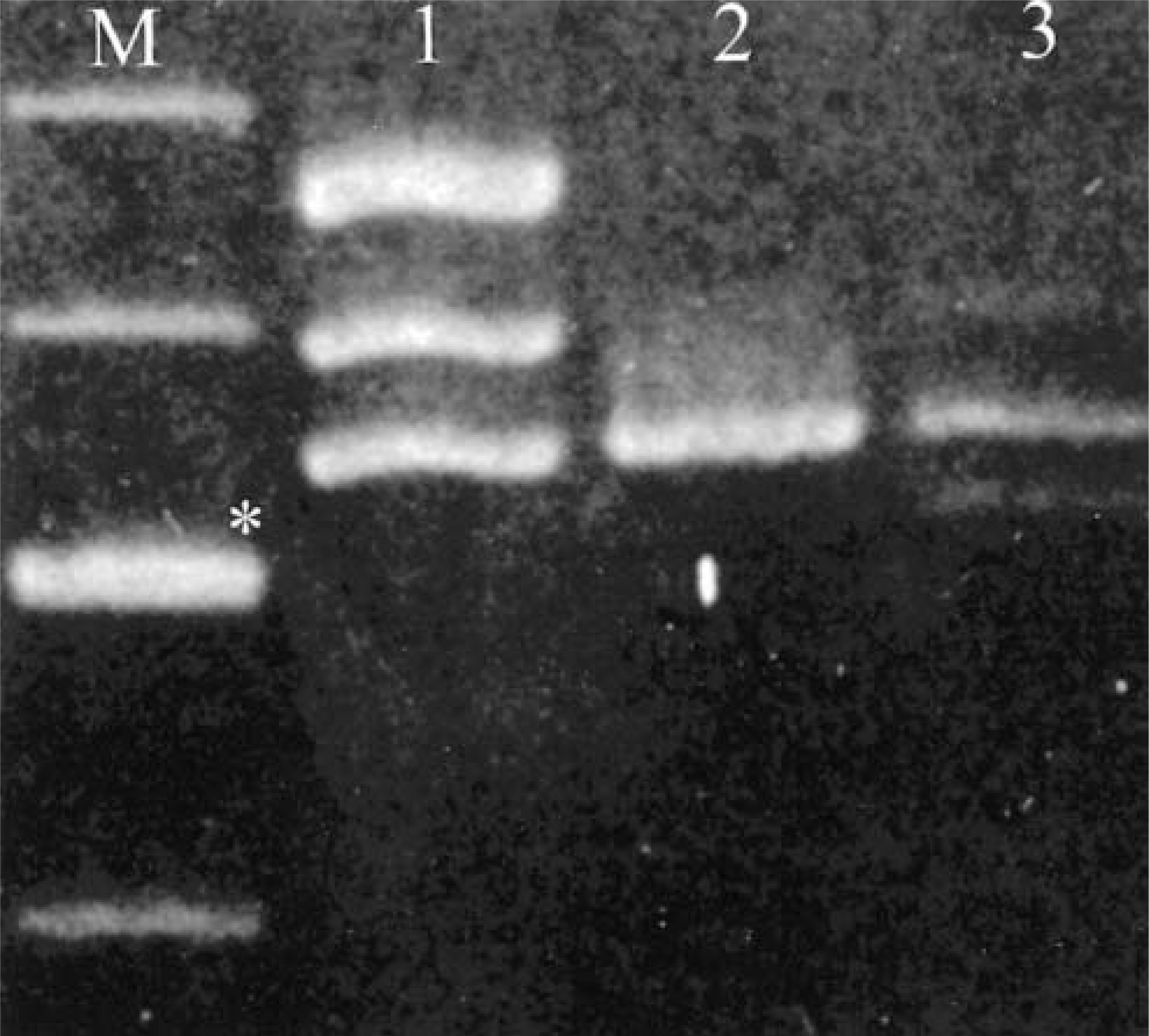
Agarose gel showing products obtained by amplification of the juxtamembrane region of c-KIT from different sources. Lane M contains a 100-bp DNA ladder. The 600-bp band is marked with an asterisk. For lane 1, the lower band contains normal sequence and the middle band contains a 45-bp duplication (Dup2). The upper band is a heteroduplex of normal and mutant sequence. Lane 2 is an example of amplification from normal tissue. In lane 3, the lower band contains a 30-bp deletion (Del1), the middle band is normal, and the upper band is a heteroduplex of normal and mutant sequence.
Grade distribution of mast cell tumors in various dog breeds.
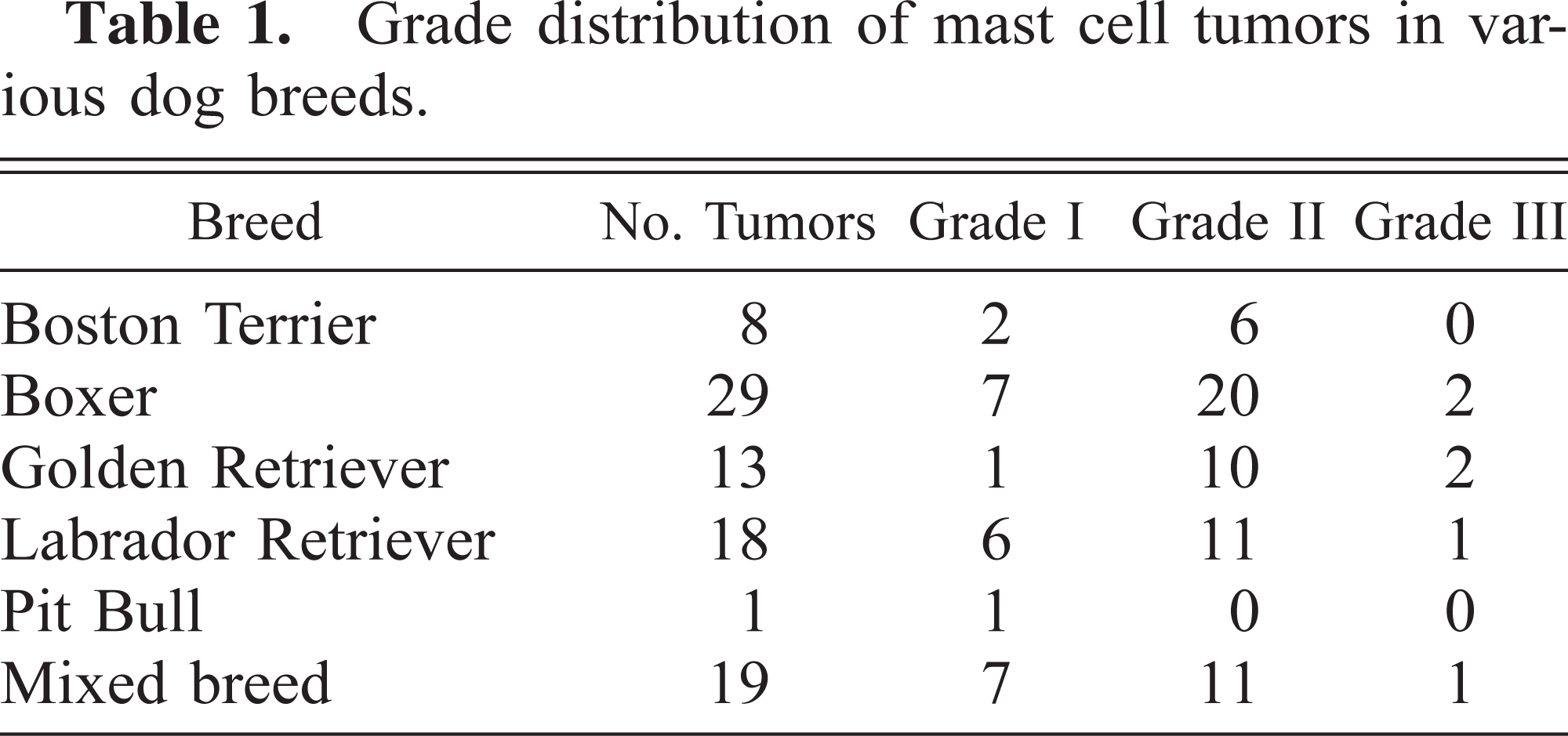
Figure 1 shows a portion of the juxtamembrane domain sequence (GenBank accession Nos. AF448146, AF448147, and AF448148), with the positions of the duplications indicated. The duplications were all in frame, ranged in size from 44 to 69 base pairs (bp), and were located near the 3′ end of exon 11, with five of them (Dup3, Dup4, Dup6, Dup7, Dup8) extending into the neighboring intron 11. In two of the dogs, the duplicated sequences (Dup7 and Dup8) were identical. Three of the duplications caused tandem repeats within the protein sequence: Dup1, residues Asp575–Arg589; Dup2, residues Pro576–Asn590; Dup5, residues Pro576–Arg591. Dup4 resulted in the insertion of a Gly residue after Phe594, followed by a direct repeat of residues Pro576–Phe594. The numbering of the amino acid residues is based on the normal canine c-KIT cDNA sequence (GenBank accession No. AF448148). The remaining four duplications involve duplication of part or all of the consensus splice sequence at the 5′ end of intron 11, creating the potential for alternative splicing events. For all four duplications, no change to the protein is made if the upstream site is used. If the downstream site is used, then Dup3 causes an insertion of Gly-Gln-Tyr plus a repeat of residues Asp575–Phe594, Dup6 causes an insertion of Gly plus a repeat of residues Gln578–Phe594, and Dup7/Dup8 cause an insertion of Gly-Gln plus a repeat of residues Tyr581–Phe594, with the insertion occurring after Phe594 in all four cases. RNA studies are required to determine the splicing pattern in these tumors.
Figure 1 also shows the location of the identified deletions. Del1 encompasses 30 bp, including the last few bases of intron 10 and part of the 5′ end of exon 11. Because the deletion removes part of the 3′ splice site sequence for intron 10, the exact effect of this deletion on the protein is not known. If the first AG following the deletion is used for splicing, the result would be a deletion of Lys553–Lys561; however, other potential splice acceptors are present in the area. A study of the RNA from such cases is necessary to determine which of these sites, if any, is used. Del2 removes 6 bp and causes the substitution of a Phe residue for Trp560–Val562 in the protein sequence. Del3 and Del4 remove 7 bp but are followed by the insertion of a single G, so no frameshift is created. They cause the substitution of an Arg residue for Gln559–Lys561.
The distributions of the different mutation types by breed and tumor grade are shown in Tables 2 and 3. None of the mutations identified were from grade I tumors. All of the deletions were found within grade II tumors, and equal numbers of duplications were found in grade II and grade III tumors. The mutation frequencies for the three grades were significantly different from each other as determined by a minimum chi-square analysis (χ2 = 21.68, 95% confidence level = 9.49). With respect to breed, two of the four deletions were found in Labrador Retrievers (Table 3). Half of the duplications were identified in Boxers, and the remaining half consisted of single tumors in each of the other breeds. No significant association was seen between presence of a mutation and the breed of dog (χ2 = 6.06, 95% confidence level = 18.31).
Grade distribution of duplications and deletions in c-KIT among canine mast cell tumors.

Distribution of duplications and deletions in c-KIT among different dog breeds.
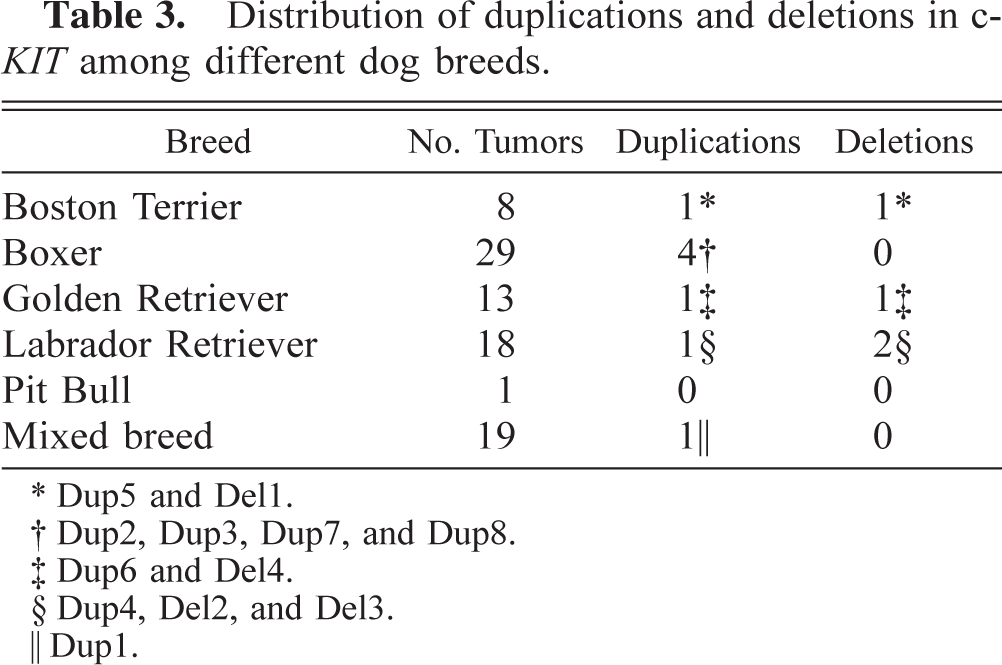
Dup5 and Del1.
Dup2, Dup3, Dup7, and Dup8.
Dup6 and Del4.
Dup4, Del2, and Del3.
Dup1.
During the course of sequencing work on c-KIT for this and other projects, a number of single nucleotide polymorphisms (SNPs) were found throughout the gene, six of which are located in the region shown in Figure 1. Five of these SNPs are located in introns and have no effect on the protein. The allele frequencies of the SNPs among 61 of the dogs in this study were evaluated and determined to be 35.2% A + 64.8% G for SNP1, 49.2% T + 50.8% C for SNP2, 48.4% C + 51.6% T for SNP3, 48.4% G + 51.6% T for SNP4, and 47.5% C + 52.5% T for SNP5. The sixth SNP is located within exon 11 but is silent, causing no amino acid change. The allele frequency of this SNP was 24.6% T + 75.4% C.
Discussion
Duplications and deletions of sufficient size can readily be distinguished by agarose gel electrophoresis (Fig. 2). The smaller 6 or 7-bp deletions were only identifiable by direct sequencing. All of the duplications and the largest of the deletions identified in this study produced a pattern of three bands similar to the examples in Figure 2. The lower two bands represent the normal and mutant sequences, indicating that the tumors were heterozygous for their respective mutations. The uppermost band represents heteroduplexes containing one strand each of normal and mutant DNA, as determined by sequencing. In heteroduplexes, unpaired bases in one of the strands form a bulge in the DNA, which retards the migration of the heteroduplexes during electrophoresis and causes them to appear as if they were larger fragments.2
The results of this study suggest that there is a relationship between tumor grade and the presence of c-KIT juxtamembrane domain mutations, particularly duplications. No mutations were found in any of the grade I tumors tested, and <10% of the grade II tumors had mutations (Table 2). Among grade III tumors, however, four of six contained mutations in the juxtamembrane domain. Equal numbers of duplications were seen in grade II and grade III tumors, in spite of the fact that nearly 10 times more grade II than grade III tumors were tested. Juxtamembrane duplications appear to be associated with only the most aggressive tumors.
With respect to breed, there does not appear to be an association between the presence of mutations and the breed of dog in which they were identified (Table 3), and no significant association was observed between breed and tumor grade (χ2 = 7.97, 95% confidence level = 18.31). This finding is in contrast to those of previous studies in which a higher percentage of mast cell tumors in Boxers were of a low grade as compared with other breeds. A compilation of records for all tumors submitted to AHDL during 1999 revealed that the tumor grade distribution in Boxers was nearly identical to that seen in mixed-breed dogs. Of 69 mast cell tumors in Boxers, 29.0% were grade I, 62.3% were grade II, and 8.7% were grade III. The distribution for 99 mixed-breed dogs was 29.3% grade I, 62.6% grade II, and 8.1% grade III.
In both of the previous studies, the canine mastocytoma cell line C2 containing a c-KIT juxtamembrane domain duplication exhibited constitutive phosphorylation of c-KIT.9,10 Because the duplications identified in this study all overlap with the duplication found in that cell line, the present duplications probably also would cause activation of c-KIT. Mouse cell lines with c-KIT-activating mutations can induce growth behavior typical of tumor formation.8 Uncontrolled growth seen in tumors should be expected when a receptor regulating cell growth is constitutively active. This finding implicates the duplications found in this study as potential causes of the aggressiveness of the tumors in which they were identified. The role of the deletions requires further investigation.
In the present study, approximately 13.6% of canine mast cell tumors tested contained mutations within the coding region for the juxtamembrane domain of c-KIT. This frequency is much lower than those reported by Ma et al.,10 who found mutations in three of seven tumors and two of three cell lines, and London et al.,9 who found duplications in 5 of 11 tumors. Our study was based upon a larger sample set, which is less susceptible to ascertainment bias, and tumors of all grades were included. London et al.9 studied only grade II tumors, some of which may have been at the higher end of that range (borderline grade III). If that were the case, our finding of mutations in four of six grade III tumors would agree more closely with the results of London et al. The grades of the tumors studied by Ma et al. were not reported.10
A significant portion of canine mast cell tumors currently have no identified genetic defect in the juxtamembrane domain of c-KIT. In addition to the possibility that other genes are involved, there is also the chance that some of these tumors may contain other mutations in c-KIT. In this study and that of London et al.,9 only the juxtamembrane domain was screened for mutations; however, Ma et al.10 also tested the phosphotransferase domain. The phosphotransferase domain of c-KIT has been reported to harbor mutations in some human7 and rodent15–17 mast cell tumors. In addition to the duplications and deletions, our identification of numerous SNPs in c-KIT, both in the juxtamembrane domain and in other regions of the c-KIT gene, suggests that c-KIT as a whole may be a hotspot for mutations in dogs, with the potential for mutations in mast cell tumors outside of the juxtamembrane domain.
Our finding of c-KIT mutations in grade II and grade III but not grade I tumors indicates that c-KIT may be a good marker for tumor status and suggests a role for c-KIT in at least part of the tumorigenic process, providing a potential target for intervention. Tyrosine kinase inhibitors are currently in development for treatment of a number of cancer types. Very recently, one of these tyrosine kinase inhibitors, STI-5716 (Gleevec, Novartis Pharmaceuticals), has been approved by the Food and Drug Administration for the treatment of chronic myologenous leukemia (CML) in humans. In CML, a translocation causes a hybrid tyrosine kinase made up of parts of the genes bcr and abl to be constitutively active.6 This drug may also be a good inhibitor of c-KIT.4 c-KIT inhibitors such as these and others could be used on tumors both with and without mutations to evaluate their effect on tumor growth and their potential as therapeutic agents. We are planning to launch such studies at the molecular and cellular level, followed by clinical studies. If reduced tumor growth were observed, c-KIT activation, such as that caused by these mutations, would be considered important for the development of mast cell tumors. These findings would pave the way for an additional medical treatment for the management of these tumors.
Footnotes
Acknowledgements
We thank Dr. Joseph Hauptman for assistance with the statistical analyses. This work was funded in part by the Jaqua Foundation, the Animal Health and Diagnostic Laboratory, and the Elizabeth Eddy Fund of Michigan State University.