
Abstract
Canine osteosarcoma (OS) cell lines contain mutations that directly or indirectly inactivate the tumor suppressor genes p53 and retinoblastoma. Another important tumor suppressor, PTEN, is mutated in many human cancers. To determine whether inactivation of PTEN plays a role in the pathogenesis of canine OS, we studied its expression in canine OS cell lines and tumors. Four of five canine OS cell lines (CO2, CO3, CO5, and CO7) constitutively express high levels of the phosphorylated form of Akt, an indirect indicator of aberrant PTEN expression. PTEN protein is essentially absent from three of these cell lines (CO2, CO5, and CO7), whereas CO3 contains a potentially inactivating amino acid substitution in PTEN at codon 340. Genomic hybridization experiments indicate that CO2, CO5, and CO7 contain large deletions within the PTEN gene. Ten of 15 OS tumors exhibit variable or negative PTEN staining. Evaluation of a PTEN-negative staining tumor by Southern blotting indicates that the PTEN gene is deleted in this tumor. These results indicate that PTEN is mutated or downregulated in a high percentage of canine OS cell lines and tumors and likely plays an important role in the pathogenesis of the disease.
Canine osteosarcoma (OS) is a prevalent, highly metastatic tumor, occurring primarily in large and giant breeds. Although canine and human OS are histologically similar, tumor prevalence and age of occurrence are dissimilar.24,31,39 Similarities and differences also exist at the molecular level. We and others have shown that canine OS cell lines and tumors, like their human counterparts, frequently contain mutations that inactivate p53. 17,19,23,40 However, whereas a high percentage of human OS cell lines and tumors contain mutations that block retinoblastoma (Rb) mRNA accumulation, protein expression, or both, little evidence of similar inactivating mutations has been found in canine OS cell lines or tumors.23 Instead, canine OS cell lines contain mutations that indirectly inactivate the three Rb family members, Rb, p107, and p130, simultaneously.19
The tumor suppressor gene, PTEN, encodes a dual specificity protein–lipid phosphatase that plays an important role in regulating cell proliferation, migration, and survival, as well as tumor cell invasion and tumor-directed angiogenesis.1,5,7,10,37 The lipid phosphatase activity of PTEN plays a crucial role in mediating the tumor suppressive effects of PTEN by downregulating phosphatidylinositol 3,4-bisphosphate and phosphatidyl-inositol 3,4,5-trisphosphate (PIP3) levels within the cell.11,21,27 PIP3 activates phosphoinositol-dependent kinase-1 (PDK-1), which in turn activates by phosphorylation several important protein kinases that stimulate cellular proliferation and enhance cell survival. Most notably, the PDK-1 substrate Akt blocks apoptosis by inactivating proapoptotic proteins BAD, FKHR, and caspase-9.4,6,8,9 In addition, Akt stabilizes hypoxia-inducible factor 1-α, thereby stimulating VEGF-mediated angiogenesis.45 PTEN has also been shown to inhibit cell proliferation, migration, and tumor cell invasion by associating with and dephosphorylating focal adhesion kinase.35–37
PTEN is mutated in a wide range of human tumors, including carcinomas of the endometrium, prostate, kidney, and breast, as well as in glioblastomas.2 PTEN loss of heterozygosity frequently is observed in melanoma cell lines and tumors.14,38 Germline mutations of PTEN are associated with several autosomal dominant disorders, most notably Cowden disease.2,20,28 To our knowledge, PTEN mutations have not been identified in human OS. We examined the expression and genomic integrity of PTEN in several canine OS cell lines and tumors. Our results indicate that aberrant expression of PTEN in canine OS cell lines and tumors can at least in part be explained by mutations in the PTEN gene.
Materials and Methods
Osteosarcoma cell lines and tumors
The isolation and maintenance of canine OS cell lines CO2, CO3, CO5, CO7, and CO8 have been described previously.19 Formalin-fixed paraffin-embedded OS tissues were obtained from the collection of specimens kept by the Cornell University College of Veterinary Medicine Section of Pathology. Individual tumor samples are described in Table 1.
Selected characteristics of canine osteosarcoma cases
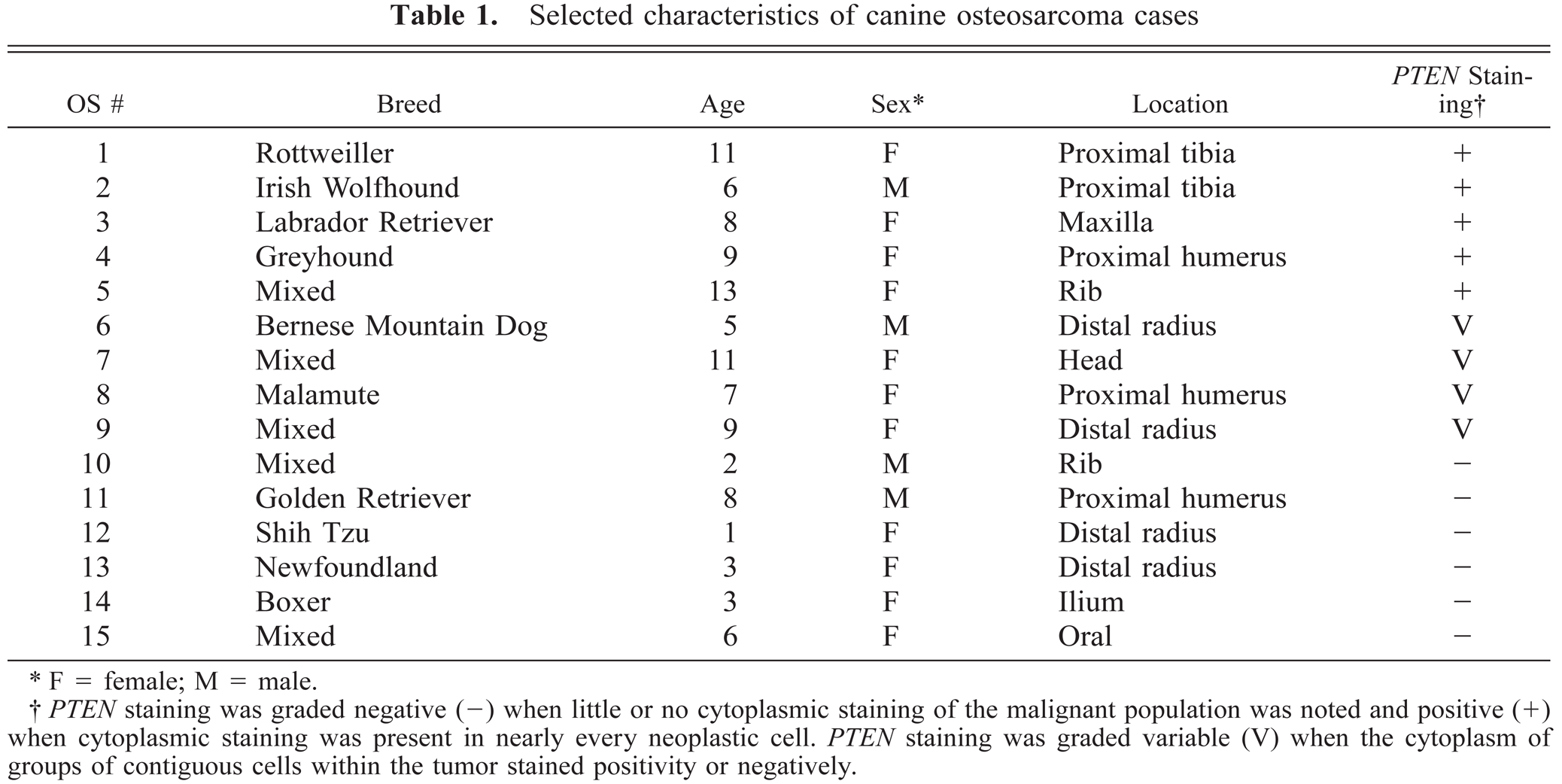
F = female; M = male.
PTEN staining was graded negative (−) when little or no cytoplasmic staining of the malignant population was noted and positive (+) when cytoplasmic staining was present in nearly every neoplastic cell. PTEN staining was graded variable (V) when the cytoplasm of groups of contiguous cells within the tumor stained positivity or negatively.
Antibodies and cloned DNAs
PTEN antibody (A2B1) was obtained from Santa Cruz Biotechnology Inc. (Santa Cruz, CA). Akt (9272) and phospho-Akt (Ser 473, 9271S) antibodies were obtained from New England Biolabs (Beverley, MA). DNA probes for full-length murine PTEN were obtained from Dr. J. Guan (Department of Molecular Medicine, Cornell University, Ithaca, NY). A DNA probe for Jnk 1 was obtained by polymerase chain reaction (PCR) with oligonucleotides (forward primer CATGAGCAGAAGCAAACGTG starting at nucleotide 24 and reverse primer ATTGATCATTGCTGCACCTGTG ending at nucleotide 1164) derived from murine Jnk 1 cDNA published sequences (GenBank accession No. AB005663).
DNA analysis
DNA was extracted from cells in culture or freshly frozen tissue samples by lysis in 100 mM NaCl, 10 mM Tris 8.0, 25 mM, ethylenediametetraacetic acid, 0.5% sodium dodecyl sulfate (SDS), and 100 µg/ml proteinase K at 50C overnight. Contaminants were removed by phenol:CHCl3 extraction and DNA was precipitated in 70% ethanol. DNA was quantitated spectrophotometrically at 260 nm. DNA was analyzed by Southern blotting and hybridization. Briefly, 20 µg of genomic DNA was digested with EcoRI and HindIII and the digest was separated on a 0.8% agarose gel. DNA was depurinated in situ for 15 minutes at room temperature in 0.125 N HCl. The gel was rinsed with H2O and the DNA was denatured in 0.5 M NaOH and 0.5 M NaCl twice for 15 minutes at room temperature. The gel was rinsed in 10× standard saline citrate (SSC) and DNA was transferred to a nitrocellulose membrane (Schleicher & Schuell, Keene, NH). The membrane was baked for 2 hours at 80C and DNA was prehybridized in 6× SSC, 5× Denhardt's, 0.5% SDS, and 100 µg/ml of single-stranded calf thymus DNA for 6 hours at 65C. The DNA was then hybridized in prehybridization buffer containing 1.5 × 106 deca s per minute/ml of 32P-labeled probe at 65C for 16 hours. After hybridization, the membrane was washed twice in 2× SSC and 0.1% SDS for 30 minutes at 65C and then twice in ×X SSC and 0.1% SDS for 30 minutes at 65C. Air-dried blots were autoradiographed onto Kodak MS XAR film (Eastman Kodak, Rochester, NY) with an intensifying screen.
RNA analysis
PTEN mRNA was assayed by northern blotting and hybridization. Briefly, equal amounts of total cellular RNA (20 µg) were separated on a 6% formaldehyde and 1% agarose gel. RNA was transferred to a nylon membrane (ICN, Costa Mesa, CA) and baked for 2 hours at 80C. Murine PTEN cDNA was radioactively labeled by random priming and hybridized in 50% formamide at 42C for 36 hours. Blots were washed twice in 2× SSC and 0.1% SDS for 30 minutes at 65C and then twice in 1× SSC and 0.1% SDS for 30 minutes at 65C. Air-dried blots were autoradiographed as described for Southern blotting.
Protein analysis
For western blotting, control fibroblasts and canine OS cell lines were harvested in ice-cold RIPA buffer (150 mM NaCl, 1% nonidet-P40, 0.5% deoxycholate, and 0.1% SDS, 50 mM Tris, pH 8.0) containing phenylmethyl–sulfonylfluoride (1 mM), aprotinin (1%), leupeptin (20 µg/ml), and sodium orthovanidate (1 mM). Equal amounts of protein (15 µg) were electrophoresed through a 10% SDS polyacrylamide gel. Protein bands were transferred to nitrocellulose by means of a Bio Rad (Hercules, CA) semidry electroblotter. Filters were blocked in 4% milk powder in Tris-buffered saline (pH 7.6) containing 0.05% Tween 20 and incubated with specific antibodies overnight at 4C. After washing, filters were incubated with the appropriate horse radish peroxidase–conjugated secondary antibody, washed, and then processed for detection by the Renaissance Western Blot Chemiluminescence Reagent (NEN Life Science Products, Boston, MA) and Biomax ML film (Eastman Kodak, Rochester, NY).
Immunohistochemistry
Indirect immunoperoxidase staining was performed by the streptavidin–biotin technique with a commercial kit from Zymed Laboratory (South San Francisco, CA) and the MicroProbe staining system (Fisher Scientific, Pittsburgh, PA). Two- to 4-μm sections of formalin-fixed paraffin-embedded tissue were deparaffinized in xylene, rinsed in graduated ethyl alcohols (100%, 95%, and 70%), and transferred from 70% ethanol to 0.5% H202 in methanol for 10 minutes to block endogenous peroxidase. Sections were rinsed in 0.01 M phosphate-buffered saline (PBS), pH 7.2, containing 0.4% BRIJ-35 (Sigma Chemical Co., St. Louis, MO) and incubated for 10 minutes with 10% normal goat serum. The normal serum was tapped off and the sections were incubated with mouse (immunoglobulin [IgG]2a) monoclonal PTEN antibody (A2B1, 1 µg/ml Santa Cruz Biotechnology Inc., Santa Cruz, CA) for 2 hours at 37C. After rinsing with PBS-BRIJ, sections were incubated with biotinylated anti-mouse immunoglobulins for 10 minutes at room temperature. Sections were rinsed with PBS-BRIJ and incubated with streptavidin–biotin complex for 15 minutes at room temperature. Sections were rinsed with PBS-BRIJ and incubated with the DAKO amplification reagent (DAKO Corporation, Carpinteria, CA) for 15 minutes at room temperature. Sections were again rinsed with PBS-BRIJ and incubated with the streptavidin–peroxidase conjugate at room temperature for 10 minutes. After a final rinse in PBS-BRIJ, sections were reacted with aminoethyl carbazole and 0.01% H2O2 (Zymed Laboratory) for 10 minutes at room temperature. The reaction was stopped by rinsing in distilled water. Sections were counterstained with Gill's No. 2 Hematoxylin (Fisher Scientific) for 30 seconds, rinsed in distilled water, and cover slipped with glycerol vinyl alcohol aqueous mounting solution (Zymed Laboratory). All incubations were carried out in a humid chamber. For the negative control, Control Mouse Ascites Fluid Clone NS-1 (M 8273, Sigma Chemical Co.) was substituted for the antibody at a concentration of 1 µg/ml. Blocking peptide experiments were performed with PTEN blocking peptide (sc-7974 P, Santa Cruz Biotechnology Inc., Santa Cruz, CA). Antibody (2 µg/ml) was incubated with a 30-fold stoichiometric excess of blocking peptide for 1 hour at room temperature with gentle stirring. The mixture was applied to tissue sections in place of antibody alone in the protocol described above. PTEN-specific staining was absent in sections pretreated with PTEN-blocking peptide.
Sequencing of PTEN cDNA
RNA isolated from CO3 was used to synthesize cDNA as described previously with primers specific for canine PTEN cDNA (GenBank accession No. U92435; forward primer GCTATGGGGTTTCCTGCAG starting at nucleotide 207 and reverse primer TCTCTGGGTCAGAGTCAGTG ending at nucleotide 1273).19 The 1,066 base pair (bp) PCR product, representing amino acids 34–388, was cloned into the T vector (Promega, Madison, WI) and the DNA was sequenced by the Cornell University Bioresource Center with BIG DYE terminators from PE Biosystems and run on ABI 3700 and ABI 377 DNA Sequencers. DNA sequences were verified by sequencing at least three independent clones.
Results
PTEN is mutated in canine OS cell lines
Western blotting analysis indicated that PTEN protein was absent or expressed at very low levels in OS cell lines CO2, CO5, and CO7, as compared to primary canine fibroblasts (Fig. 1). OS cell lines deficient in PTEN protein contain elevated levels of phosphorylated Akt, as does CO3. The level of phosphorylated Akt (Ser 473) often is elevated in PTEN-deficient cells and tumors and is used here as an indirect assay of Akt and PTEN activity.32,43 Levels of total Akt protein are similar in all of the OS cell lines and slightly higher in control fibroblasts. Only CO8 exhibits a wild-type expression pattern: high levels of PTEN protein and low levels of activated Akt. Consistent with the protein data, CO2 and CO5 do not express PTEN mRNA, whereas CO7 expresses low levels of a normal-sized and a larger molecular weight PTEN RNA (Fig. 2).
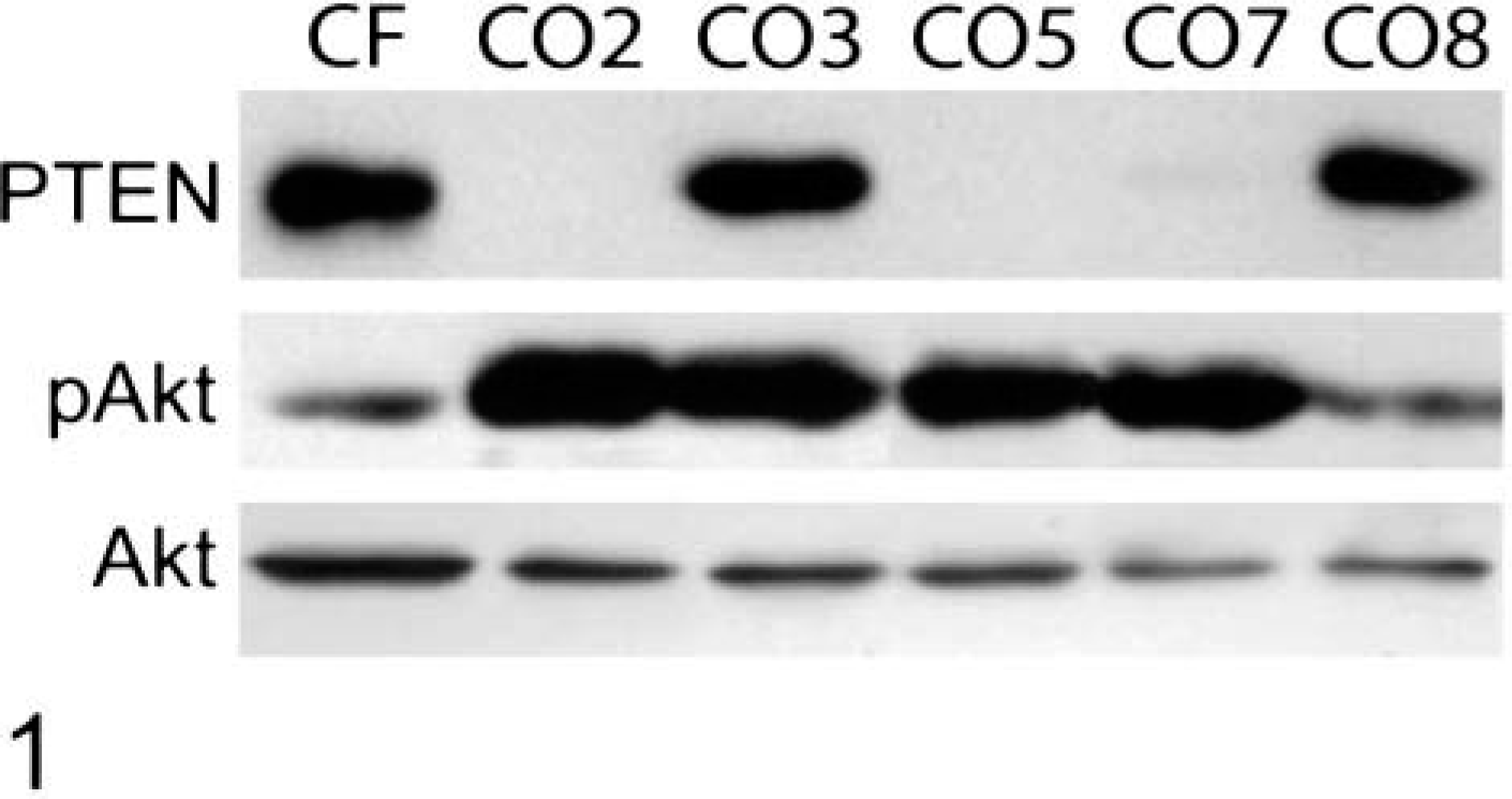
Inactivation of PTEN in canine OS cell lines. Protein extracts were prepared from exponentially growing cells and analyzed by western blotting (15 µg/lane) with antibodies to PTEN, phosphorylated (ser 473) Akt (pAkt), and total Akt.
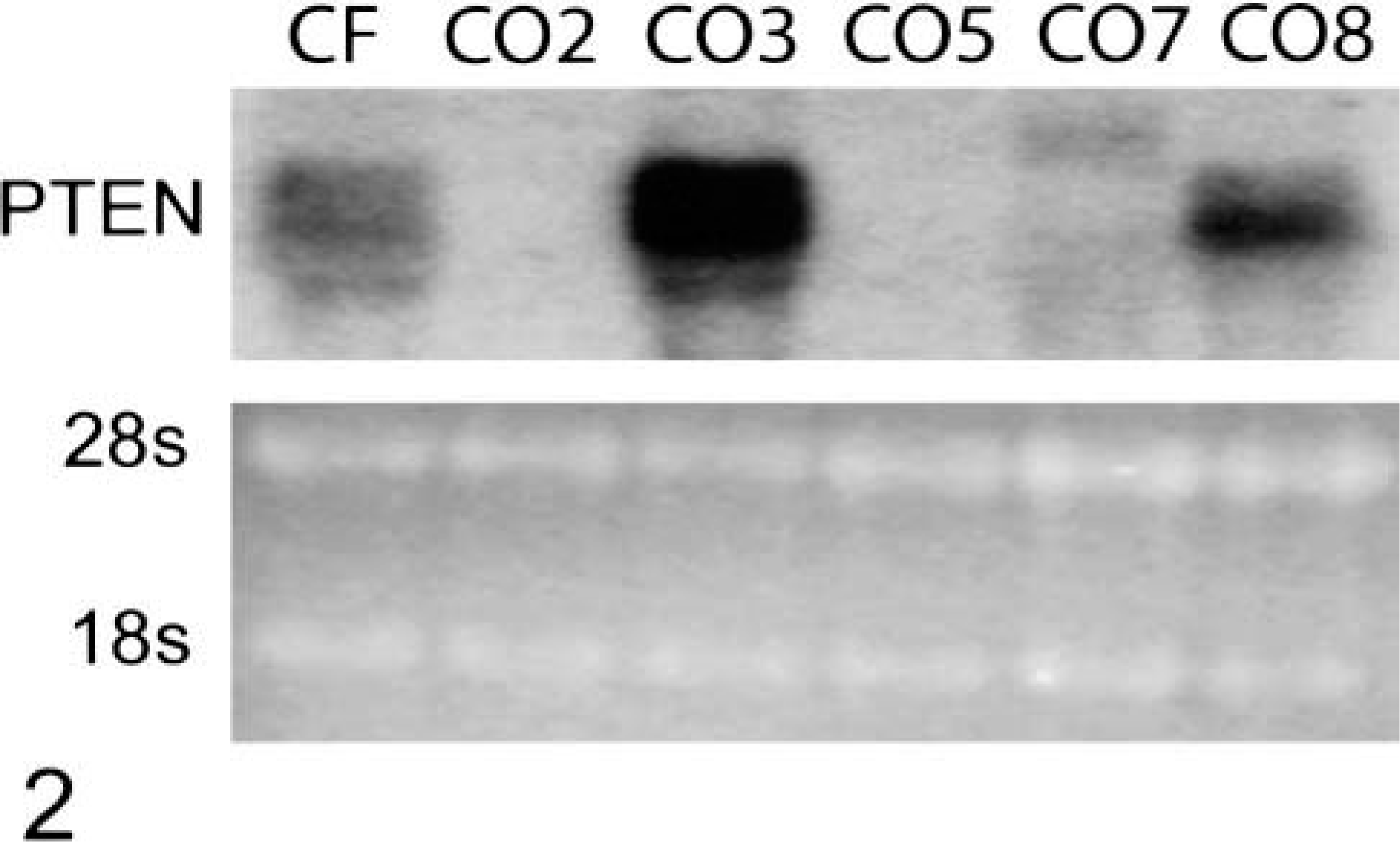
Abnormal PTEN RNA expression in canine OS cell lines. Twenty micrograms of total RNA from each cell line was analyzed by northern blotting using the murine PTEN cDNA probe. Ethidium bromide–stained ribosomal RNA (28s and 18s) was used to control for equal loading.
To determine whether gross alterations in PTEN gene structure are responsible for the observed aberrant PTEN expression, high molecular weight DNA was isolated and subjected to Southern blotting, with a full-length murine PTEN cDNA probe (Fig. 3). Four major and three minor hybridizing bands are apparent in the control, CO3, and CO8 lanes. In contrast, three of the four major bands (9.2 kb, 1.5 kb, and 0.8 kb) and several of the minor bands (6.4 kb, 5.8 kb, and 1.9 kb) are absent from CO2 and CO5. CO7 contains the normal pattern of hybridization; however, the reduced band intensities relative to the control lane are consistent with a monoallelic deletion of the gene. These results indicate that a deletion of a large part of the PTEN gene is responsible for the inability of CO2 and CO5 to express PTEN protein and contributes to the mutant phenotype of CO7.
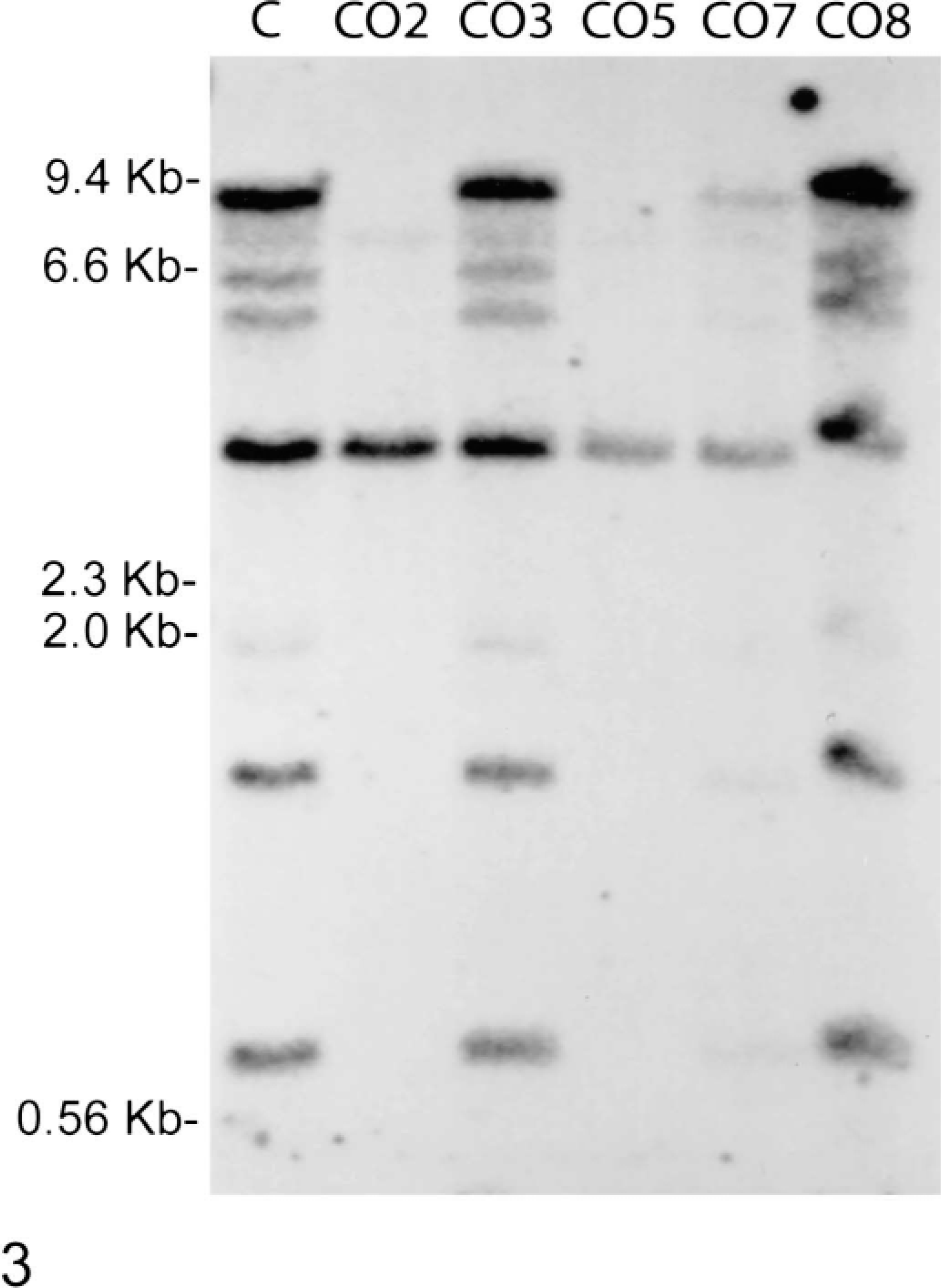
PTEN is deleted in canine OS cell lines. Fifteen micrograms of genomic DNA from a control dog (C) and each cell line was digested with EcoRI and HindIII and analyzed by Southern blotting with a PTEN cDNA probe.
The results presented above indicate that Akt is activated in CO3. To determine whether CO3 contains a mutation that inactivates PTEN without affecting PTEN protein levels, PTEN cDNA was cloned and the DNA was sequenced. CO3 PTEN contains an A to T substitution at nucleotide 1126, codon 340, which substitutes a tyrosine for an asparagine.
Inactivation of PTEN in canine OS tumors
To examine PTEN expression in primary tumors, tissue sections were prepared from 15 canine osteosarcomas and stained with PTEN antibody (Table 1). Only five tumors were uniformly positive for PTEN staining. Six tumors were negative and four tumors exhibited variable PTEN staining. Representative PTEN-positive–staining (OS 1) and negative–staining (OS 10) tumors are shown in Figs. 4 and 5.
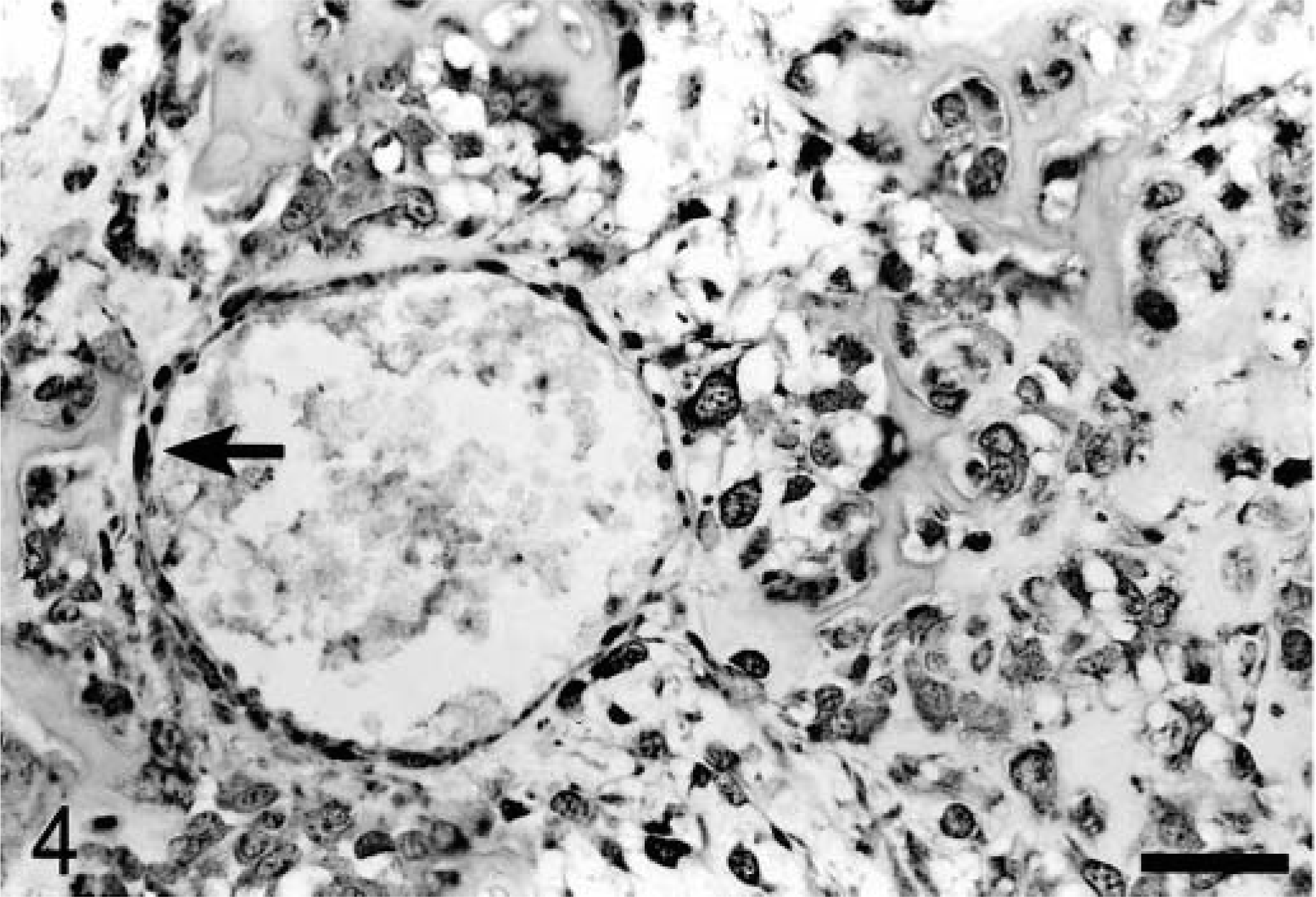
Osteosarcoma; dog; OS No. 1. Section of tumor and vessel exhibiting diffuse cytoplasmic staining of tumor cells. The arrow points to positive-staining endothelial cells (internal positive control). DAB. Bar = 50 μm.
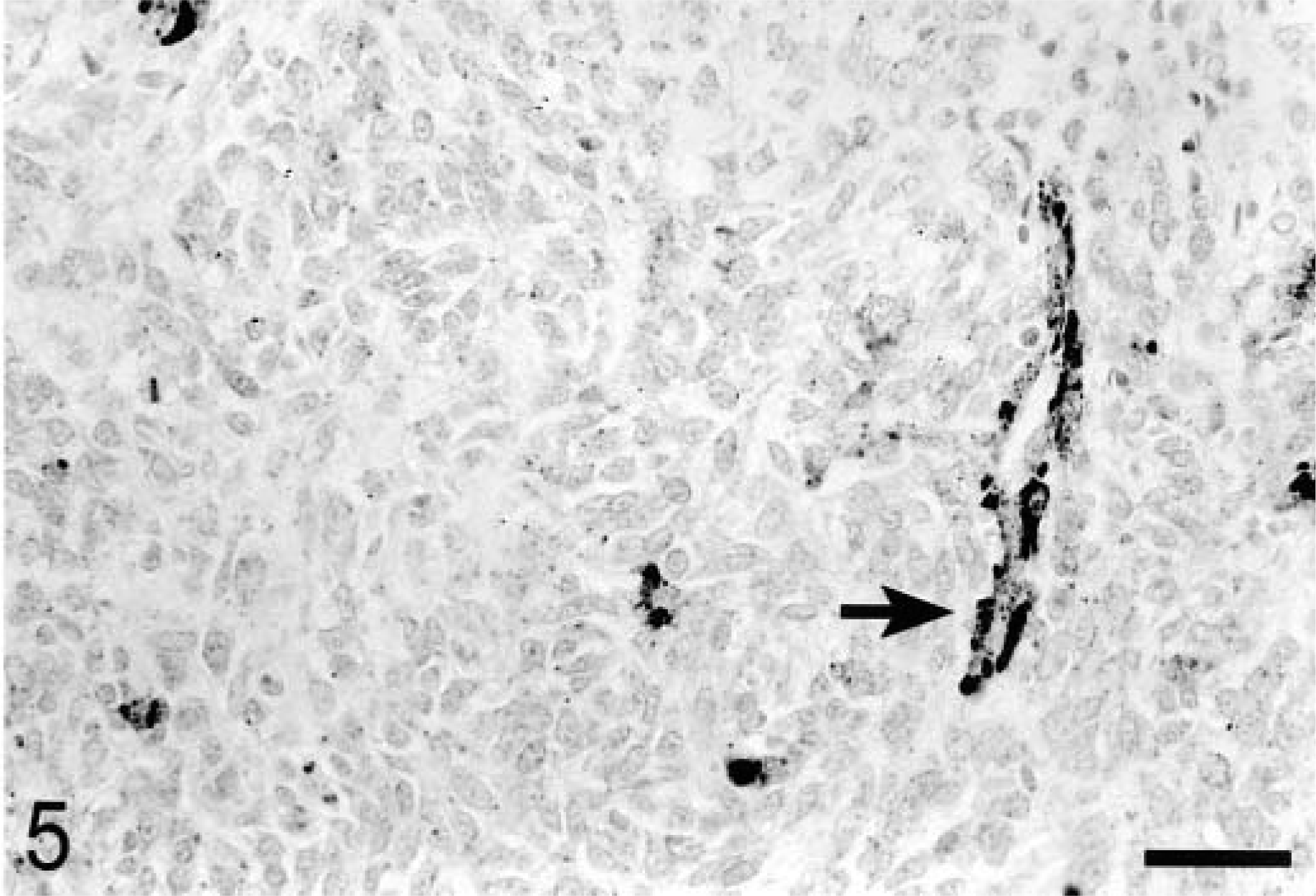
Osteosarcoma; dog; OS No. 10. Section of tumor and vessel exhibiting a diffuse lack of positive-staining. The arrow points to positive staining endothelial cells (internal positive control). DAB. Bar = 50 μm.
Genomic DNA samples from a control dog, a dog with a PTEN-positive–staining tumor (OS 1), and a dog with a PTEN-negative–staining tumor (OS 13) were subjected to Southern blotting and analyzed for the integrity of PTEN gene structure (Fig. 6). OS 13 essentially lacks PTEN hybridizing bands of 9.2, 6.4, 5.8, 1.9, and 0.8 kb. Very faint 9.2 and 0.8 kb bands are visible, but likely represent contamination of tumor samples with reactive fibroblasts and endothelial cells. A duplicate blot probed with Jnk1 produced identical banding patterns in all three lanes. These results indicate that OS 13 contains a homogenous deletion of a large part of the PTEN gene.

PTEN is deleted in canine OS 13. Genomic DNA was isolated from fresh samples of OS 1 and OS 13 and analyzed by Southern blotting with a PTEN or JNK1 cDNA probe. Small differences in band migration between samples likely are due to incomplete removal of salt during DNA purification.
Discussion
Canine OS cell lines and tumors contain mutations that directly or indirectly inactivate p53 and Rb family genes.17,19,23 These tumor suppressors act at cell cycle checkpoints primarily to regulate cell proliferation, differentiation and survival in response to internal and external environmental signals.18,29,41 Additional mutations likely are required to confer other essential cellular properties characteristic of tumor cells, including self-sufficiency in growth signals, sustained angiogenesis, tissue invasion, and metastasis.15 The tumor suppressor PTEN plays an important role in regulating many of the cellular behaviors that commonly are deregulated during tumorigenesis.7,10 Therefore, it is not surprising that PTEN is inactivated in a large number of human tumors.
Mutations in the PTEN gene are present in at least three of five canine OS cell lines and are coincident with increased levels of phosphorylated Akt. PTEN deletions are frequently associated with the loss of PTEN protein in sporadic human tumors.2 Two of the OS cell lines, CO2 and CO5, contain large deletions in both PTEN alleles. CO7 contains a deletion of one PTEN allele and likely contains a mutation in the remaining allele, which results in low levels of an aberrantly sized PTEN mRNA and very low levels of protein.
The remaining OS cell line, CO3, contains a misregulated phosphoinositide 3-kinase/Akt signaling pathway, as indicated by the constituitively elevated levels of phosphorylated Akt. We identified a single base change in PTEN cloned from CO3 that results in a tyrosine to asparagine amino acid substitution at codon 340. Although this specific mutation has not been identified in human cancers, mutations within this region of the C-terminal end of human PTEN have been shown to alter protein folding and impair phosphatase activity.12 Whether this substitution represents a mutation that alters PTEN function or a polymorphism within the gene must await the results of functional studies.
In contrast to the other OS cell lines, CO8 expresses normal levels of PTEN protein and activated Akt. Given the importance of the phosphoinositide 3-kinase/Akt signaling pathway in regulating cell proliferation and survival, it is reasonable to speculate that a mutation in a distinct pathway antagonizes PTEN action, providing an alternative mechanism to bypass this important tumor suppressor.
As a tumor suppressor gene, PTEN expression is downregulated in tumors and tumor cell lines by genetic and epigenetic mechanisms.2,44 Unlike the p53 tumor suppressor, mutations in the PTEN gene do not generally result in elevated levels of PTEN protein.2,3 The fact that PTEN immunostaining of OS tumor sections is variable or negative in 10 of 15 tumors argues that PTEN expression is downregulated in a high percentage of canine osteosarcomas. The mechanisms leading to PTEN downregulation in most of these tumors is unknown, but at least one PTEN-negative tumor (OS 13) contains a deletion of a large portion of the PTEN gene. The mutational inactivation of PTEN in at least three of five OS cell lines provides additional support for the hypothesis that PTEN mutations occur frequently in canine OSs.
Canine OS occurs primarily in older dogs, although a significant proportion of affected animals are less than 5 years of age. Early-onset human cancers often are associated with the inheritance of mutated tumor suppressor genes, including PTEN. 2,13 All four of the early-onset OSs in dogs less than 5 years of age that we examined were negative for PTEN immunostaining. However, our preliminary results indicated that the one early-onset tumor for which the appropriate genomic DNA samples were available (OS 13) does not contain a corresponding germline mutation in the PTEN gene (data not shown).
In humans, the majority of mutations in PTEN have been identified in epithelial cell-derived tumors, including cancers of the breast, endometrium, prostate, kidney, and glia.2,7 PTEN(±) mice develop a variety of tumors that are similar, but not identical to, the types of tumors observed in humans.30,33 In humans and mice, PTEN inactivating mutations therefore are linked to the development of tumors of specific tissues and histologic types. To our knowledge, PTEN mutations have not been associated with the occurrence of OS. Therefore, it is significant that PTEN is mutated or downregulated in a high percentage of canine OS cell lines and tumors. These data underscore the important role that PTEN plays in negatively regulating tumorigenesis in mesenchymal as well as epithelial cell types.
The expression of exogenous PTEN in PTEN-deficient epithelial cell–derived tumor cells has multiple effects depending on the cell type that is used. PTEN can inhibit proliferation; block anchorage-independent growth; inhibit cell spreading, migration, and invasiveness; and stimulate apoptosis.16,22,25,26,36,42 Reintroducing PTEN into PTEN-deficient mouse embryo fibroblasts sensitizes cells to various proapoptotic stimuli, but does not alter the rate of cell proliferation under normal growth conditions.32,34 An understanding of the role that PTEN plays in the pathogenesis of canine OS must await further experiments in which PTEN activity is restored in OS cell lines and tumors.
Footnotes
Acknowledgements
We are grateful to Sean Mc Donough and Clive Huxtable for helpful discussions and Margaret McEntee, Rodney Page, Susan Ettinger, and the oncologists and surgeons at Cornell Veterinary College for supplying fresh OS specimens.