
Abstract
Cyclooxygenase-2 (COX-2) was detected and localized in 15 pigs with naturally occurring pleuropneumonia using a 437–base pair digoxigenin-labeled cDNA probe in an in situ hybridization protocol. Histopathologic changes in the acute stage were characterized by coagulative necrosis of lung parenchyma, hemorrhage, vascular thrombosis, edema, fibrin deposition, and infiltration of lung parenchyma by neutrophils and alveolar macrophages in nine pigs. In chronic lesions, a thick layer of granulation tissue surrounded foci of pulmonary necrosis in six pigs. All 15 pigs infected with Actinobacillus pleuropneumoniae, confirmed by bacterial isolation, had distinct positive hybridization signals for COX-2 in bronchial, bronchiolar epithelial cells, alveolar macrophages, neutrophils, and type I pneumocytes. COX-2 expression was detected primarily in neutrophils from pigs with acute lesions and primarily in alveolar macrophages from pigs with chronic lesions. The results suggest that a prostanoid product of COX-2 is an important component of the inflammatory response to acute and chronic A. pleuropneumoniae infection.
Porcine pleuropneumonia, caused by Actinobacillus pleuropneumoniae, is a contagious, and often fatal pneumonia in swine. Histologic lesions are characterized by fibrinous pleuritis, coagulation necrosis of lung parenchyma, hemorrhage, vasculitis, vascular thrombosis, edema, fibrin deposition, and infiltration of the lung parenchyma by neutrophils and mononuclear cells. 3,30 Vasculitis and thrombosis are important to the pathogenesis in porcine pleuropneumonia. 1,23 In gram-negative bacterial diseases, these vascular abnormalities are thought to be partly mediated by lipopolysaccharide (LPS) up-regulation of prostaglandins. 5,13
Arachidonic acid–derived prostaglandins are potent inflammatory mediators, and they also modulate immune responses and physiologic processed such as bronchoconstriction, vasodilation, and mucus secretion in the lung. 12,28 In addition, prostaglandins increase transvascular hydrostatic pressure and promote increased microvascular permeability by synergizing with and stimulating the release of secondary mediators. 11,24 Prostaglandins are synthesized from arachidonic acid by two cyclooxygenase (COX) isoforms. COX-1 facilitates the synthesis of prostaglandins involved in homeostatic functions, whereas COX-2 is an inducible enzyme predominantly expressed at sites of inflammation. COX-2 expression is induced by LPS and inflammatory cytokines such as interleukin-1 (IL-1) and tumor necrosis factor-α (TNF-α). 16,21,25 The objective of this study was to determine whether COX-2 was expressed in the lungs of pigs naturally infected with A. pleuropneumoniae.
Materials and Methods
Animals
Samples were obtained at necropsy from pigs submitted to the Department of Veterinary Pathology of Seoul National University from January 1994 to December 1997. Fifteen pigs (Nos. 1–15) approximately 87–140 days old, from 15 different herds, were selected on the basis of clinical signs, characteristic lesions, bacteria isolation, and serotype. All 15 pigs were negative for Mycoplasma hyopneumoniae, Pasteurella multocida, and porcine reproductive and respiratory syndrome virus by culture and in situ hybridization. 6,22
Positive control sections were from pigs that had been naturally infected with A. pleuropneumoniae. 8,27 Two additional positive control samples at 24 and 48 hours after inoculation were from pigs that had been experimentally infected with A. pleuropneumoniae. Negative control sections were prepared from an 1-day-old colostrum-deprived pig that had not been exposed to any viral and bacteria pathogens, and conventional 60-day-old and 80-day-old pigs that were negative for bacteria isolation. Lung section from a 3-month-old calf with naturally occurring Pasteurellosis was also used for negative controls.
Tissue processing
Samples of lung, liver, kidney, palatine tonsil, mediastinal lymph node, and large and small intestine specimens were collected from infected and noninfected animals, fixed in 10% (w/v) neutral buffered formalin for 24–48 hours, and embedded in paraffin according to standard laboratory procedures. Sections were cut 4-μm thick, floated on a water bath containing diethylpyrocarbonate-treated (DEPC) water and mounted on positively charged slides (Superfrost/Plus slide, Erie Scientific Co., Portsmouth, NH).
RNA extraction
LPS-stimulated alveolar macrophages, and pleuropneumonic lung specimens from 15 pigs naturally infected with A. pleuropneumoniae and 2 positive control pigs experimentally infected with A. pleuropneumoniae were used for extraction of COX-2 RNA, respectively, as previously described. 8 RNA was extracted from these cells with Trizol LS reagent (Gibco BRL, Grand Island, NY) according to the manufacturer's instructions. RNA extracts were treated with DNase I (Gibco BRL) to eliminate genomic DNA contamination.
Primer
The primers were designed based on the porcine COX-2 cDNA sequence (GenBank accession number AF207824). The forward and reverse primers were 5′-GGAGAGACAGCATAAACTGC-3′ (nucleotides 413–432) and 5′-GTGTGTTAAACTCAGCAGCA-3′ (nucleotides 830–849), respectively. The primers amplified a 437–base pair (bp) cDNA fragment.
Reverse transcription-polymerase chain reaction
For the first-strand cDNA synthesis, 1 μl of the COX-2 RNA (5 ng/μl) was supplemented in a total reaction volume of 20 μl with 1× RT buffer (50 mM Tris-HCl, 8 mM MgCl2, 30 mM KCl, 1 mM dithiothreitol [pH 8.3]), 0.5 mM (each) deoxynucleotide triphosphates (dNTPs) (GeneClone, Suwon, Korea), 2.5 μM random hexanucleotide mixture (Applied Biosystems, Foster City, CA), 20 U of RNase inhibitor (Applied Biosystems), and 50 U of Moloney murine leukemia virus reverse transcriptase (Applied Biosystems). After incubation for 15 minutes at 42 C, the mixture was incubated for 5 minutes at 99 C to denature the products. The mixture was then chilled on ice.
The composition of the polymerase chain reaction (PCR) mixture (150 μl) was 30 μl of cDNA (5 ng/μl), 2 μl of each primer (250 nM), 15 μl of 10× PCR buffer (10 mM Tris-HCl, 40 mM KCl, 1.5 mM MgCl2 [pH 8.3]), 1.2 μl of each dNTP (0.2 mM), 2.5 unit of Taq polymerase (GeneClone) in a volume of 29 μl, and 67.2 μl of distilled water. The PCR reaction for COX-2 was done under the following conditions in a thermal cycler (Perkin-Elmer-Cetus, Norwalk, CT): 35 cycles of denaturation at 94 C for 30 seconds, annealing at 56 C for 30 seconds, and elongation at 72 C for 30 seconds.
Preparation of labeled probe
PCR products of COX-2 were purified using a 30-kD cutoff membrane ultrafiltration filter. The nucleotide sequences of the purified PCR products were determined by use of BigDye chemistry with the ABI Prism Sequencer (Applied Biosystems). Sequencing was performed in the purified PCR products before PCR products were labeled by random priming with digoxigenin-dUTP (Boehringer Mannheim, Indianapolis, IN) according to the manufacturer's instructions.
In situ hybridization
Sections were deparaffinized in xylene and rehydrated in phosphate-buffered saline (PBS) (pH 7.4, 0.01 M) for 5 minutes. Deproteinization was carried out in 0.2 N HCl for 20 minutes at room temperature. Tissues were then digested at 37 C for 20 minutes in 100 μg/ml proteinase K (Gibco BRL) in PBS (pH 7.4, 0.01 M). Serial sections of each tissue section examined were treated with RNase A (Boehringer Mannheim) at 100 μg/ml in 10 mM Tris-HCl (pH 7.4) for 30 minutes at 37 C to remove target RNA as a specificity control. After digestion, tissues were fixed in 4% paraformaldehyde in PBS for 10 minutes. After rinsing twice with PBS, the slides were acetylated in 300 ml of 0.1 mM triethanolamine-HCl buffer (pH 8.0) to which 0.75 ml of acetic anhydride (0.25%) had been added. After 5 minutes, an additional 0.75 ml of acetic anhydride was added, and 5 minutes later the slides were rinsed in 2× saline sodium citrate (SSC) (1× SSC contains 50 mM NaCl and 15 mM sodium citrate pH 7.0). The slides were allowed to equilibrate for 60 minutes in a standard hybridization buffer that consisted of 5× SSC with 50% deionized formamide, 10× 2% buffered blocking solution (Boehringer Mannheim), 0.1% N-lauroylsarcosine, and 0.02% sodium dodecyl sulfate.
Hybridization was done overnight at 45 C for COX-2. The digoxigenin-labeled probe (0.1 ng/μl) was diluted in 300 μl of the standard hybridization buffer, heated for 10 minutes at 95 C on a heating block, and quenched on ice before being applied to the tissue sections. Approximately, 50 ng of the digoxigenin-labeled probe was added to the standard hybridization buffer (50 μl), which was then layered over the section. Fluid was held in place by a coverslip, and the edges were sealed with rubber cement. After hybridization, sections were thoroughly washed twice in 4× SSC for 5 minutes at room temperature, twice in 2× SSC for 10 minutes at 37 C, twice in 0.2× SSC for 5 minutes at room temperature, and once in maleic acid buffer (100 mM maleic acid and 150 mM NaCl, pH 7.5) for 5 minutes at room temperature.
For detection of hybridization, sections were incubated with anti-digoxigenin conjugated with alkaline phosphatase (Boehringer Mannheim) diluted 1:250 in 0.1 M Tris-HCl (pH 7.4), 0.15 M NaCl with 1% blocking reagent (Boehringer Mannheim). After three washes in buffer, substrate consisting of nitroblue tetrazolium and 5-bromocresyl-3-indolylphosphate was layered over the sections. Color was allowed to develop for 5–8 hours in the dark, and the reaction was stopped by dipping slides briefly in tri-ethylenediaminetetraacetic acid buffer (10 mM Tris-HCl and 1 mM EDTA, pH 8.0). Sections were counterstained with 0.5% methyl green, and the slides were then washed with distilled water for 1 minute, allowed to dry completely, dipped into the absolute xylene, and coverslipped with Canada balsam mounting medium (Hayashi Pure Chemical Industries Ltd., Osaka, Japan).
Results
Reverse transcription-polymerase chain reaction
Amplification of template cDNA with primers of COX-2 resulted in amplified products corresponding to those of the predicted size. PCR products were sequenced, and their identity was confirmed as COX-2. To investigate whether A. pleuropneumoniae was able to express COX-2, RT-PCR analyses were performed with RNA extracted from lung tissues of pigs that had been infected with A. pleuropneumoniae. COX-2 was consistently detected in lung tissues from the 15 pigs with naturally occurring pleuropneumonia (Fig. 1). COX-2 was also detected in lung tissues from the two pigs experimentally infected with A. pleuropneumoniae. In contrast, COX-2 was not detected in lung tissues from the negative control pigs.
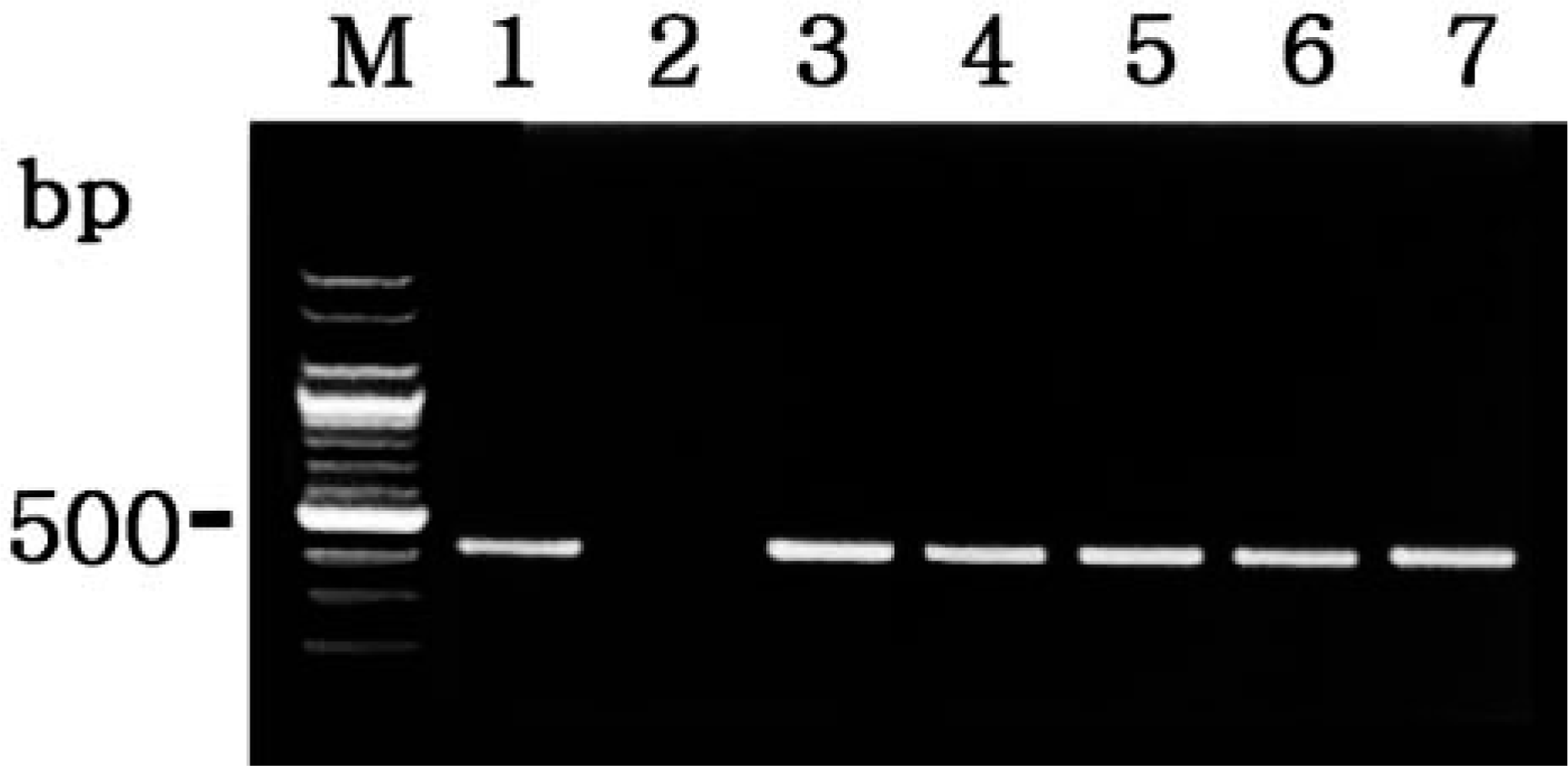
Agarose gel electrophoresis of PCR-amplified COX-2 cDNA products. From left to right: M = 100-bp DNA ladder; lane 1 = positive control from LPS-treated macrophages; lane 2 = negative control from normal macrophages; lane 3 = positive COX-2 cDNA from pig No. 1; lane 4 = positive COX-2 cDNA from pig No. 7; lane 5 = positive COX-2 cDNA from pig No. 8; lane 6 = positive COX-2 cDNA from pig No. 11; lane 7 = positive COX-2 cDNA from pig No. 14; lane 8 = positive COX-2 cDNA from pig No. 15.
Microscopic lesions
Histopathologic changes in the acute stage were characterized by coagulative necrosis of lung parenchyma, hemorrhage, vascular thrombosis, edema, fibrin deposition, and infiltration of lung parenchyma by neutrophils and alveolar macrophages in nine pigs (Nos. 1, 3, 7, 8, 11–15). In chronic lesions, a thick layer of granulation tissue surrounded foci of pulmonary necrosis in six pigs (Nos. 2, 4–6, 9, 10). There was no association between serotype and the characteristics of the histopathology.
In situ hybridization
The 15 pigs naturally infected with A. pleuropneumoniae had distinct and positive hybridization signals for COX-2 (Table 1). The morphology of host cells was preserved despite the relatively high temperature required during the incubation procedure. The signal intensity varied within and between anatomical structures in any one section and between pigs. Positive cells had dark brown or black reaction product in the cytoplasm without background staining.
In situ hybridization in 15 pigs naturally infected with Actinobacillus pleuropneumoniae.
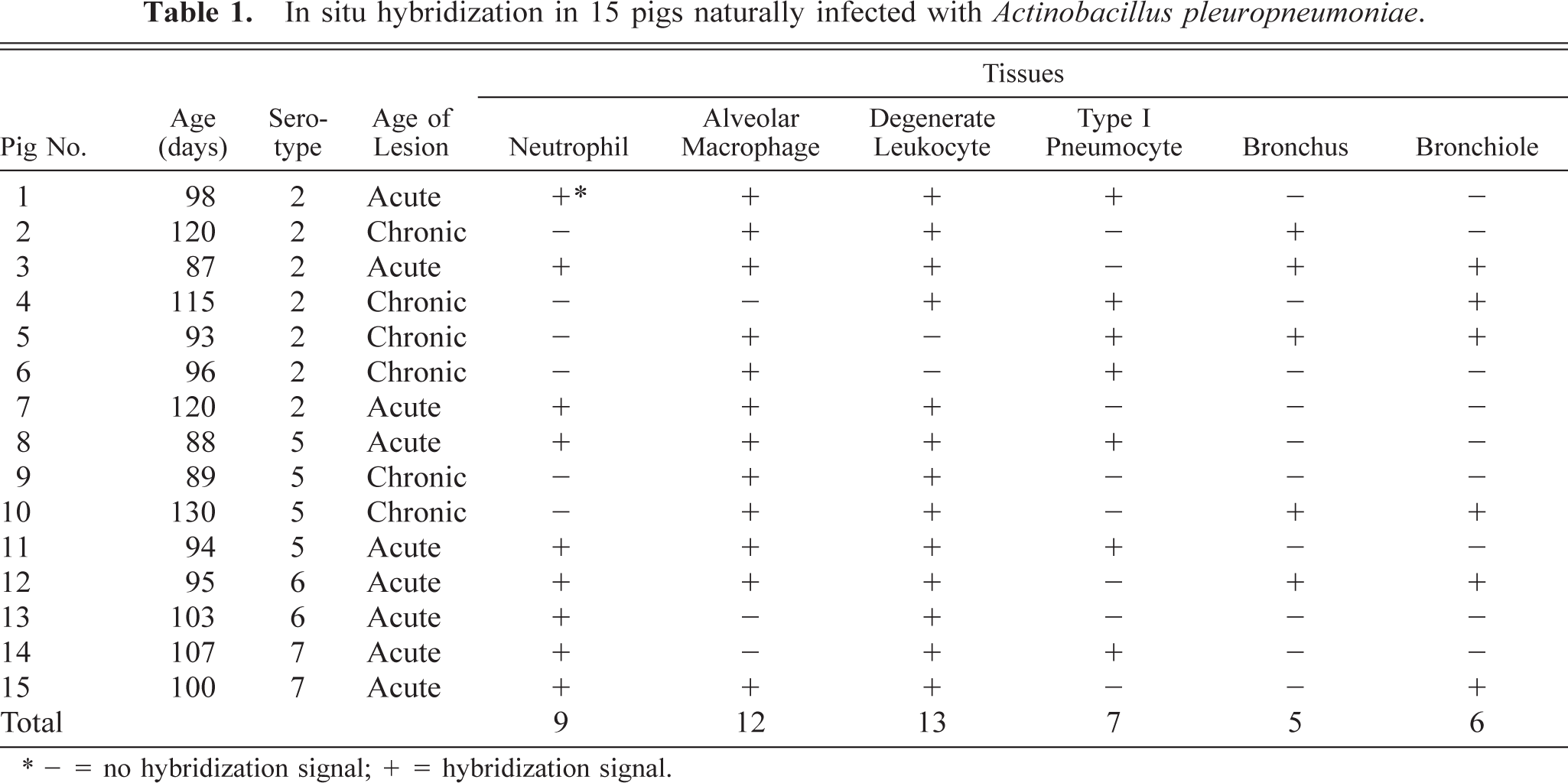
∗ - = no hybridization signal; + = hybridization signal
Bronchial, bronchiolar epithelial cells, alveolar macrophages, neutrophils, and type I pneumocytes had positive hybridization signals. A strong hybridization signal for COX-2 was seen in degenerate alveolar leukocytes (“oat cells”) adjacent to foci of coagulative necrosis, and in alveolar spaces (Fig. 2) and also in neutrophils (Fig. 3) and macrophages that had infiltrated alveolar spaces. Expression of COX-2 was consistently associated with lung lesions, but it was negligible in unaffected portions of lung from the A. pleuropneumoniae–infected pigs. Hybridization signals were seen in type I pneumocytes in seven pigs (Nos. 1, 4–6, 8, 11, 14), bronchial epithelial cells in five pigs (Nos. 2, 3, 5, 10, 12) and bronchiolar epithelial cells (Fig. 4) in six pigs (Nos. 3–5, 10, 12, 15). Hybridization signals were frequently detected in inflammatory cells in the lumina of bronchioles (Fig. 4). Positive in situ hybridization signals for A. pleuropneumoniae were detected in neutrophils from all naturally infected pigs (Nos. 1, 3, 7, 8, 11–15) with acute lesions. In contrast, neutrophils were negative but positive alveolar macrophages were observed in 83% (5/6) of the naturally infected pigs with chronic lesions.
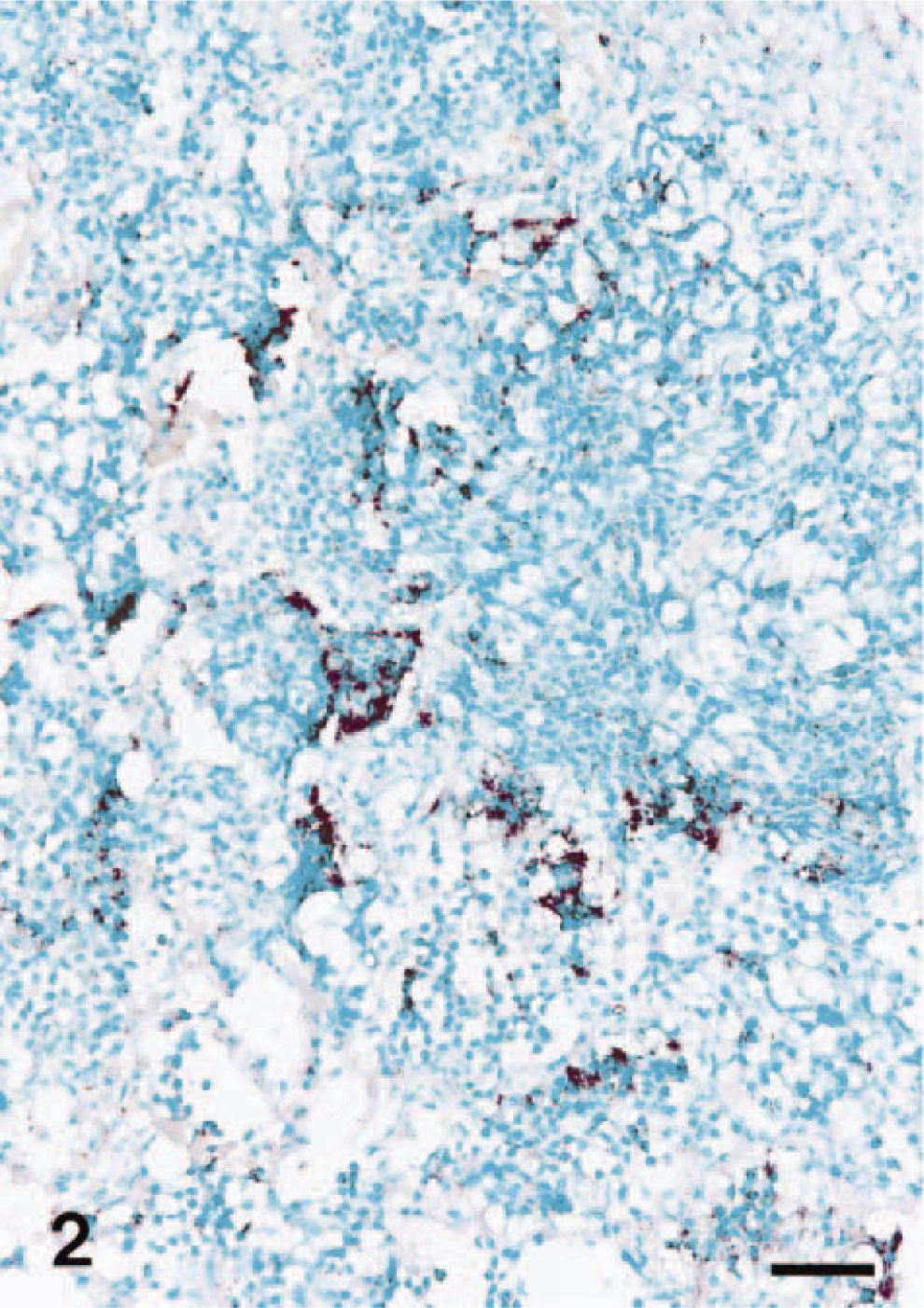
Lung; pig No. 1, naturally infected with Actinobacillus pleuropneumoniae serotype 5. COX-2 RNA (dark brown reaction) was detected in streaming degenerate alveolar leukocytes. In situ hybridization; cDNA probe; nitroblue tetrazolium/5-bromocresyl-3-indolylphosphate, methyl green counterstain. Bar = 55 μm.
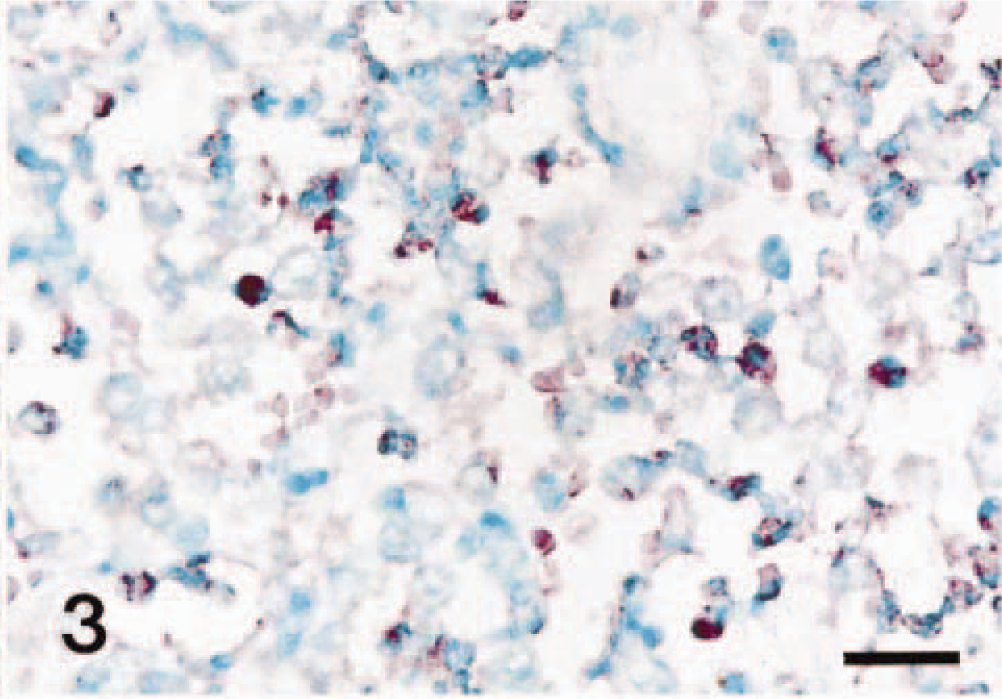
Lung; pig No. 3, naturally infected with Actinobacillus pleuropneumoniae serotype 2. COX-2 RNA (dark brown reaction) was detected in neutrophils of alveolar spaces. In situ hybridization; cDNA probe; nitroblue tetrazolium/5-bromocresyl-3-indolylphosphate, methyl green counterstain. Bar = 30 μm.
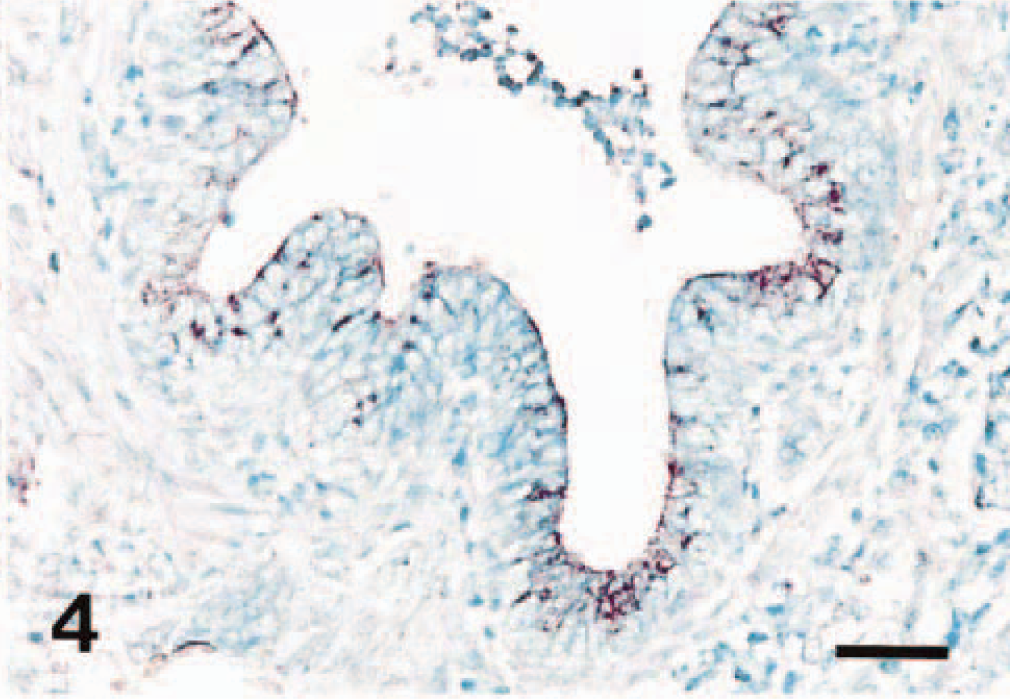
Lung; pig No. 10, naturally infected with Actinobacillus pleuropneumoniae serotype 5. COX-2 RNA (dark brown reaction) was detected in bronchiolar epithelial cells Positive signals were seen in inflammatory cells in lumen of bronchiole. In situ hybridization; cDNA probe; nitroblue tetrazolium/5-bromocresyl-3-indolylphosphate, methyl green counterstain. Bar = 55 μm.
Pretreatment with RNase A eliminated hybridization signal from 15 pigs naturally infected with A. pleuropneumoniae and positive control pigs. Sections from negative control pigs showed no hybridization signals for COX-2. No hybridization signal was detected in lung sections from a calf with Pasteurellosis.
Discussion
The major site of COX-2 expression in lungs of pigs naturally infected with A. pleuropneumoniae are neutrophils and macrophages. These data are supported by results of other studies where observations of increased COX-2 expression was detected in isolated alveolar macrophages or neutrophils after stimulation with LPS or inflammatory cytokines. 21,25,26,29 Besides these inflammatory cells, expression of COX-2 was found occasionally in bronchial epithelial cells and type I pneumocytes. COX-2 induction in bronchial epithelial cells and pneumocytes has also been reported in a mouse model of oxygen-induced acute respiratory distress syndrome and in severe bronchopneumonia of nonhuman primates. 1,23
Traditionally, neutrophils have not been known to release products of the COX pathway of arachidonic acid metabolism. Although the COX isoforms have been described in a variety of cells and tissues, there have been few reports regarding the expression of these products in neutrophils. 26,29 Human neutrophils express COX-2 and release PGE2 after LPS stimulation. 14 Neutrophils that produce prostanoids such as PGE2 could modify the inflammatory process. PGE2 produced by stimulated neutrophils may be involved in the formation of edema at sites of acute inflammation. 18,24
COX-2 regulation may occur in a complex and cell-specific manner. COX-2 may be proinflammatory during the early stage of inflammation dominated by neutrophils, but it might also aid resolution in the later mononuclear cell–dominated stage by facilitating the synthesis of an alternative set of antiinflammatory prostaglandins. 16,17 Our results are in agreement with these previous reports in other species with COX-2 expression primarily in neutrophils of acute lesions and in alveolar macrophages of chronic lesions. It is well accepted that PGE2 formed by COX-2 contributes to lesions of acute inflammation by causing vasodilation, and increased vascular permeability. 10 In chronic infection where the macrophage is the dominant inflammatory cell, which contain COX-2 may exert an additional proinflammatory effect by contributing to angiogenesis. 15 Thus, a prostanoid product of COX-2 is an important component of the inflammatory response to acute and chronic A. pleuropneumoniae infection.
Two virulence factors of A. pleuropneumoniae are the LPS and Apx. 19,32 Previous studies indicate that the LPS is responsible for acute pulmonary inflammation and that the Apx is responsible for the necrosis seen in the acute pneumonia. 7,9,32 Prostaglandins are important mediators of LPS-induced inflammation, 25 and satisfactory clinical resolution of LPS-induced inflammation is often seen in pigs with endotoxin-induced acute respiratory failure when nonsteroidal antiinflammatory therapeutic agents are administered. 20 Moreover, indomethacin, a nonselective COX inhibitor, not only lessened the severity of pulmonary lesions but also prevents further death losses in pigs challenged with A. pleuropneumoniae. 4 These observations suggest that COX-2 expression may play a role in the pathophysiology of pleuropneumonia.
The increased expression of COX-2 may be a direct response to A. pleuropneumoniae LPS because the expression of COX-2 was greater in lesions of pleuropneumonia than in unaffected portions of lung from infected pigs although the association between COX-2 expression and histopathologic findings does not prove cause and effect. Furthermore, in situ hybridization of serial sections of lung indicated that the majority of areas containing numerous COX-2-positive cells also have numerous A. pleuropneumoniae DNA-positive cells (data not shown). Alternatively, A. pleuropneumoniae–stimulated expression of COX-2 may be secondary to the effects of cytokines produced in response to A. pleuropneumoniae. COX-2 is expressed after host cells are exposed to proinflammatory cytokines (e.g., TNF-α, IL-1). 31 Expression of proinflammatory cytokines are detected in porcine pleuropneumonia caused by A. pleuropneumoniae. 2,8 Although the manner in which these cytokines interact is not completely understood, it is likely that TNF-α and IL-1 act synergistically with A. pleuropneumoniae LPS to induce COX-2 expression by inflammatory cells in infected lungs.
In this study, in situ hybridization with nonradioactive digoxigenin-labeled probes was successfully applied to A. pleuropneumoniae–infected lung to detect COX-2 and to identify the sites at which it was expressed. But the detection of COX-2 nucleic acid does not necessarily equate with functional activity of prostanoid production in vivo. Further study is needed to determine the biological role of COX-2 in the pathophysiologic process of pleuropneumonia.
Footnotes
Acknowledgements
The research reported here was supported by a grant of the Korea Health 21 R&D Project (02-PJ1-PG3-20799-0001), the Ministry of Health and Welfare, Republic of Korea.