
Abstract
Veterinary pathologists engaged in basic research use a variety of methods to study disease pathogenesis at the light microscopic and submicroscopic (protein and mRNA) levels. The ribonuclease protection assay is a sensitive and accurate method to measure mRNA expression. The major advantages of the assay are that multiple mRNA species can be measured simultaneously in a single total RNA sample and that the assay has relatively high throughput. The major disadvantage is that the assay requires moderate technical skill.
Keywords
All veterinary pathologists are trained to recognize changes in diseased tissue at the gross, light microscopic, and ultrastructural levels, and in many cases (e.g., trauma, infarct) these standard morphologic techniques are sufficient to determine the pathogenesis of a lesion. In other cases (e.g., immunologic injury, genetic defect), tissue injury results from complex events that occur at the molecular level, so the pathologist must study temporal changes in protein and messenger RNA (mRNA) expression to really understand disease pathogenesis.
Techniques that allow one to study protein expression within a lesion (e.g., immunohistology, enzyme-linked immunosorbent assay, western analysis, flow cytometry) are extremely powerful, but they require that specific antibodies (polyclonal or preferably monoclonal) be raised against proteins of interest, a process that could take a year or more. Molecular techniques that allow one to study mRNA expression (e.g., northern analysis, reverse transcription polymerase chain reaction [RT-PCR], in situ hybridization, cDNA microarrays, ribonuclease protection assay) require only knowledge of the gene sequence, and then specific reagents (primers, probes) can be made rapidly in the laboratory or purchased. However, mRNA is extremely labile, and because of factors such as mRNA half-life, posttranscriptional processing, and rate of mRNA translation to protein, mRNA levels may or may not actually correspond to the level of protein expressed within a tissue.
Clearly, each of the aforementioned methods has various advantages and disadvantages when used as a single test. A combination of all the morphologic, protein, and mRNA expression data associated with a lesion should be used to elucidate the pathogenesis of the disease. To make sense of the available data, pathologists involved in basic discovery research must have a working knowledge of the various assays used to measure protein and mRNA expression. This mini-review is the first in a series that will discuss some of the newer techniques currently being used by veterinary pathologists to study the pathogenesis of disease at the submicroscopic level.
Ribonuclease Protection Assay Procedure
In its simplest form, the ribonuclease protection assay (RPA) is useful for measuring expression of a single target mRNA species in a complex mixture of total RNA. 3 A DNA fragment that codes for a portion of the target mRNA is cloned into a plasmid adjacent to an SP6 or T7 bacteriophage promoter (Fig. 1). The enzyme DNA-dependent RNA polymerase is then added to a reaction mixture containing the plasmid, buffers, nucleotides (ATP, GTP, CTP, and UTP), and 32P-UTP. The polymerase binds to the SP6 or T7 promoter and begins to reverse transcribe the DNA probe fragment into single-stranded RNA (hence, reverse transcription), incorporating 32P-UTP. 15 In addition to reverse transcribing the desired probe sequence, the polymerase invariably continues downstream, reverse transcribing additional irrelevant plasmid-derived sequence until it reaches a stop codon (signal). The product of the probe synthesis reaction is a radioactive single-stranded RNA molecule that is antisense (the opposite coding sequence) to the target mRNA to be detected. Typically, this process produces an RNA sequence a bit longer than necessary, but the “extra” irrelevant RNA sequence in the probe performs a valuable function later in the assay.
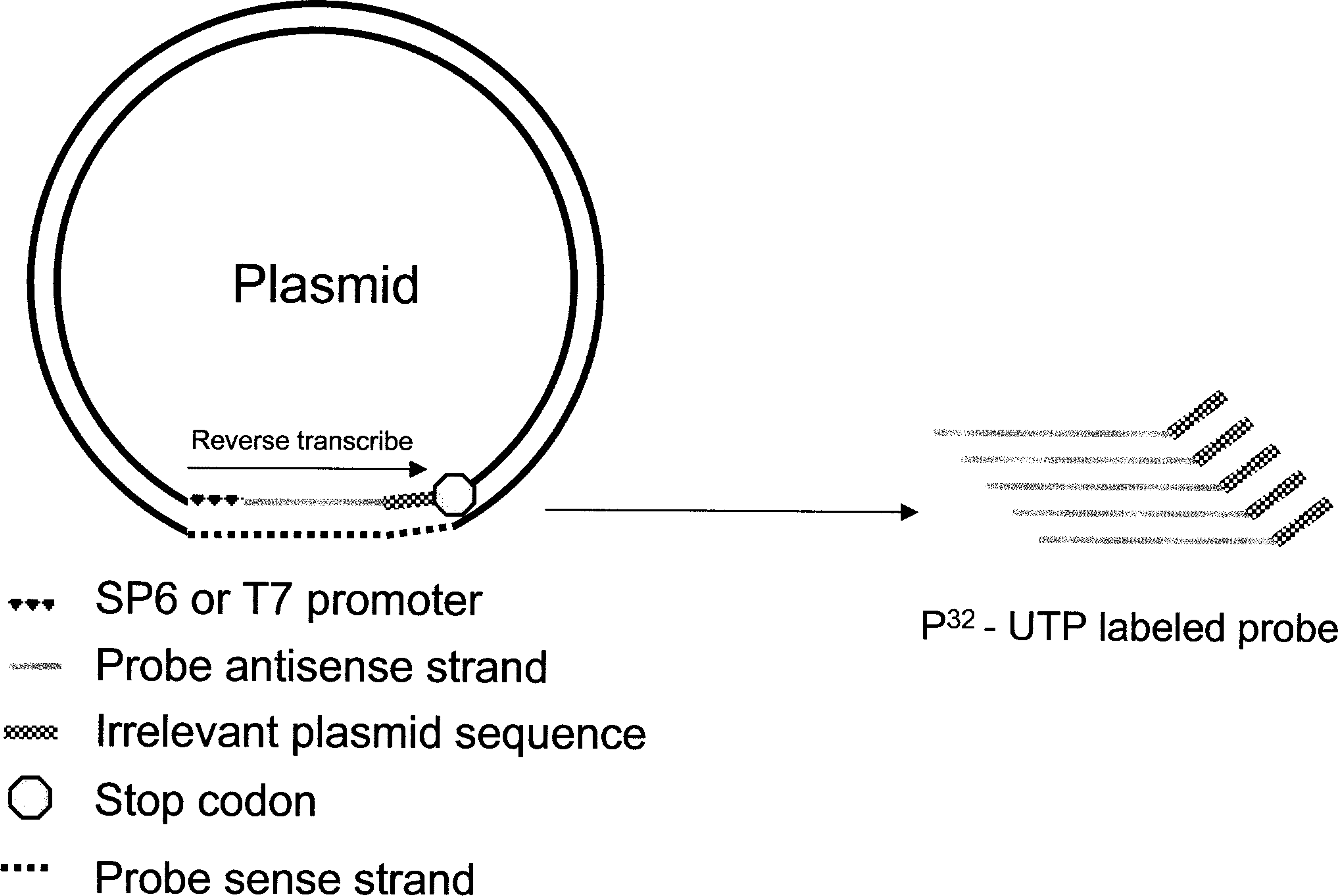
Probe synthesis for the ribonuclease protection assay. A plasmid containing the target mRNA probe sequence adjacent to an SP6 or T7 bacteriophage promoter is added to a reaction mixture containing the enzyme DNA-dependent RNA polymerase, buffers, nucleotides (ATP, GTP, CTP, and UTP), and 32P-UTP. The polymerase binds the SP6 or T7 promoter and transcribes the DNA probe fragment gray line into single-stranded RNA. In addition to reverse transcribing the desired probe sequence, the polymerase continues downstream, reverse transcribing additional irrelevant plasmid sequence hatchal line until it reaches a stop codon (hexagon). The product of the probe synthesis reaction is a radioactive single-stranded RNA molecule antisense to the target mRNA to be detected, which is a bit longer than necessary because of the presence of the “extra” RNA.
Using standard methods, total RNA is then isolated from the tissue or cells of interest (Fig. 2A). The radioactive probe is added to the RNA sample, and the probe seeks out and binds to the target mRNA (Fig. 2B), thus forming partially double-stranded RNA (dsRNA). The dsRNA hybrids are then digested with RNase A and T1 (Fig. 2C), which remove any nonhybridized single-stranded portions of the dsRNA (including the “extra” irrelevant probe RNA), resulting in dsRNA fragments (protected fragments), which are subsequently purified (Fig. 2D). The protected dsRNA fragments and a small sample of the original probe are then added to different lanes of a denaturing polyacrylamide gel and separated by electrophoresis (Fig. 2E). The denaturing conditions cause the dsRNA to dissociate into single strands. After electrophoresis, the gel is dried, and a phosphorimage screen is exposed to quantitate radioactive signal. The protected fragment is identified by its molecular weight (Fig. 2E); the intensity of the radioactive signal generated is directly proportional to the amount of specific target mRNA in the original total RNA sample. The protected fragment of the probe (lane 2) is significantly shorter than the original probe (lane 1) because of digestion of the “extra” irrelevant RNA.
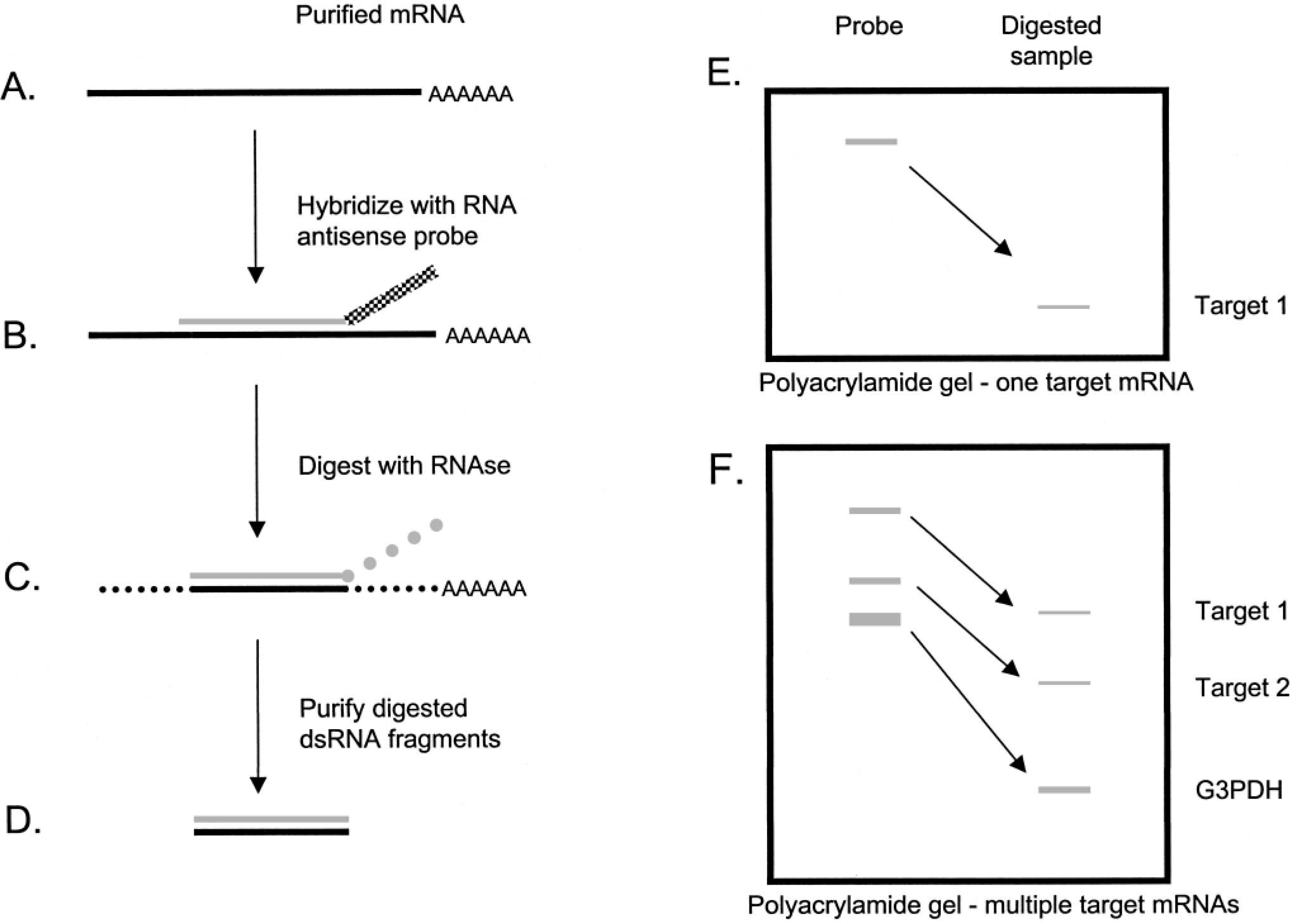
Ribonuclease protection assay procedure. Fig. 2A RNA is isolated from a tissue sample or cells by standard techniques. Fig. 2B The radioactive probe is added to an RNA sample, and the probe binds to the target mRNA, thus forming partially double-stranded RNA (dsRNA). Fig. 2C The dsRNA hybrids are digested with RNase A and T1. Fig. 2D The resulting dsRNA fragments (protected fragments) are subsequently purified. Fig. 2E The protected dsRNA fragments and a small sample of the original probe are then separated on a denaturing polyacrylamide gel, causing the dsRNA to dissociate into single strands. After electrophoresis, the gel is dried, and radioactive signals are measured. The intensity of the radioactive signal generated is directly proportional to the amount of specific target mRNA in the original total RNA sample. Fig. 2F In the more complex form of the assay, multiple mRNAs, including the housekeeping gene G3PDH, are detected in the same sample, and protected fragments are identified by their molecular weights. To control for sample-to-sample variation in mRNA loading, the radioactive signals for each target are expressed as the target:G3PDH mRNA ratio.
In the more common commercially available versions of the assay, multiple target mRNA species are measured at once. BD Pharmingen manufactures a series of kits (Riboquant) specific for humans, pigs, mice, and rats that allow measurement of mRNA coding for chemokines, cytokines, various receptors, cell cycle proteins, apoptosis pathway proteins, leukocyte phenotype (CD) markers, etc. The major difference in the commercially available kits is that the original probe synthesis reaction contains up to 10 or 15 different plasmids, each encoding a unique probe sequence. The absolute requirement in this more complex assay is that the probes must be designed so that the protected fragments are of significantly different molecular weights so that they can be separated and easily distinguishable on the gel. Typically, at least one of the plasmids contains a probe sequence for the housekeeping gene glyceraldehyde-3′-phosphate dehydrogenase (G3PDH). G3PDH is expressed at constant levels in tissue regardless of the activation state of the cells in the sample and, therefore, is used as an internal control for sample-to-sample variation in RNA loading (Fig. 2F). The radioactive signals generated by the target mRNA and G3PDH mRNA are measured, and the target/G3PDH mRNA ratio is calculated for each target. This ratio, expressed in arbitrary units, reflects the relative abundance of the target mRNA compared with the G3PDH mRNA in each sample. These ratios are then used to determine whether differences in target mRNA expression exist between samples. Table 1 contains a list of some of the different ways the RPA is utilized in the laboratory.
Some uses of the RPA as previously reported.

Advantages and Disadvantages
Table 2 provides a comparison of the RPA with other common molecular techniques used to measure mRNA expression (northern analysis and RT-PCR). The major advantage the RPA has over the other techniques is that the expression of up to 10 or 12 mRNAs can be studied simultaneously in a given sample. The RPA is also a relatively sensitive assay. Using the RPA, it is possible to detect as few as 105 copies of a particular mRNA sequence in a complex total RNA sample. This makes the RPA more sensitive than northern analysis because there must be at least 106 mRNA copies present in a sample to be detected by the latter technique. One possible reason for the increased sensitivity of the RPA is that probe hybridization to the target mRNA occurs in a liquid matrix that allows optimal three-dimensional interaction between the two molecules. In contrast, perhaps a fraction of the RNA molecules adherent to the (two-dimensional) membrane in northern analysis are in the correct orientation to bind probe.
Comparison of RPA and other molecular methods used to measure mRNA in a complex total RNA sample.
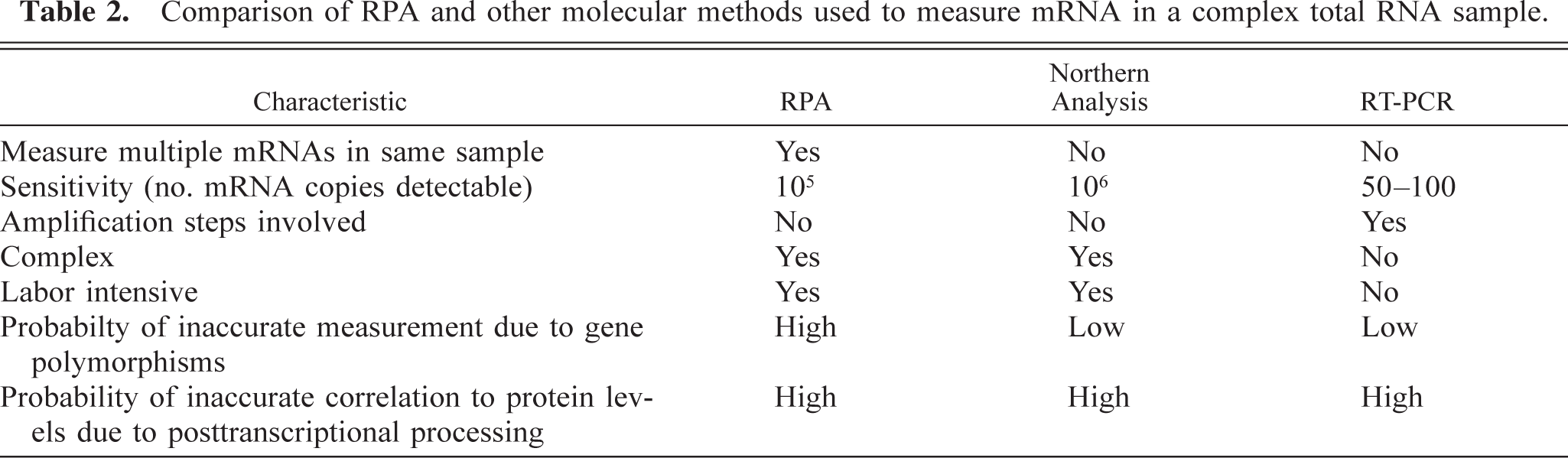
The RPA is not as sensitive as RT-PCR, which can detect 50–100 copies of mRNA. However, the big advantage that RPA has over RT-PCR is that there are no amplification steps involved in ribonuclease protection, and therefore quantitative data are more reliable. In contrast, the RPA is more complex than RT-PCR and takes several days to complete by an experienced technician.
One caveat with the RPA is that gene polymorphisms between strains of mice may also lead to inaccurate mRNA measurements. 8 Any differences between the probe and target mRNA sequences will yield short runs of single-stranded RNA, which will be digested by the RNases used in the assay. This event will result in smaller protected fragments that cannot be distinguished based upon molecular weight. The other techniques for measuring mRNA are not as sensitive to gene polymorphisms because RNase digestion is not involved. A final but important characteristic common to all three techniques is that because of issues such as posttranscriptional processing, the levels of mRNA in a sample may or may not be directly proportional to the level of protein expression in the same sample. Thus, one must weigh the various advantages and disadvantages when deciding which technique is best for the task at hand.
Specific Examples of RPA Use
A common application of the RPA is to study changes in gene expression in inflamed versus noninflamed tissues. In a recent experiment, central nervous system (CNS) chemokine mRNA expression was examined in B6/129 mice that had the chemokine receptor CCR1 genetically deleted (CCR1−/−) and in wild-type (CCR1+/+) B6/129 mice during the course of experimental allergic encephalomyelitis (EAE). 13 CCR1 is expressed principally on monocyte/macrophages and microglia within the CNS and binds the chemokines RANTES and MIP-1α, two chemokines previously demonstrated to be important in the pathogenesis of multiple sclerosis and EAE. 4,5,16 The hypothesis was that CCR1−/− animals that cannot bind these two chemokines would be protected from developing disease. After immunization with myelin oligodendrocyte glycoprotein (MOG) peptide in complete Freund's adjuvant, CCR1+/+ mice developed maximal clinical disease by day 14 postimmunization (PI). The CCR1−/− mice also developed clinical disease, albeit at a decreased incidence and severity (Fig. 3), suggesting that disease susceptibility was not dependant on CCR1, MIP-1α, and RANTES alone. To determine which chemokines may promote leukocyte recruitment to the CNS of CCR1−/− animals, we set up an additional series of in vivo studies. Groups of two CCR1+/+ and CCR1−/− mice were euthanatized at 0, 7, 14, 21, and 28 days PI, and RPA of brain mRNA was performed using a commercially available kit from BD Pharmingen (Fig. 4). After calculating the chemokine: G3PDH ratios (Table 3), we determined that the chemokines IP-10, RANTES, and MCP-1 were upregulated in CCR1+/+ mice prior to onset of disease (day 7 PI) and expression persisted through day 28 PI. In contrast, the only chemokine that was upregulated in CCR1−/− mice prior to disease onset was IP-10; RANTES and MCP-1 were only present in significant quantities on day 14 PI, after disease onset. The receptor for IP-10 (CXCR3) is expressed on activated memory T cells and a subset of B cells. 12 Thus, these data suggest that CXCR3+ lymphocytes participate in the original recruitment of lymphocytes to the brain of both CCR1+/+ and CCR1−/− animals via IP-10–mediated chemotaxis. Furthermore, disease may be less severe in CCR1−/− mice because of deficient expression of RANTES and MCP-1 and thus decreased CNS recruitment of other effector cells such as lymphocytes, monocyte/macrophages, and microglia that express receptors for these chemokines (RANTES = CCR3 and CCR5; MCP-1 = CCR2). In this case, the RPA helped elucidate temporal expression of chemokine mRNA and led to proposal of a novel disease pathogenesis.
CNS chemokine:G3PDH mRNA ratio in CCR1-/- and CCR1+/+ mice with EAE.∗
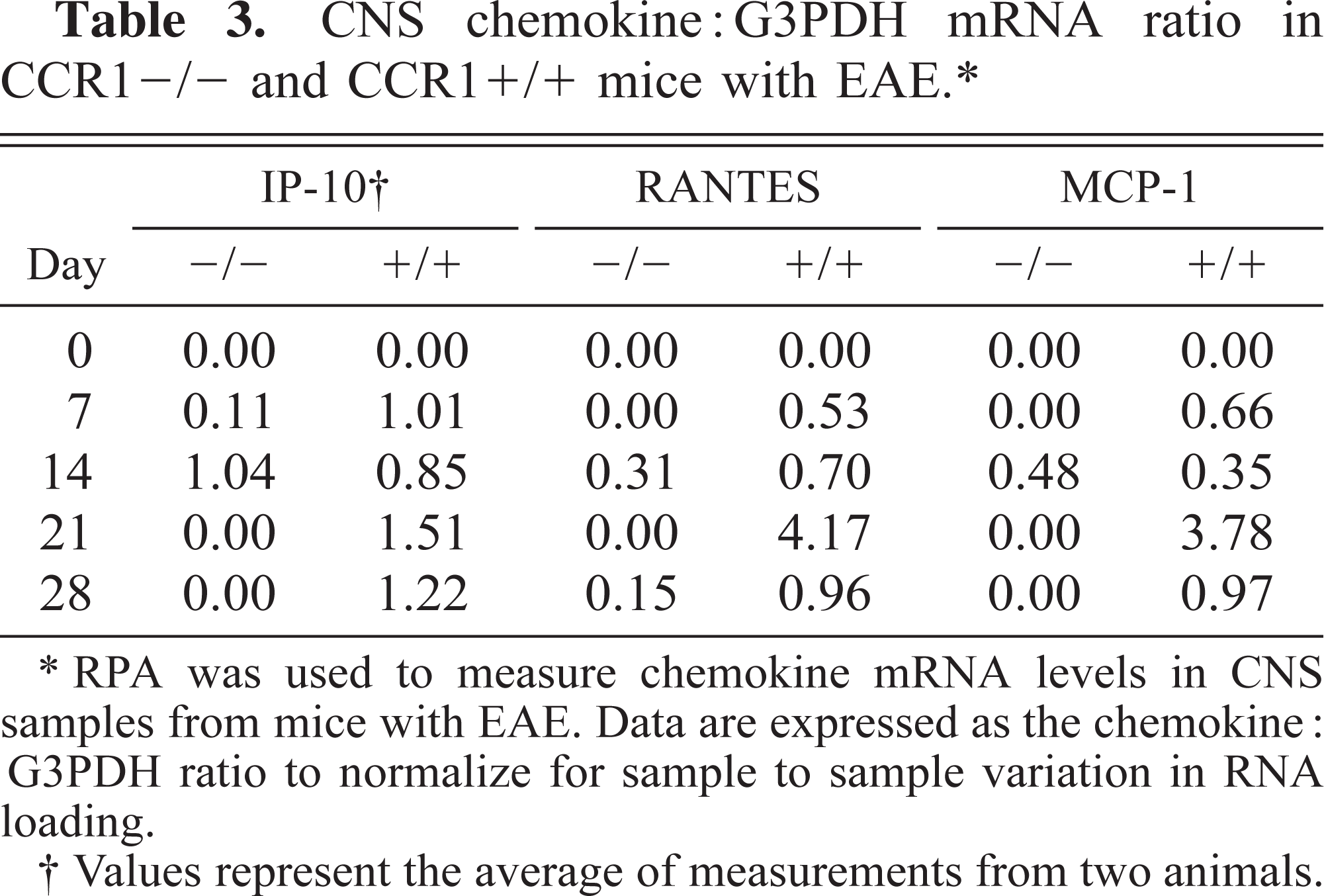
∗ RPA was used to measure chemokine mRNA levels in CNS samples from mice with EAE. Data are expressed as the chemokine:G3PDH ratio to normalize for sample to sample variation in RNA loading.
† Values represent the average of measurements from two animals.
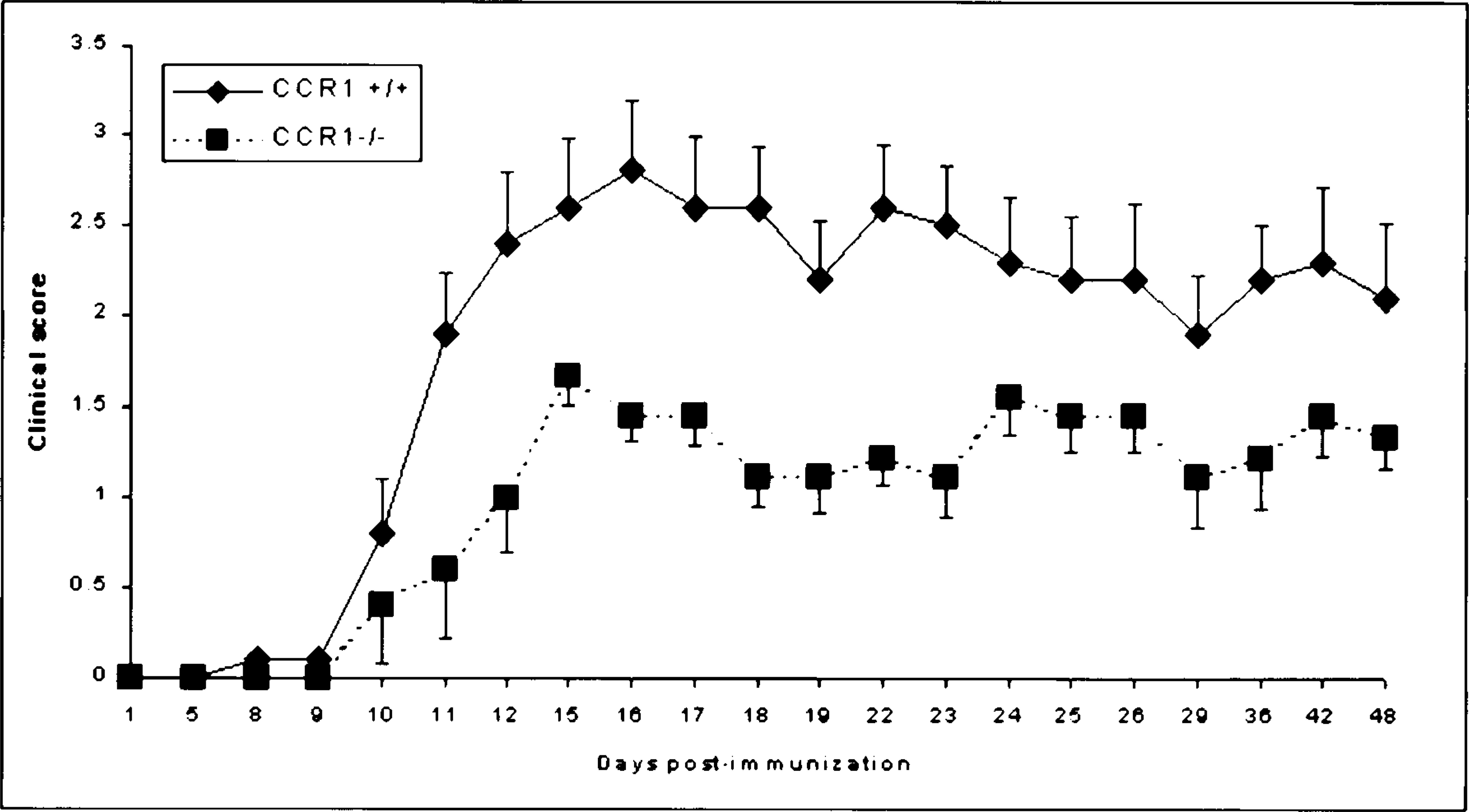
Incidence and severity of clinical disease in mice with EAE. Following immunization with 100 µg MOG peptide in complete Freund's adjuvant, B6/129 CCR1+/+ and CCR1−/− mice (n = 10 mice/group) were scored clinically each day by researcher blinded to the identity of the animals using the following scoring system: 0 = normal; 1 = loss of tail tone; 2 = posterior paresis; 3 = posterior paralysis and loss of righting response; 4 = tetraparesis/moribund. Data are expressed as mean ± SEM. The clinical score was always less in CCR1−/− than in CCR1+/+ mice, and this difference was significant (P < 0.05) on days 11–23 and day 36. 13
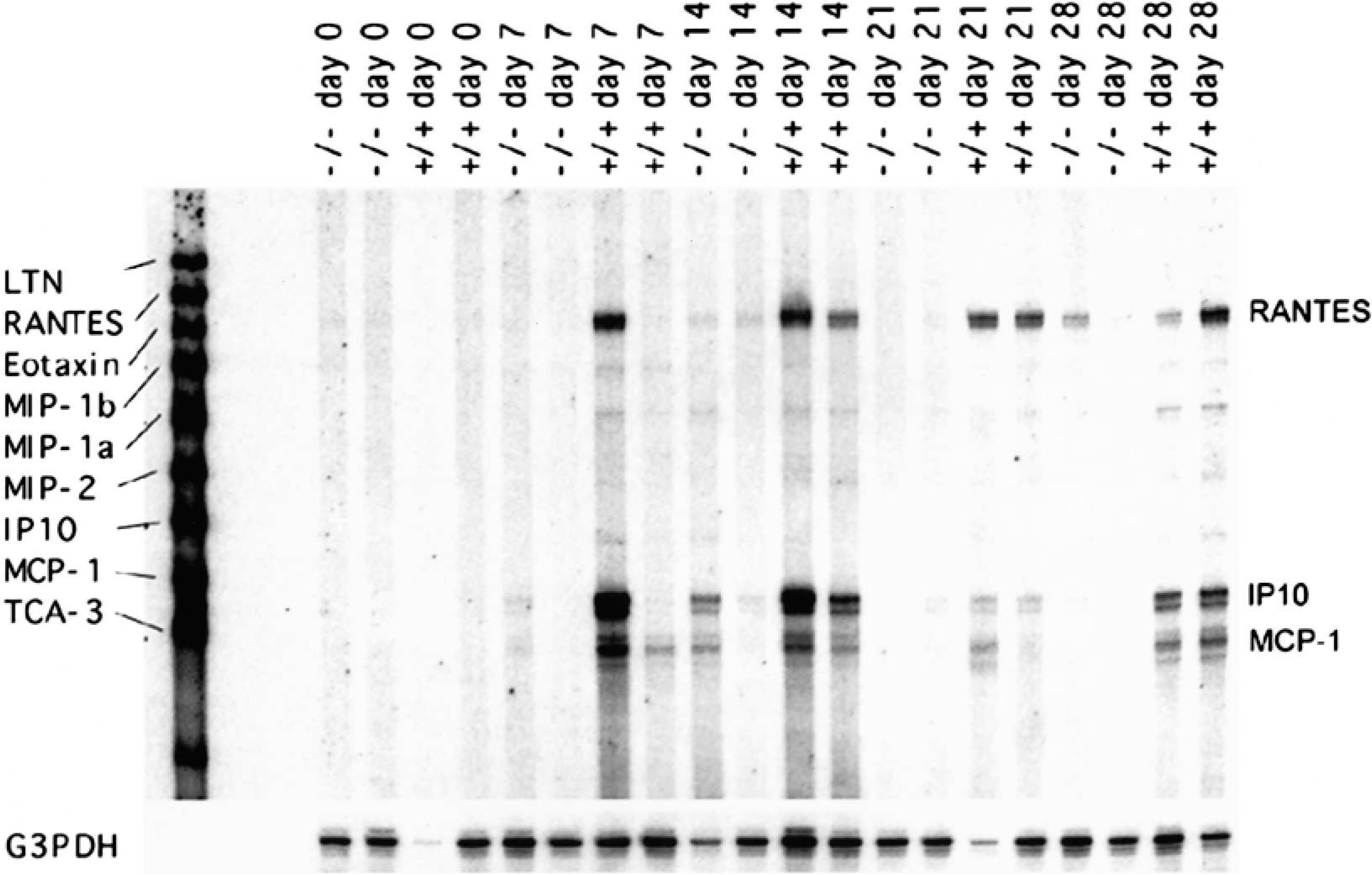
Representative ribonuclease protection analysis of brain chemokine mRNA expression from CCR1+/+ and CCR1−/− mice with EAE. Following immunization with 100 µg MOG peptide in complete Freund's adjuvant, B6/129 mice (n = 2 mice/group) were serially euthanatized on days 0, 7, 14, 21, and 28 PI. Brain mRNA was harvested, and chemokine mRNA was analyzed using a commercially available ribonuclease protection assay kit (BD Pharmingen). Radioactive signals on this polyacrylamide gel were measured, and the data are presented in Table 3.
Another way the RPA can be used is to study the temporal infiltration of leukocyte subsets into an inflamed tissue. Typically, this type of study is accomplished by careful morphometric analysis of serial tissue sections stained using an immunoperoxidase technique. The RPA has the advantage that the mRNA is isolated from large samples, if not the entire tissue specimen (e.g., brain). Thus, the variation in target expression (or sampling error) from one histologic section to the next is eliminated. In a recent study, my colleagues and I wished to determine the time during the pathogenesis of EAE when CD4+ lymphocytes first began to enter the brain in relation to the onset of clinical symptoms. 14 SJL mice were immunized with a peptide component of myelin, proteolipoprotein (PLP) 35–55 in complete Freund's adjuvant. Groups of three mice were euthanatized at various times following immunization, brains were collected, and each sample was divided, one half for RNA isolation and the other for immunohistochemistry (IHC). Using a commercially available RPA kit (BD Pharmingen), we measured CD4 and G3PDH mRNA expression in the serially collected samples and expressed the data as the CD4:G3PDH ratio to control for RNA loading (Fig. 5). We then performed IHC analysis of the other half of the brain specimens to directly compare CD4 protein expression with the corresponding mRNA expression data (Fig. 6). Although clinical disease was not detectable until day 14 PI, by RPA we could detect CD4 mRNA expression in the brain as early as day 10 PI, whereas CD4+ cells were not detectable by IHC until day 12 PI. Thus, in this application, the RPA can identify tissue infiltration by small numbers of CD4+ cells before such infiltration is detectable by IHC. In summary, the RPA is useful for studying the kinetics of tissue infiltration by specific subsets of leukocytes.
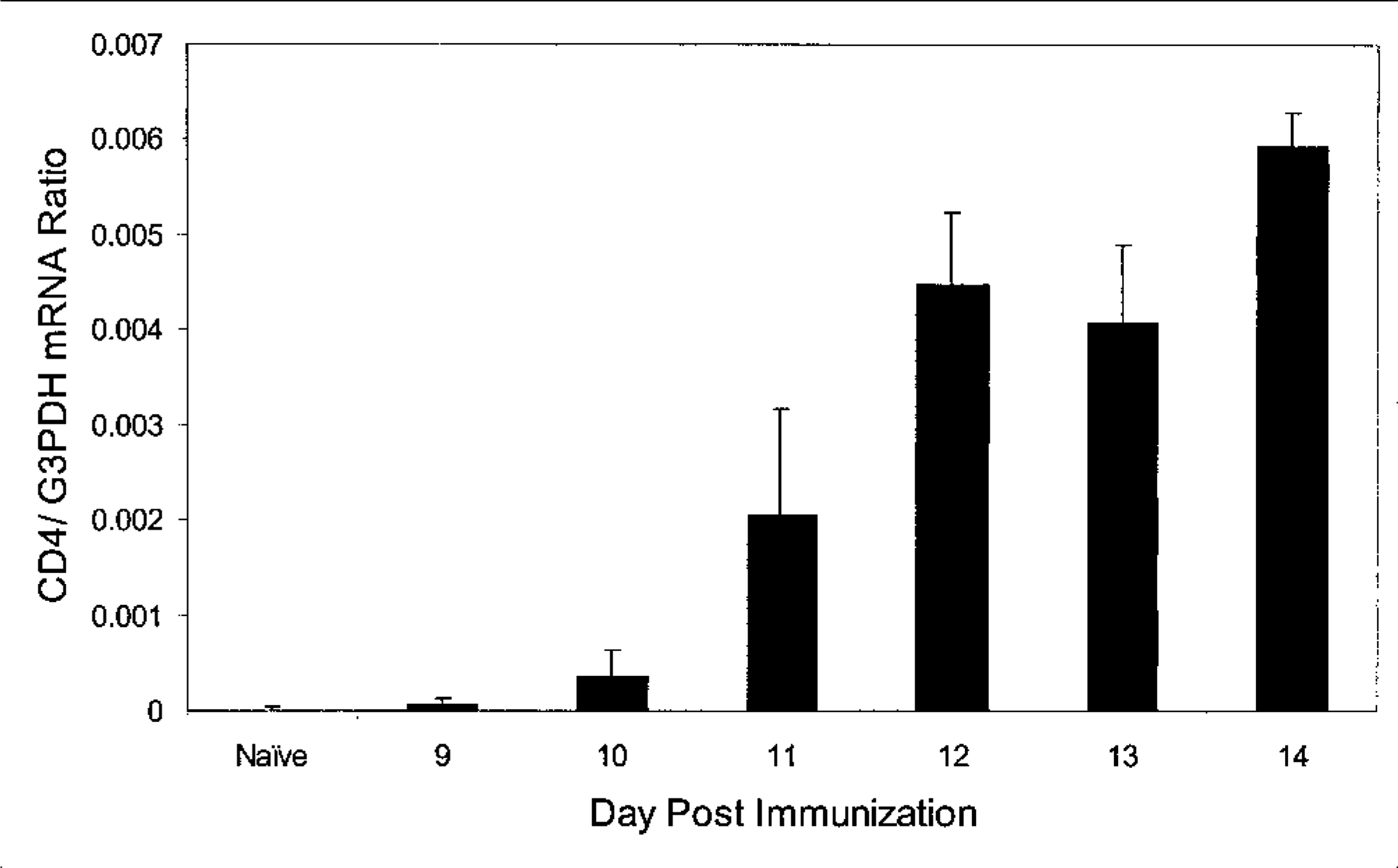
Ribonuclease protection analysis of brain CD4 lymphocyte infiltration in mice at various times during the clinical course of EAE. Following immunization with 100 µg PLP peptide in complete Freund's adjuvant, SJL mice (n = 3 mice/group) were serially euthanatized on days 9–14 PI. Brain RNA was harvested, and CD4 mRNA was analyzed using a commercially available ribonuclease protection assay kit (BD Pharmingen). CD4 mRNA was first detectable in the brain on day 10 PI and reached maximal levels on day 12, 2 days before onset of clinical disease in this model.
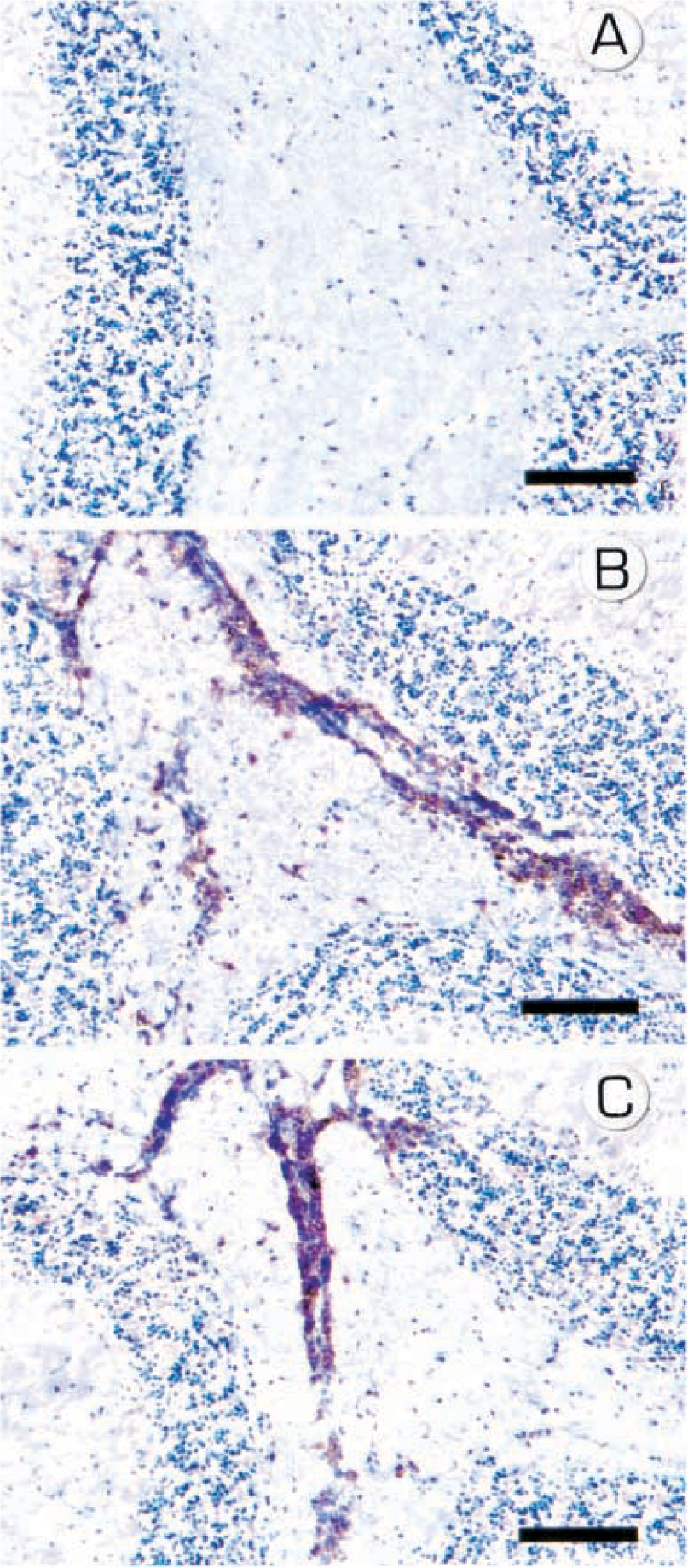
Immunohistochemical analysis of brain CD4 lymphocyte infiltration in mice during the clinical course of EAE. 14 Fig. 6A CD4+ cells were not detectable by IHC on day 10 PI. Fig. 6B CD4+ cells easily observed on day 12 PI. Fig. 6C CD4+ cells easily observed on day 14 PI. Immunoperoxidase technique, hematoxylin counterstain. Bar = 100 µM.
Conclusions
The RPA is a powerful tool that can be used in a variety of configurations to measure expression of specific mRNA molecules in a complex mixture of total RNA. Although laborious, it can be used to analyze as many as 25 samples in a single assay and to examine up to 10–15 genes in each sample. The combination of sensitivity and high throughput makes this assay useful for discovery biology applications. Data generated with the RPA are best used in combination with those generated by standard morphologic techniques to elucidate the pathogenesis of a disease process.
Footnotes
Acknowledgements
I thank Drs. Patricia Rao and Christopher Horvath for critical review of this manuscript.