
Abstract
An approximately 8-week-old pet Syrian hamster (Mesocricetus auratus) with a 1-week history of dyspnea, hyporexia, and ataxia was submitted for necropsy. On gross examination, the hamster had multiple abdominal adhesions and enlargement of the mesenteric lymph node. Histologic evaluation revealed multicentric lymphoma of the liver, jejunum, mesenteric lymph node, testicular fat pad, and epididymis. Based on the hamster's age and the type and distribution of the lymphoma, a presumptive diagnosis of hamster polyomavirus- induced lymphoma was made. A specific polymerase chain reaction (PCR) was developed, which confirmed the diagnosis. An in situ PCR demonstrated hamster polyomavirus DNA within lymphocytes of the multicentric lymphoma and renal tubular epithelial cells and within clusters of enterocytes in the jejunum. These data are consistent with environmental dissemination of hamster polyomavirus virions through the renal tubular epithelium and into the urine and with fecal shedding of hamster polyomavirus virions; however, additional studies will be needed to confirm these observations.
Hamster polyomavirus (HaPV) is a naturally occurring, highly transmissible virus of Syrian hamsters (Mesocricetus auratus) that has been associated with disease outbreaks in research hamster colonies in both the United States and Europe. 1,5,11 HaPV was first isolated from the nuclei of keratinizing epithelial cells in trichoepitheliomas from an endemically infected colony of Syrian hamsters in Berlin-Buchs, Germany. 11 Epitheliomas and skin lesions develop in hamsters from endemically infected hamster colonies or when adult hamsters are infected with HaPV. The lesion progression in epitheliomas from affected hamsters is strikingly similar to those seen in papillomavirus infections; however, the target tissue tropisms are different. Papillomaviruses infect the interfollicular epidermal keratinocytes, and HaPV infects the hair follicle keratinocytes. 23 A second disease course is seen when HaPV infects naive young hamsters in which it readily induces multicentric lymphoma originating from the mesentery and metastasizing to the liver, kidney, and thymus. 3,10,19,22 The ability of HaPV to infect both undifferentiated keratinocytes and lymphocytes is unique among the papovaviruses and led to its initial misclassification as a papillomavirus. However, characterization of virus genomic sequence and gene structure definitively identifies HaPV as a polyomavirus. 18,23
Hamster polyomavirus is in the genus Polyomavirus of the virus family Papovaviridae. 18 HaPV virions are nonenveloped and approximately 40 nm in diameter and have icosahedral symmetry. 18 The genome of HaPV is composed of a single closed circular molecule of histone-associated double-stranded DNA that is 5,366 base pairs in length. 7,18 The genomic organization of HaPV is similar to that of the prototypic mouse polyomavirus strain A2, in which the genes are divided into early and late transcriptional units that are transcribed on opposite DNA strands that are separated by a noncoding region. 18,23 Early phase transcription includes the small t, middle T, and large T antigens. T antigens act singly or in concert to regulate replication of the virus genome, activation of the late phase transcriptional unit, and autoregulation of the early promoter. 20 In addition to these functions, the T antigens are also responsible for tumor induction by polyomaviruses. 8,20 In simian virus 40, a polyomavirus of nonhuman primates, the large T antigen interacts with the host cell proteins p53 and Rb to transform virus-infected cells. 8,14 Mouse polyomavirus and HaPV are the only polyomaviruses that produce the middle T antigen. In mouse polyomavirus, the middle T antigen is responsible for virus transformation. 2,26 However, recent in vitro studies of HaPV indicate that cooperation between the small t and middle T antigens is required to transform cultured F111 rat cells. 9 The late coding region encodes the virus capsid structural proteins, including the major capsid protein VP1 and the capsid proteins VP2 and VP3. 18,23
The pathogenesis of HaPV infections is believed to be similar to that of the prototypic murine polyomavirus. 19 When a polyomavirus infects an animal, it absorbs to the cell's surface, is taken up by endocytosis, and is transported to the nucleus, where virus uncoating occurs. Once in the nucleus of the infected cell, polyomaviruses initiate replication via transcription and translation of early phase proteins, including the T antigens. When a permissive cell is infected, the virus initiates a lytic replication cycle, resulting in lysis of the infected cell and release of complete virions. Polyomavirus infection of mouse kidney results in release of virus particles in urine, and persistent viruria is the most likely mechanism for environmental dissemination of infective virus particles. 19 Persistent viruria has been suggested as a mechanism of environmental dissemination of HaPV virions. 19 The presence of HaPV virions in keratinocytes suggests that the virus may be spread by sloughing or removal of infected cells through grooming or biting behaviors. When polyomavirus infects nonpermissive cells, genomic integration or episomal amplification with nonrandom deletions of the late coding regions occur, resulting in virus transformation of cells to a neoplastic phenotype. 22 In HaPV infection, lymphocytes represent a nonpermissive cell type, with infection leading to transformation to lymphoma. Recent work with HaPV has contributed to the understanding of virus-induced transformation, but there is little information on the basic pathogenesis of HaPV infection.
Here, we describe a case of HaPV infection in a pet Syrian hamster. HaPV has been previously reported in laboratory hamsters in the United States and Europe; however, this is the first report of HaPV infection in a Syrian hamster outside of the laboratory environment. 1,5,11 HaPV infection was suspected in this case because of the unusual clinical history and the presence of lymphoma in a young hamster. Diagnosis was established by amplification of HaPV-specific amplicons by a polymerase chain reaction (PCR) and was documented to be associated with tumor cells and other target organs by direct in situ PCR.
An approximately 8-week-old male Syrian hamster was presented to the referring veterinarian with a 1-week history of dyspnea, hyporexia, and ataxia. The owner reported that the hamster's dam had been donated to a local school and was subsequently found to be pregnant. When the pups were 4 weeks of age, they were adopted by various owners, and this hamster was the last surviving member of the litter of five or six pups. The hamster expired while being examined by the referring veterinarian.
The juvenile Syrian hamster was thin (41.75 g) with several large palpable abdominal masses. Gross examination revealed the cecum adhered to the left abdominal wall, the right testicle adhered to the jejunum, marked enlargement of the mesenteric lymph node, and multiple abdominal adhesions. Brain, lung, liver, kidney, stomach, duodenum, cecum, jejunum, and right testicle were collected for histologic evaluation and preserved in 10% neutral buffered formalin. After a 12-hour fixation period, the tissues were routinely processed for paraffin embedment, sectioned, and stained with hematoxylin and eosin (HE).
Multicentric lymphoma was identified in the liver, jejunum, mesenteric lymph node, testicular fat pad, and epididymis. Large neoplastic lymphocytic infiltrates associated with the biliary tree were found in several lobes of the liver. Two large masses with necrotic centers were associated with the mesenteric lymph node and a large jejunal adhesion. Additional abdominal adhesions were characterized by effacement of the jejunal muscularis by invading lymphocytes and attachment of the jejunum to the abdominal wall and right testicle. There was neoplastic lymphocytic invasion of the right testicular fat pad and extension of neoplastic lymphocytes into the epididymis. Sheets of neoplastic lymphocytes invaded the serosa and muscularis of the jejunum in multiple places and surrounded and compressed the crypts of Lieberkühn (Fig. 1). Neoplastic lymphocytes were 5–7-µm-diameter mononuclear cells with round to irregular vesicular nuclei, scant to moderate amounts of amphophilic cytoplasm, and four to six mitotic figures per 400× field (Fig. 2). Because of the young age of the hamster and the type and distribution of the lymphoma, a presumptive diagnosis of HaPV-induced lymphoma was made.
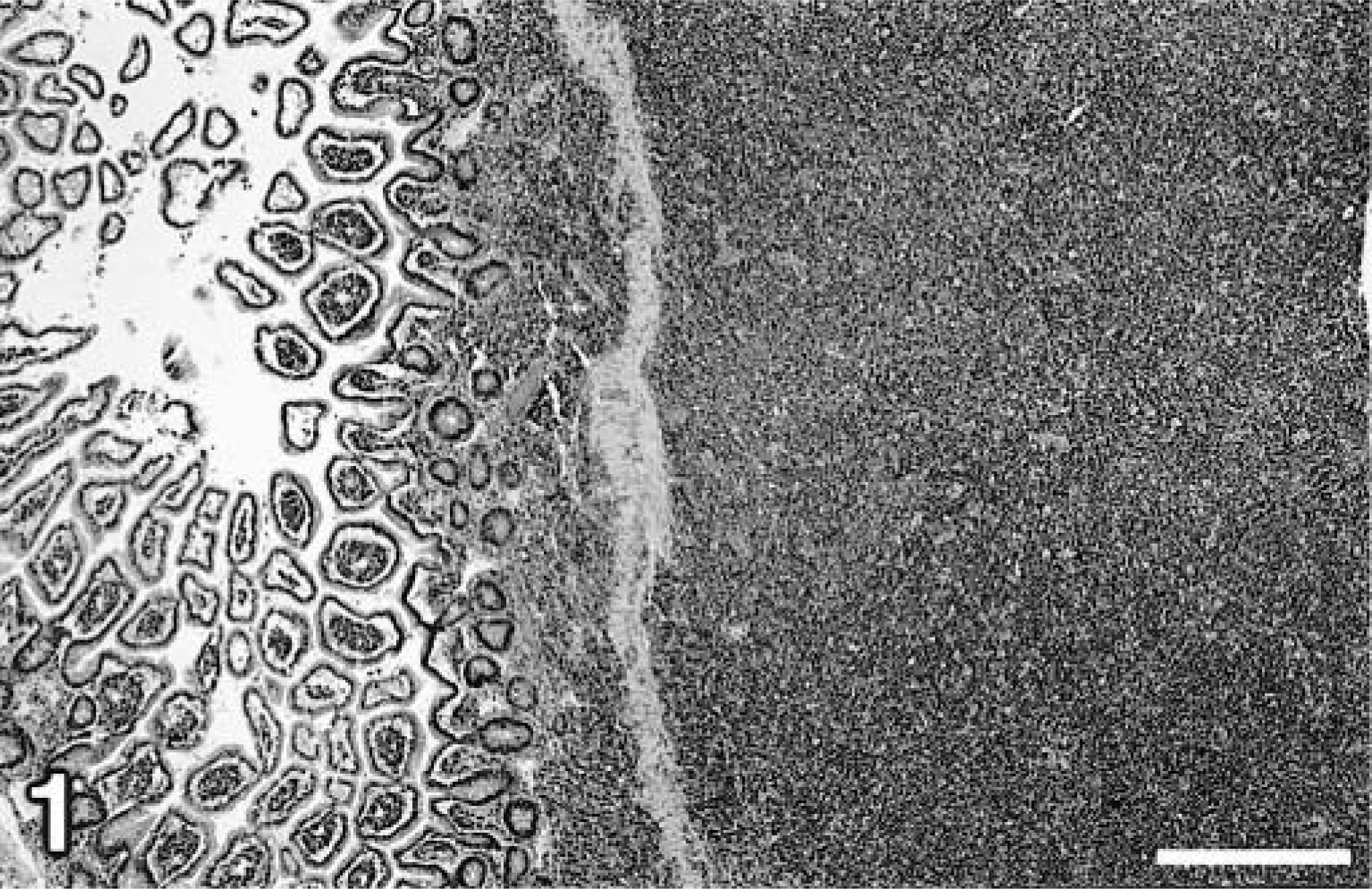
Jejunum; hamster. Sheets of neoplastic lymphocytes infiltrate the serosa and muscularis of the jejunum and surround and compress the crypts of Lieberkühn. HE. Bar = 400 µm.
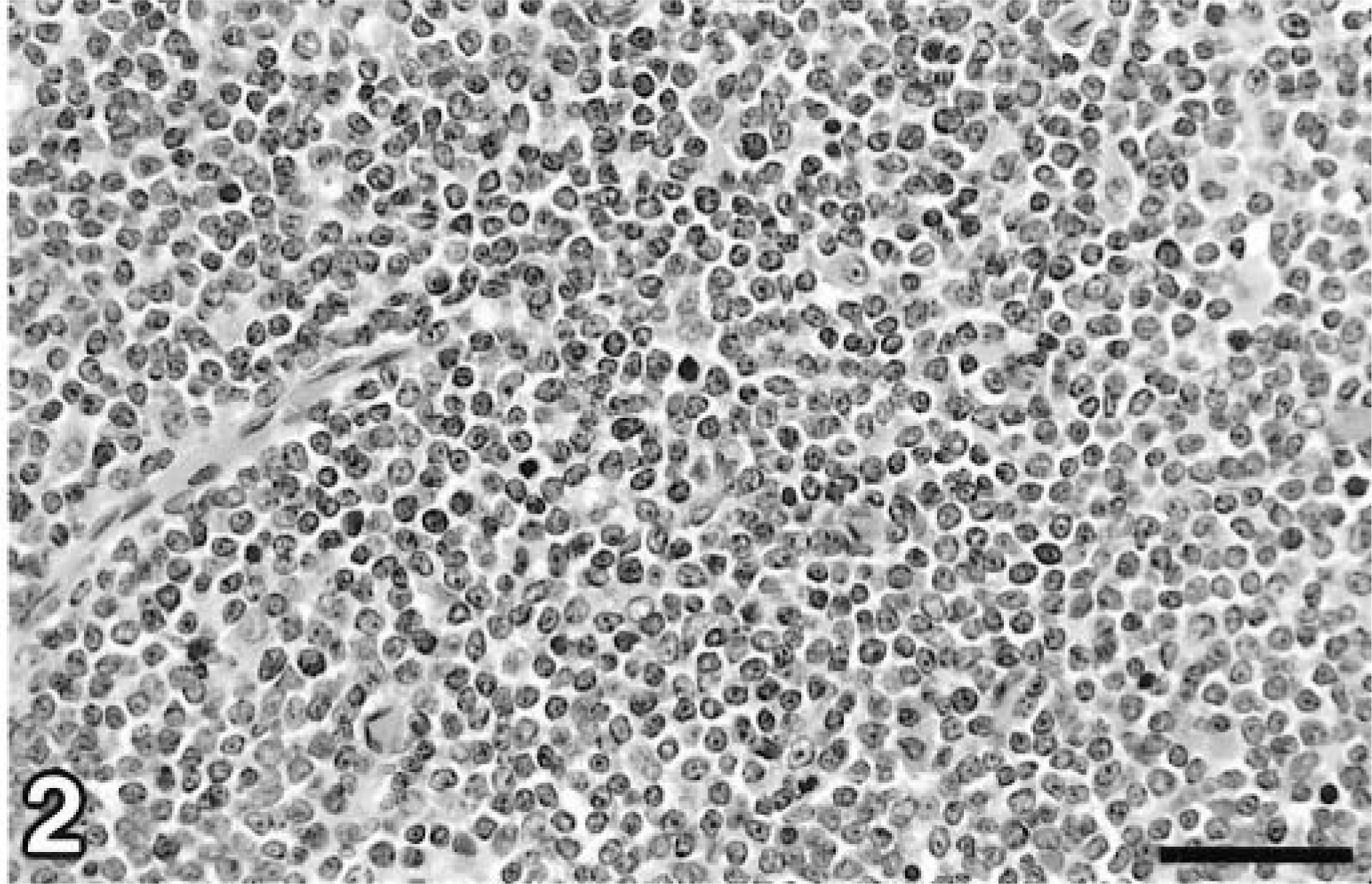
Jejunum; hamster. Neoplastic lymphocytes from the affected hamster were 5–7-µm-diameter mononuclear cells with round to irregular vesicular nuclei, scant to moderate amounts of amphophilic cytoplasm, and four to six mitotic figures per 400× field. HE. Bar = 50 µm.
To confirm the diagnosis of HaPV infection, a PCR test was developed. The majority of the gene sequence of HaPV is available via GenBank accession number M26281. 7 This sequence was used to generate the PCR primers (Table 1) HaPVf and HaPVr. Viral DNA was isolated from paraffin-embedded tissue sections using stringent techniques to avoid contamination. 13 For each PCR, five 5-µm tissue sections were cut with a microtome in a room separate from the thermocycler. Tissue sections were transferred to an autoclaved 2.0-ml microcentrifuge tube, and the paraffin was removed from the tissue sections by solvent extraction. DNA isolation was performed with a QiAmp Tissue Kit (Qiagen, Chatsworth, CA) used according to the manufacturer's instructions. PCR mixtures contained 1 µM of each primer, 200 µM of each dNTP (dATP, dCTP, dGTP, and dTTP), PCR buffer (10 mM Tris, 1.5 mM MgCl2, 50 mM KCl, pH 8.3 at room temperature), 1.25 U of Taq polymerase, and 5 µl of template DNA. PCR mixtures were heated to 94°C for 30 seconds once, followed by 45 cycles of denaturation at 94°C for 3 seconds, primer annealing at 55°C for 5 seconds, and primer extension at 72°C for 1 minute in a Perkin-Elmer 2400 thermocycler (Perkin Elmer, Foster City, CA). PCR controls included a no-template reaction and DNA isolated from the paraffin-embedded tissues of a clinically normal hamster. PCR products were subjected to electrophoresis in 2% NuSieve agarose gels (FMC BioProducts, Rockland, ME) containing 0.25 µg/ml ethidium bromide. Gels were visualized on an ultraviolet light box and photographed. The molecular weight of PCR products was assessed by comparison to commercially available molecular weight standards (Molecular Weight Marker VI, 0.15–2.1 kbp, Boehringer Mannheim, Indianapolis, IN).
Oligonucleotide primers used to amplify hamster polyomavirus DNA.
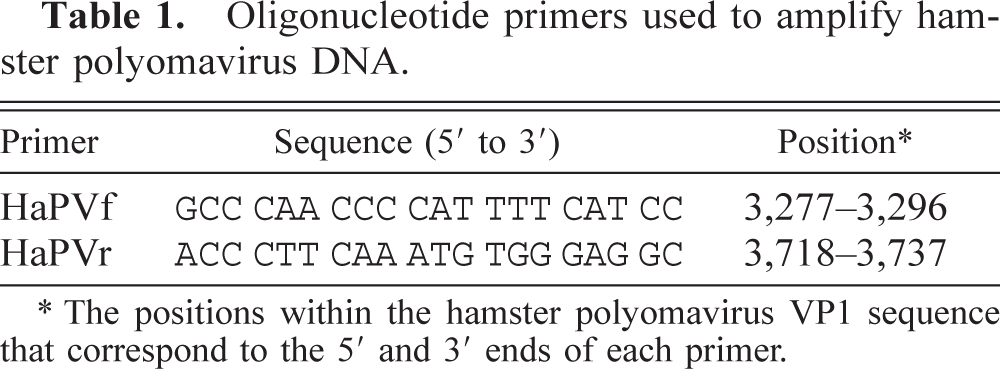
∗ The positions within the hamster polyomavirus VP1 sequence that correspond to the 5′ and 3′ ends of each primer.
An electrophoretogram of PCR amplification products made using primers from Table 1 and template DNA from the affected hamster and a clinically normal (control) hamster is shown in Fig. 3. PCR analysis of DNA from the affected hamster (lane 2) revealed the expected 460-bp PCR amplicon.
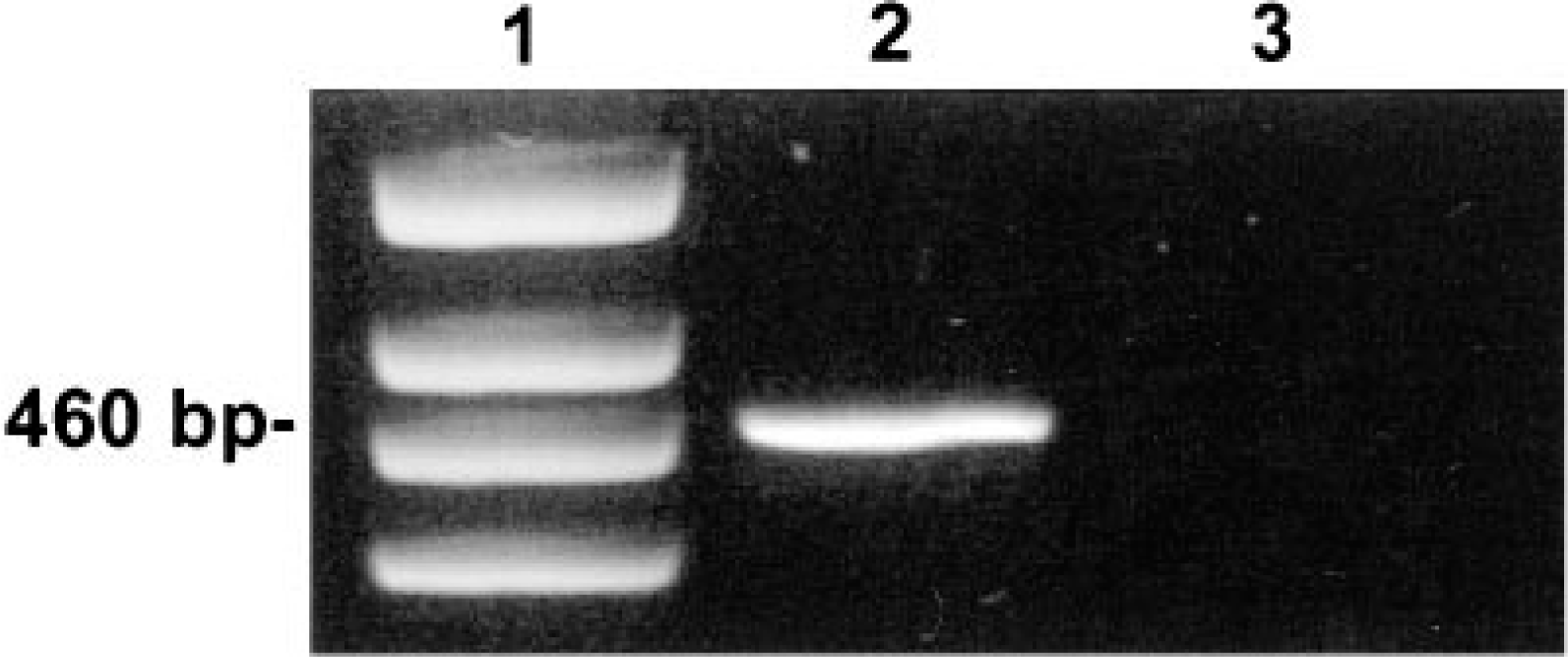
Agarose gel electrophoretogram of PCR amplicons using the hamster polyomavirus–specific primers HaPVf and HaPVr. Lane 1 = DNA molecular weight markers; lane 2 = PCR of DNA isolated from the jejunal lymphoma of the affected hamster demonstrating the expected 460-bp PCR amplicon; lane 3 = PCR of DNA harvested from a clinically normal control hamster.
The identity of the PCR amplification product was established by sequencing the 460-bp PCR amplicon. DNA templates were prepared for sequencing by isolation of the PCR product from a 3.5% polyacrylamide gel as previously described. 24 Sequencing reactions were then performed by PCR using 100 ng of gel-purified HaPV template DNA, 1 µM of each of the primers in Table 1, and a commercially available Taq dideoxy chain termination sequencing kit (Taq Dye Terminator Cycle sequencing kit; Applied Biosystems, Foster City, CA) used according to the manufacturer's instructions. Sequence analyses were performed with the GCG software package (Genetics Computer Group, Madison, WI).
Sequence analysis of the 460-bp PCR product from the affected hamster was consistent with the published HaPV sequence. 7 The GenBank HaPV sequence (accession number M26281) is incomplete; it has a contiguous segment of 60 indeterminate nucleotides within the VP1 gene (nucleotides 3,361–3,420). This section of indeterminate nucleotides is within the PCR amplicon that was sequenced from the affected hamster. Of the remaining 400 nucleotides, the sequence from the affected hamster was 99.75% (399/400) homologous with the published HaPV sequence. The sequenced PCR product is available from GenBank under accession number AF073287.
To demonstrate HaPV tissue distribution, a direct in situ PCR was performed on tissue sections from the HaPV-infected hamster and sections from a clinically normal (control) hamster, using a modification of a previously described technique. 17 Paraffin-embedded tissues were cut into 5 µm sections and transferred to sialinized glass in situ PCR slides (Perkin Elmer). 13 Paraffin was removed from the tissue sections, and the slides were air dried and rehydrated with phosphate-buffered saline (PBS), followed by proteinase K treatment to expose template DNA within the fixed tissue sections. An in situ PCR was performed using the GeneAmp® In Situ PCR Core Kit (Perkin Elmer). Fifty microliters of an in situ PCR master mix containing the primers HaPVf and HaPVr (Table 1) at 1 µM each, 200 µM dATP, 200 µM dCTP, 200 µM dGTP, 125 µM dTTP, 5 µM Dig-11-dUTP (Boehringer Mannheim), PCR buffer, and 10 U AmpliTaq IS (Perkin Elmer) was pipetted onto the air-dried tissue sections. The in situ PCR was performed for 30 cycles of denaturation at 94°C for 1 minute, annealing at 55°C for 1 minute, and primer extension at 72°C for 1 minute, followed by a final extension at 72°C for 10 minutes in a GeneAmp In Situ PCR System 1000 thermocycler (Perkin Elmer). After cycling was completed, the slides were rinsed in 10 mM Tris, pH 8.3 at room temperature, 50 mM KCl, and 1.5 mM MgCl2 containing 0.001% gelatin (weight/volume). The slides were then rinsed in 2× standard saline citrate (SSC)–50% formamide buffer and subsequently rinsed twice in 2× SSC.
An alkaline phosphatase immunoassay system was used to visualize the in situ PCR products. The slides were soaked in maleic acid buffer (100 mM maleic acid, 150 mM NaCl, pH 7.5) and then incubated in blocking reagent (maleic acid buffer containing 1% blocking reagent; Boehringer Mannheim). Levamisole (Sigma Chemical Co., St. Louis, MO) and sheep's serum were added to the blocking reagent to inhibit endogenous tissue alkaline phosphatases and nonspecific binding of detection antibodies, respectively. 21 The slides were incubated with a 1:5,000 dilution of anti-digoxigenin–alkaline phosphatase Fab fragments from sheep (Boehringer Mannheim) followed by rinses in maleic acid buffer and detection buffer (100 mM Tris, pH 9.5 at room temperature, 100 mM NaCl, 50 mM MgCl2). A color substrate solution was then prepared by adding 20 µl of nitroblue tetrazolium/bromocresyl-indolyl phosphate stock solution (Boehringer Mannheim) to 1 ml of detection buffer. Approximately 200–250 µl of the color substrate solution was pipetted onto each in situ PCR slide. Once sufficient color developed, the slides were soaked for 5 minutes in deionized water to terminate the color reaction. To aid in visualization of normal tissue architecture, the slides were counterstained with eosin and coverslipped.
HaPV DNA was amplified and labeled by direct in situ PCR within several tissues. Neoplastic lymphocytes showed a specific, diffuse light purple staining, indicating that HaPV DNA was present in the transformed lymphocytes. The majority of cells within the kidney showed no specific staining; however, there was modest labeling of several nuclei within the renal tubular epithelium of the papillary collecting ducts (Fig. 4). The most intense nuclear label was observed within jejunal enterocytes. Labeled enterocytes were distributed in multiple areas and usually in discrete clusters of adjacent cells (Fig. 5). Localized nonspecific staining was seen surrounding the necrotic centers of large lymphomas. This generalized staining is most likely due to the leakage of HaPV DNA from necrotic lymphocytes. Similar tissues from uninfected control hamsters did not exhibit any specific nuclear staining from the in situ PCR reaction.
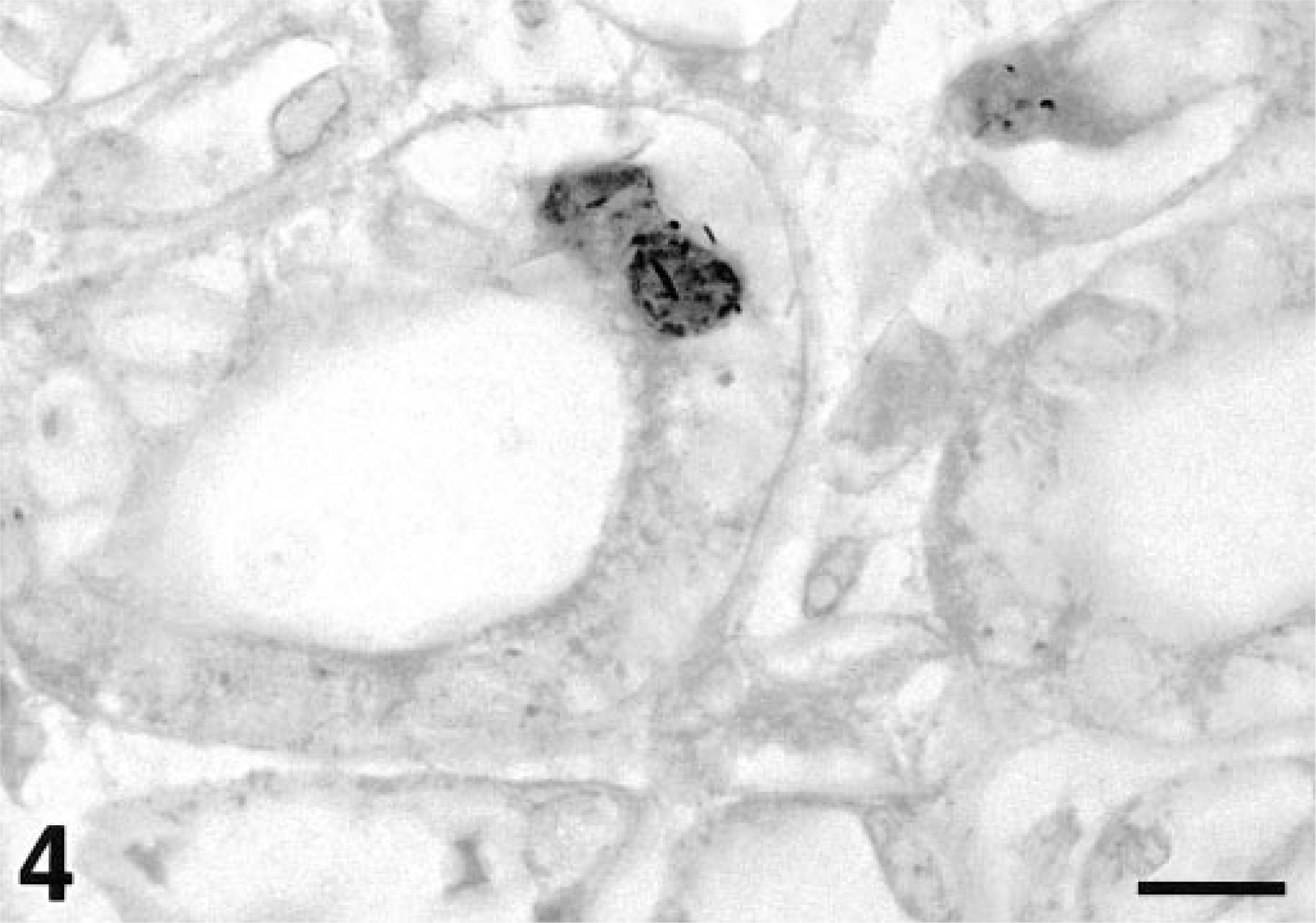
Kidney; hamster. Renal tubular epithelium of the papillary collecting ducts. Specific renal tubular epithelial nuclei are modestly stained, and thus contain HaPV DNA. In situ PCR. Counterstained with eosin, which gives the erythrocytes a very dark color. Bar = 10 µm.
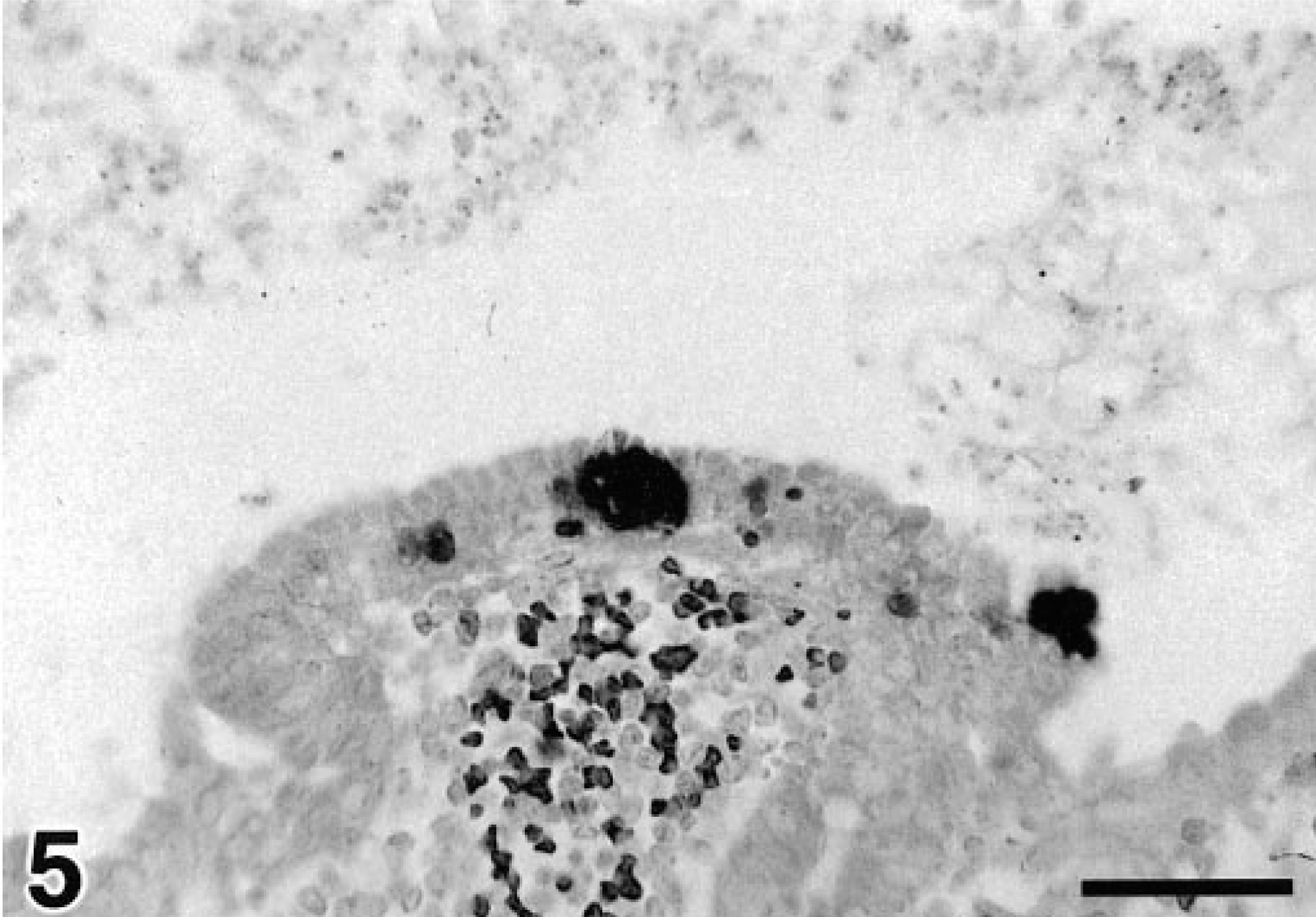
Jejunum; hamster. Specific intense labeling of the clustered nuclei of apical jejunal enterocytes, indicating that HaPV DNA is present in these clusters of adjacent jejunal enterocytes. In situ PCR. Counterstained with eosin, which gives the erythrocytes a very dark color. Bar = 50 µm.
A classic case of HaPV-associated juvenile lymphoma occurred in a pet Syrian hamster. As a part of this investigation, we developed a PCR assay for diagnosis of HaPV infection. The HaPV diagnostic PCR assay is more rapid and specific than other commonly used diagnostic techniques, including histology, immunohistochemistry, and electron microscopy. We also developed a direct in situ PCR assay that can be used to specifically amplify HaPV DNA within infected cells. The in situ PCR assay was used to demonstrate the tissue distribution of HaPV-infected cells, including lymphocytes, renal tubular epithelial cells, and jejunal enterocytes. The in situ PCR data add to the growing body of information regarding HaPV pathogenesis, including a potential new mechanism of virus shedding.
Polyomaviruses have two distinct courses of interaction with cultured cells: 1) they can productively infect permissive cells, undergo cytolysis, and release infectious virions or 2) they can abortively infect nonpermissive cells, leading to cellular transformation. 25 A similar virus–host interaction seems to occur between HaPV and hamsters in vivo. In endemically infected colonies or when HaPV infects older hamsters, infection is typically productive, resulting in virus-containing epitheliomas, primarily around the face and feet, with persistent virus shedding in the urine. 19 When HaPV infects immunologically naive young hamsters, it causes epizootics of lymphoma with morbidity as high as 80%. 19 Yet, the search for HaPV virions within lymphomas from affected hamsters has been called “vacuous and unrewarding” because no virions have been demonstrated by electron microscopy. 3 Transformation and immortalization of primary cell cultures by polyomaviruses is commonly associated with integration of virus DNA into the host genome. 6 However, in an experimental infection study using hamsters, integration of HaPV DNA into the host genome was found in only 2 of 22 lymphomas. 16 In the remaining 20 lymphomas, the HaPV DNA was present as multiple extrachromosomal episomal copies. 16 Thus, extrachromosomal amplification or chromosomal integration represent alternative mechanisms of transformation and oncogenesis in HaPV-infected hamsters. Regardless of the mechanism of transformation, nonrandom deletions have been found in the late coding regions of HaPV DNA. 16,22 Oncogenesis is a direct consequence of and has been systematically associated with deletion of the late coding regions of the HaPV genome. 16
Specific amplification of HaPV DNA from neoplastic lymphocytes is consistent with previous reports indicating that HaPV DNA is present as abundant nonrandomly deleted extrachromosomal copies or as a single integrated copy within HaPV-induced lymphomas. 16 The relatively light staining of neoplastic lymphocytes suggests there is less HaPV template DNA within infected lymphocytes than there is in other infected cell types. One possible explanation for this paucity of HaPV template DNA may be low copy numbers in infected lymphocytes due to HaPV integration into the host genome; polyomaviruses are known to integrate into the genomes of transformed cells. 3,4 However, no intact virions have been identified within the transformed lymphocytes of HaPV-infected hamsters, and genomic integration by HaPV is less common than episomal amplification in lymphoma oncogenesis. 15,16 Another explanation for minimal virus sequence amplification is that episomal copies of the HaPV genome are incomplete because they have nonrandom deletions in the late coding regions. 16,22,23 The PCR primers we developed to amplify HaPV are located in the VP1 gene, which is a late gene product. Even though HaPV-induced lymphomas usually contain numerous episomal viral copies; nonrandom deletions in the late coding regions may result in DNA that serves as a poor template for the primer set that we used. Developing a primer set that would amplify early gene products, such as the T antigen genes, could potentially refine the PCR assay.
HaPV is believed to be spread by environmental contamination by urine from HaPV-infected hamsters. 19 Modest staining of several nuclei of renal tubular epithelial cells was noted, indicating that HaPV DNA was present within these specific cells. Shedding of virus from tubular epithelial cells would provide an excellent opportunity for infective virus to enter the urine. The lack of extensive specific staining within the renal tubular epithelium could be due to the stage of HaPV infection within this particular hamster, as occurs with another murine papovavirus, K virus. K virus nucleic acids were rarely detected in the renal tubular epithelium 2 months after inoculation into naive mice; however, at 6 months after inoculation the renal tubular epithelial cells represented a major source of virus persistence. 12
The most intense labeling for HaPV DNA was found within the nuclei of jejunal epithelial enterocytes. This staining was distinct from and not associated with invasion of the submucosa by transformed lymphocytes. The intensity of nuclear staining is indicative of the presence of multiple copies of the HaPV genome within affected enterocyte nuclei. Release of virus from affected enterocytes is another potential route of virus shedding. Hamsters are coprophagous rodents; thus, fecal shedding and consumption of HaPV virions could be a natural route of virus dissemination. Further study of the mechanisms by which HaPV is spread among hamsters will be required to confirm these observations.
The HaPV-induced lymphoma in this juvenile pet Syrian hamster prompted development of several tools that should be useful in future studies of HaPV pathogenesis, including a PCR assay that can be used for definitive diagnosis of virus within affected colonies and an in situ PCR to further characterize the tissue distribution of HaPV in affected hamsters.
Footnotes
Acknowledgements
We thank A. M. Fernandez and R. Knowles for technical assistance and H. Wilson for assistance with computer graphics.