
Abstract
In situ hybridization techniques that employed a nonradioactive digoxigenin-labeled probe were used to detect and localize ApxI, II and III genes in tissue sections of pneumonic lung naturally infected with Actinobacillus pleuropneumoniae. In pigs infected with either serotype 2 or 6, a hybridization signal for apxIICA, apxIIICA, apxIBD, and apxIIIBD was detected, and in pigs infected with serotype 5, a hybridization signal for apxICA, apxIICA, and apxIBD was detected in the pneumonic lesions. A hybridization signal for apxIICA and apxIBD was detected in pigs infected with serotype 7. A strong hybridization signal for apx genes was seen in streaming degenerate alveolar leukocytes bordering zones of coagulative necrosis. Simultaneous detection of hybridization signals for the apxCA and apxBD genes provided scientific evidence that the expression of the apx genes could be potential indicators of the production of corresponding Apx toxins. This study demonstrates the expression of ApxI, II, and III genes in pneumonic lesions caused by A. pleuropneumoniae.
Porcine pleuropneumonia, caused by Actinobacillus pleuropneumoniae, is worldwide in its distribution and causes severe economic losses in pig-rearing countries. Acute lung lesions are characterized histologically by coagulative necrosis, hemorrhage, vascular thrombosis, edema, fibrin deposition, and infiltration of lung parenchyma by neutrophils and mononuclear cells. 3,26 In chronic lesions, a thick layer of granulation tissue sequesters areas of pulmonary necrosis. 18 A. pleuropneumoniae is detected by in situ hybridization in neutrophils and alveolar macrophages in peripheral foci of coagulative necrosis and in granulation tissue that has sequestered foci of necrosis. 20
Based on nicotinamide adenine dinucleotide (NAD) requirements, NAD-dependent strains (biotype 1) and NAD-independent strains (biotype 2) can be distinguished. 25 To date, at least 12 A. pleuropneumoniae serotypes (1–12) have been described, and serotypes 1 and 5 have been further subdivided into subtypes A and B. 14,24 Reference strains for the 12 serotypes of A. pleuropneumoniae secrete one or two of Apx toxins: ApxI is produced by reference strains of serotypes 1, 5, 9, 10, and 11, ApxII is produced by reference strains of all serotypes except serotype 10, and ApxIII is produced by reference strains of serotypes 2, 3, 4, 6, and 8. 7 These Apx toxins are encoded by operons that consist of four contiguous apx genes designated, C, A, B, and D. The C and A genes code for the active toxin protein, and the B and D genes code for proteins involved in secretion of the toxin. 7
The Apx toxin and lipopolysaccharide (LPS) produced by A. pleuropneumoniae have been suggested as key factors in the pathogenesis of the disease process. 9,30 The LPS appears to be associated with inflammation and the expression of cytokines such as interleukin-1 (IL-1), tumor necrosis factor α, and IL-6. 1,6 The Apx toxin appears to cause coagulative necrosis early in the pathogenesis of the disease. 9,15,30 In addition, Apx toxin is cytotoxic for porcine neutrophils, alveolar macrophages, and lymphocytes. 28,29 Field isolates of A. pleuropneumoniae that produce Apx toxins or carry apx genes have been identified. 2,16,21,27
One objective of the present study was to develop an in situ hybridization technique using a nonradioactive digoxigenin-labeled DNA probe that targets specific ApxI, -II, and -III genes from chromosomal DNA of A. pleuropneumoniae. The second objective was to use this technique to detect and localize the expression of ApxI, -II, and -III genes in an effort to elucidate the role of Apx toxin in the pathogenesis of pleuropneumonia caused by A. pleuropneumoniae.
Materials and Methods
Animals
Samples were obtained at necropsy from pigs submitted to the Department of Veterinary Pathology of Seoul National University from January 1994 to December 1997. Fifteen pigs (Nos. 1–15) approximately 87–140 days of age from 15 different herds were selected on the basis of clinical signs, characteristic lesions, bacterial isolation, and serotype. All 15 pigs were negative for Mycoplasma hyopneumoniae, Pasteurella multocida, and porcine reproductive and respiratory syndrome virus by culture and in situ hybridization. 5,17
Negative control sections were prepared from 1-day-old colostrum-deprived pigs that had not been exposed to A. pleuropneumoniae. Also included as a control was lung tissue from a 3-week-old conventional pig infected with P. multocida.
Reference strains and antisera
A. pleuropneumoniae reference strains were used: serotype 1, S4074; serotype 2, S4226; serotype 3, S1421; serotype 4, M62; serotype 5, K17; serotype 6, Femo; serotype 7, WF83; serotype 8, 405; serotype 9, CVJ1361; serotype 10, D13039; serotype 11, 56153; and serotype 12, 8328. All reference serotypes and rabbit antisera against the 12 serotypes were provided by K. R. Mittal (University of Montreal, Saint-Hyacinthe, PQ, Canada).
Bacteriologic examination
All pigs were submitted alive, and immediately upon receipt they were euthanatized for necropsy. Specimens of lung were collected for bacteriologic culture on 5% sheep blood agar. Identification of isolates as A. pleuropneumoniae was based on Gram staining, positive hemolysis on 5% sheep blood agar, a positive Christie–Atkins–Munch–Petersen (CAMP) reaction, a requirement for NAD, urease production, and xylose and mannose fermentation. The CAMP reaction was determined on 5% sheep blood agar, with a beta-hemolysis–producing Staphylococcus intermedius strain. A. pleuropneumoniae isolates were subjected to slide agglutination with antisera to serotypes 1–12. 22
Polymerase chain reaction
Base sequences, locations, and predicted sizes of amplified products for the specific primers used in this study are shown in Table 1. A polymerase chain reaction (PCR) for apx genes was carried out as previously described; 7,21 however, a single PCR was performed for each apx gene instead of multiplex PCR, and the PCR conditions were also slightly modified. Chromosomal DNA of all 15 A. pleuropneumoniae isolates and the 12 reference strains was purified using a commercial kit (Pharmacia-LKB Biotech, Upssala, Sweden) according to the manufacturer's instructions. Amplification of bacterial DNA was performed with 50-µl volumes containing 5 µl of the prepared sample supernatant, the oligonucleotide primers (90 ng), 0.2 mM of (each) dATP, dGTP, dCTP, and dTTP, 10 mM Tris-HCl (pH 8.8), 1.5 mM MgCl2, 50 mM KCl, and 1 unit of Taq polymerase. The PCR profile used a denaturing step at 94 C for 1 minute followed by annealing of the primers at 55 C for 2 minutes and an extension step at 72 C for 1 minute. The 35 cycles of these three-step procedures were performed in a thermal cycler, followed by a 10-minute extension at 72 C.
Primers used in polymerase chain reaction to amplify specific fragments from apx genes.
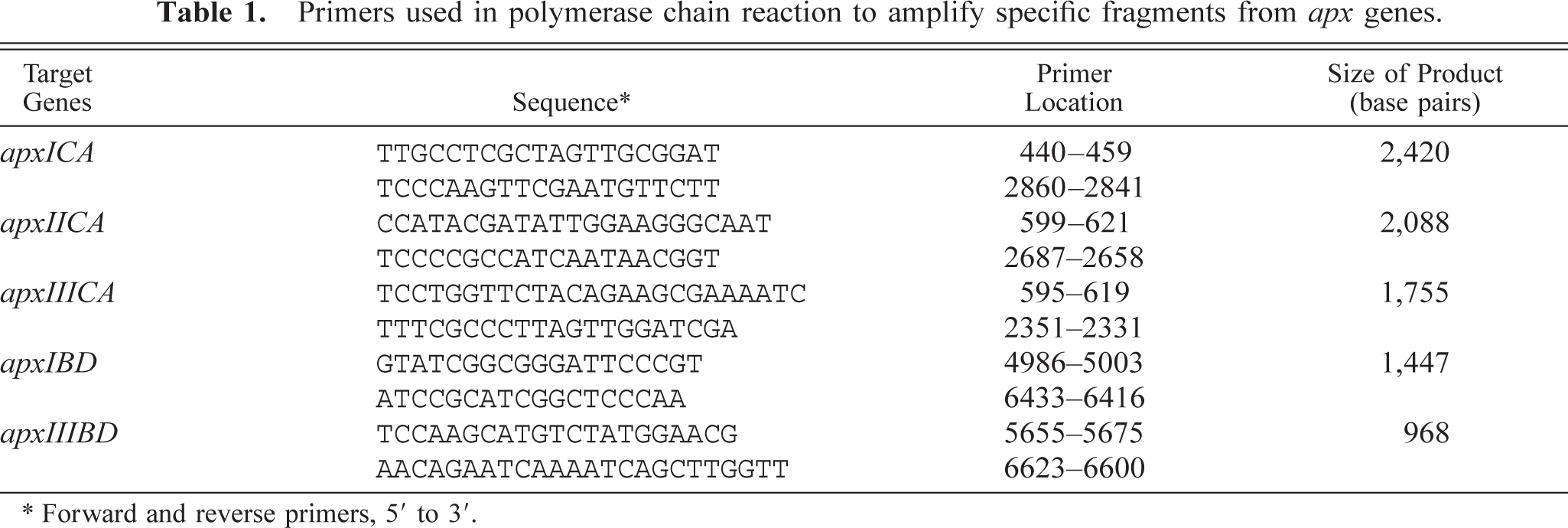
∗ Forward and reverse primers, 5′ to 3′.
Probe
PCR products for ApxI, -II, and -III genes were purified using a 30-kD cutoff membrane ultrafiltration filter. The nucleotide sequences of the purified PCR products were determined by BigDye chemistry with the ABI Prism Sequencer (Applied Biosystems, Foster City, CA). Sequencing was performed in the purified PCR products before PCR products were labeled by random priming with digoxigenin-dUTP (Boehringer Mannheim, Indianapolis, IN) according to the manufacturer's instructions.
In situ hybridization
Tissues from each pig were collected in 10% neutral buffered formalin and after fixation for 1–2 days were dehydrated through graded alcohols and a xylene step and embedded in paraffin. Sections were then cut at 4 µm, placed on Superfrost/plus slides (Fisher Scientific, Pittsburgh, PA), and stored at room temperature. Just before use, sections were deparaffinized in xylene and rehydrated in phosphate-buffered saline (PBS; pH 7.4, 0.01 M) for 5 minutes. Deproteinization was carried out in 0.2 N HCl for 20 minutes at room temperature. Tissues were then digested at 37 C for 20 minutes in 200 µg/ml proteinase K (Gibco BRL, Grand Island, NY) in PBS. One of two serial sections examined was treated with DNase I (Boehringer Mannheim) at 0.1 U/ml in 10 mM Tris-HCl (pH 7.4) for 30 minutes at 37 C to remove target DNA, as a specificity control. After digestion, tissues were fixed in 4% paraformaldehyde in PBS for 10 minutes. After rinsing twice with PBS, the slides were acetylated in 300 ml of 0.1 mM triethanolamine-HCl buffer (pH 8.0) to which 0.75 ml of acetic anhydride (0.25%) had been added. After 5 minutes, a further 0.75 ml of acetic anhydride was added, and 5 minutes later the slides were rinsed in 2× saline sodium citrate (SSC) (1× SSC contains 50 mM NaCl and 15 mM sodium citrate, pH 7.0).
Hybridization was carried out overnight at 45 C. The digoxigenin-labeled probe (1 ng/µl) was diluted in 50 µl of the standard hybridization buffer that consisted of 2× SSC containing deionized formamide 50%, 10 mg salmon sperm DNA (Oncor, Gaithersburg, MD), 0.02% sodium dodecyl sulfate, 1% Denhart's solution, and 50% dextran sulfate solution (50% concentration). Approximately, 75 ng of digoxigenin-labeled probe was added to standard hybridization buffer (70 µl), which was then layered over the section. Fluid was held in place by a coverslip (the edges of which were sealed with rubber cement) and heated for 10 minutes in a 95 C heating block. After overnight hybridization, sections were thoroughly washed: twice in 4× SSC for 10 minutes at room temperature, twice in 2× SSC for 10 minutes at 45°C, twice in 2× SSC for 10 minutes at room temperature, twice in 0.2× SSC for 10 minutes, once in maleic acid buffer (100 mM maleic acid and 150 mM NaCl, pH 7.5) for 5 minutes, and once in 1× blocking reagent (Boehringer Mannheim) for 40 minutes at room temperature.
For detection of hybridization, sections were incubated with anti-digoxigenin conjugated with alkaline phosphatase (Boehringer Mannheim) diluted 1:500 in 1× blocking reagent (Boehringer Mannheim). After three washes in buffer, substrate consisting of nitroblue tetrazolium (NBT) and 5-bromocresyl-3-indolylphosphate (BCIP) was layered over the sections. Color was allowed to develop for 3–4 hours in the dark, and development was stopped by dipping slides briefly in tri-ethylenediaminetetraacetic acid (EDTA) buffer (10 mM Tris-HCl and 1 mM EDTA, pH 8.0). Sections were counterstained with methyl green 0.5%, and the slides were then washed with distilled water for 1 minute and dried completely.
Results
Microbiologic, PCR, and in situ hybridization results for the 15 infected pigs are presented in Table 2. PCR products of ApxI, -II, and -III genes from all 15 A. pleuropneumoniae isolates and the 12 reference strains were sequenced, and their identity was confirmed as ApxI, -II, and -III genes (data not shown). A. pleuropneumoniae was isolated from the lungs of all pigs. Among the 15 isolates, 4 isolates each of serotypes 2 and 5 were identified. Three isolates were serotype 6, two were serotype 7, and two were untypable. Each specific primer pair for apxICA, apxIICA, apxIIICA, apxIBD, and apxIIIBD yielded a PCR product of the expected size for the reference strains and field isolates.
Polymerase chain reaction (PCR) and in situ hybridization (ISH) results in 15 pigs naturally infected with Actinobacillus pleuropneumoniae.
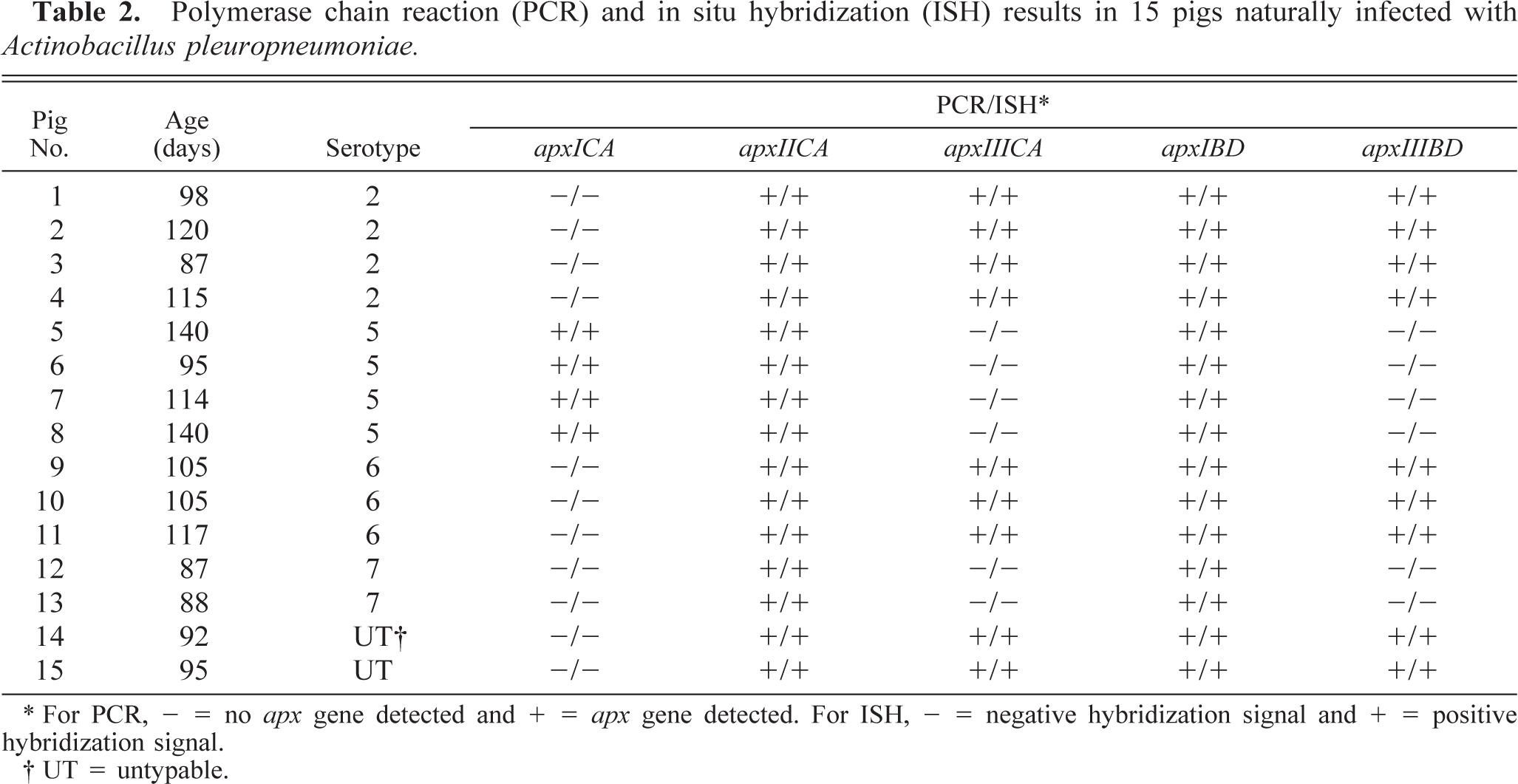
∗ For PCR, - = no apx gene detected and + = apx gene detected. For ISH, - = negative hybridization signal and + = positive hybridization signal.
† UT = untypable.
The morphology of host cells was preserved despite the relatively high temperature required during the incubation procedure. The signal intensity varied among tissue sections and among pigs. Positive hybridization typically produced a dark brown to black reaction product in intracellular and extracellular locations without background staining (Fig. 1). A very close cell-to-cell correlation among three serial sections from each of the 15 lung samples was confirmed by in situ hybridization. Coordinate expression of related apx genes was consistently seen. In seven pigs infected with either serotype 2 or 6, a hybridization signal for apxIICA, apxIIICA, apxIBD, and apxIIIBD (Fig. 1) was detected, and in four pigs infected with serotype 5, a hybridization signal for apxICA (Fig. 2), apxIICA, and apxIBD was detected in the pneumonic lesions. In two pigs infected with serotype 7, a hybridization signal for apxIICA and apxIBD was detected, and in 2 pigs infected with an untypable serotype, a hybridization signal for apxIICA, apxIIICA, apxIBD, and apxIIIBD was detected in the pneumonic lesions (Table 2).
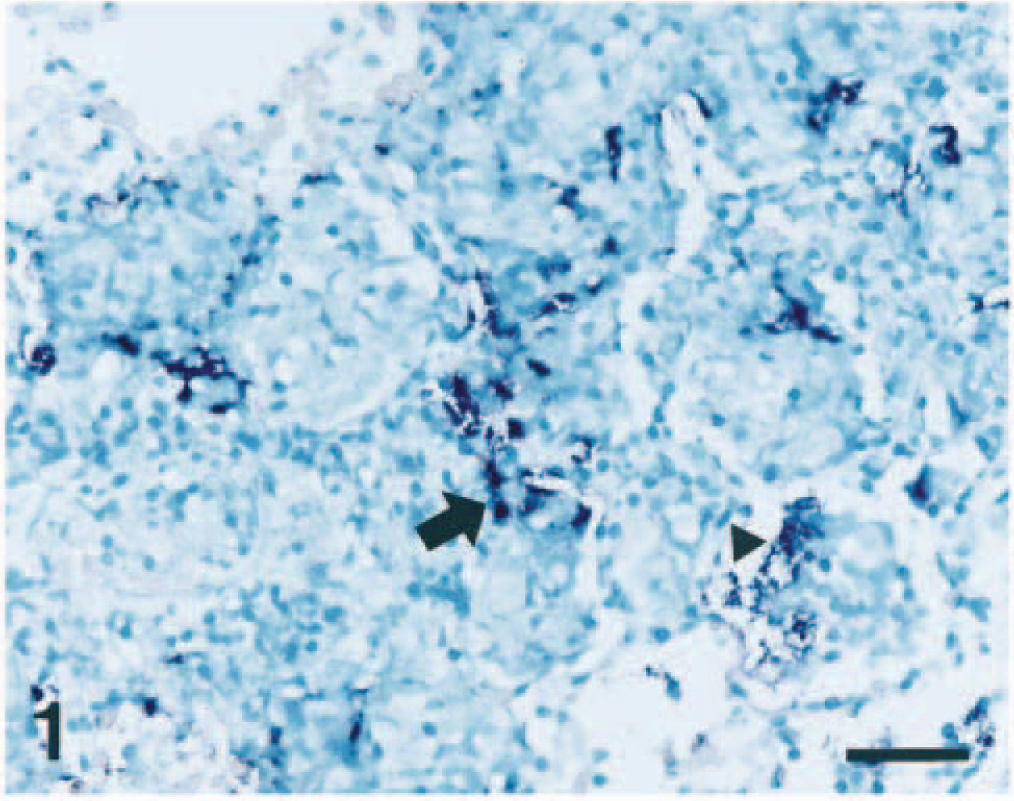
Lung; pig No. 3, naturally infected with Actinobacillus pleuropneumoniae serotype 2. The apxIIIBD gene (dark blue reaction) was detected at an intracellular location (arrow) in streaming degenerate alveolar leukocytes and at an extracellular location (arrowhead) in alveolar spaces. In situ hybridization; NBT/BCIP methyl green counterstain. Bar = 55 µm.
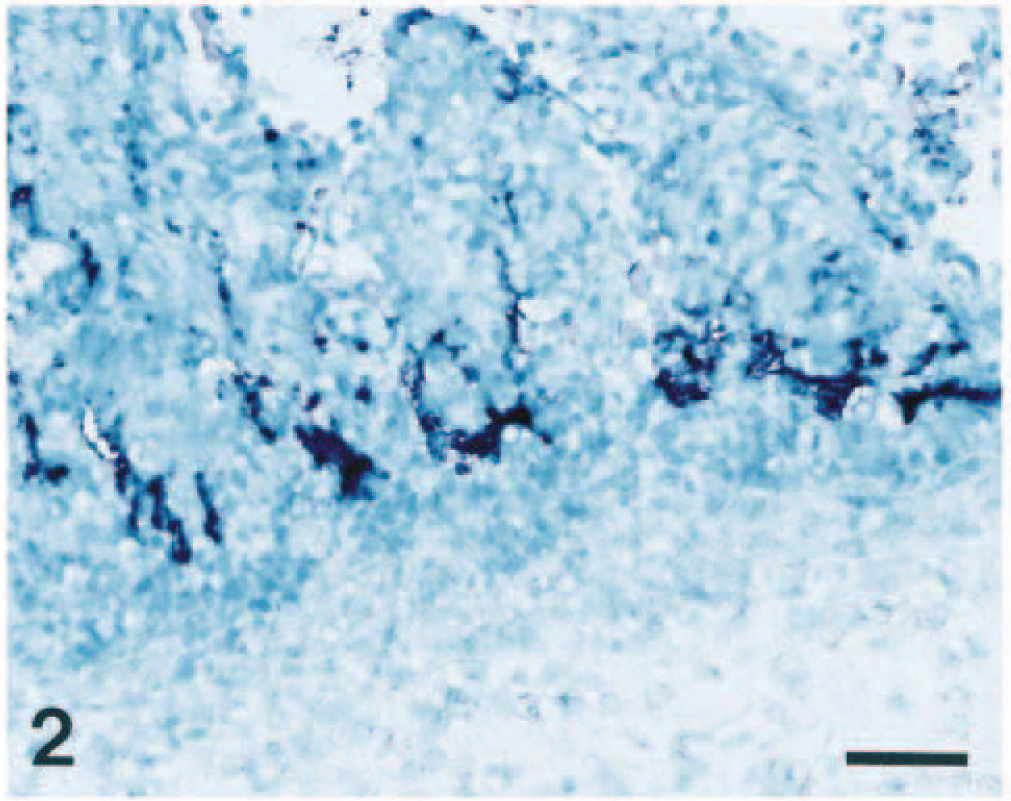
Lung; pig No. 6, naturally infected with Actinobacillus pleuropneumoniae serotype 5. The apxICA gene (dark blue reaction) was detected in streaming degenerate alveolar leukocytes bordering zones of coagulative necrosis. In situ hybridization; NBT/BCIP, methyl green counterstain. Bar = 55 µm.
A strong hybridization signal was seen in “streaming” degenerate alveolar leukocytes (“oat cells”) bordering zones of coagulative necrosis and alveolar spaces (Fig. 1). Less intense signals were seen in granulation tissues surrounding necrotic foci. Neutrophils and macrophages that had infiltrated into alveolar spaces frequently showed a strong hybridization signals. Identification of the cell types expressing apx genes was occasionally difficult but examination of adjacent sections stained with hematoxylin and eosin confirmed that apx genes were present in neutrophils and macrophages. Positive hybridization signal was also detected in the alveolar spaces with extracellular locations.
No hybridization signal was consistently seen in tissue sections treated with DNase I prior to in situ hybridization. Sections from the two control pigs lacked hybridization signal for apx genes.
Discussion
This study demonstrates the presence of ApxI, -II, and -III genes in lung lesions caused by A. pleuropneumoniae and implicates the Apx toxins as an important factors in pathogenesis of the disease. Apx toxins are encoded by operons consisting of four contiguous apx genes; C, A, B, and D. The C and A genes code for the active toxin protein, and the B and D genes code for proteins involved in secretion of the toxin. 7 Simultaneous detection of hybridization signals for apxCA and apxBD genes could suggest the production of the respective Apx toxins.
Although several bacterial components, including LPS, Apx toxins, and polysaccharide capsule, likely contribute synergistically to cause disease, 10 Apx toxins are considered the most important factor for bacterial virulence and pathogenicity. 4,10,11 The role of the Apx toxin was shown when a hemolysin-deficient serotype 5 mutant that secreted neither ApxI nor ApxII was found to be nonpathogenic in pigs. 11 Apx toxins kill neutrophils and augment inflammation in the lung. 12 Neutrophils appear to predominate in the inflammatory cellular exudate 24–48 hours after inoculation. 16 Ultrastructural examination revealed intralesional neutrophils in various stages of degeneration and deterioration in mice and pigs. 9,18 Neutrophils generate a variety of substances, such as oxygen radicals, neutral proteases, and cationic proteins, that are capable of causing severe damage to healthy host tissues when released in excessive amounts from degranulating or necrotic neutrophils. 19 ApxI, -II, and -III genes were detected mainly in the degenerate neutrophils and macrophages in this study. A. pleuropneumoniae organisms in the lung are rapidly phagocytosed by or adhere to alveolar macrophages and neutrophils and produce the ApxI, -II, and -III toxins. All Apx toxins may be potential toxic for alveolar macrophages and neutrophils. Only in areas where there was sufficient Apx toxin concentration to cause lysis of inflammatory cells was there a high enough colonization of A. pleuropneumoniae to be detected by in situ hybridization. These observations suggest that high concentration of Apx toxins could lead to host-mediated tissue damage by the lysis of inflammatory cells. This hypothesis must be tested using biochemical or in vitro methods.
Our results also revealed a high degree of correlation between the respective serotypes and the expression of apx genes. These data indicated that the apx operons are inherent to their respective serotypes. The ApxII toxin is produced by all serotypes except serotype 10, 2 and this finding could be useful in the development of better diagnostic techniques. For example, the 2-kb fragment of the apxIICA gene used in this study may be a good choice for as a sensitive and specific probe for the diagnosis of A. pleuropneumoniae infection.
An accurate diagnosis of bacterial disease has hitherto depended heavily on isolation of the etiologic agent, followed by examination of its biochemical and morphologic properties. Culture of A. pleuropneumoniae can be relatively insensitive, especially in chronic cases, and it is time consuming, 23 but the isolation of A. pleuropneumoniae is required for serotyping. Fluorescent in situ hybridization using a DNA probe designed from the 16S ribosomal RNA of A. pleuropneumoniae can be used to detect the 12 A. pleuropneumoniae serotypes. 8,13 However, fluorescent in situ hybridization cannot differentiate among the 12 serotypes. Identification of serotype is important for the control of A. pleuropneumoniae infection because current bacterins provide only serotype-specific protection. In situ hybridization methodology developed in this study is complementary to the serotyping of A. pleuropneumoniae isolates. A. pleuropneumoniae serotype 2 does not carry the apxICA gene, but it does carry the apxIIIBD gene. A. pleuropneumoniae serotype 5 does not carry apxIIIBD gene, but it does carry the apxICA gene. 2 Thus, probes from the apxIIIBD and apxICA genes may be of value for in situ hybridization used to differentiate between serotype 2 and 5 in formalin-fixed, paraffin-embedded lung specimens. In Korea, >85% of the field isolates are either serotype 2 or serotype 5. 21
The technique of nonradioactive in situ hybridization using digoxigenin-labeled probes has been successfully applied to A. pleuropneumoniae-infected lung tissue to detect and localize apx genes. By using DNA probes and in situ hybridization, there is no possibility for error that could be caused by antigenic cross-reactivity or by the alteration of binding sites caused by tissue processing. The apx genes of A. pleuropneumoniae are inherent to a given serotype. Using in situ hybridization to detect apx genes in lung lesions from pigs naturally infected with A. pleuropneumoniae is a valuable tool that is complementary to the serotyping of A. pleuropneumoniae.
Footnotes
Acknowledgements
The research reported here was supported by contract research funds from the Research Institute for Veterinary Science (RIVS), College of Veterinary Medicine, Seoul National University, and from the Brain Korea 21 Project, Republic of Korea.