
Abstract
Haemobartonella felis is an epierythrocytic bacterium suspected to be the causative agent of feline infectious anemia. Previous studies with a polymerase chain reaction assay have identified a mycoplasmal 16S rRNA gene sequence that coincides with clinical disease and the presence of organisms in the blood. Tissues from a cat experimentally infected with H. felis were used for in situ hybridization studies to physically link this 16S rRNA gene to the organisms on the red cells. A biotin-labeled probe was used in conjunction with tyramide signal amplification to visualize the hybridization signal. This study clearly demonstrates a specific hybridization signal on the red cells in the tissues of the H. felis-infected cat. This in situ hybridization study is the final step in fulfilling the molecular guidelines for disease causation and proves that H. felis, a mycoplasmal organism, is the causative agent of feline infectious anemia.
Feline infectious anemia (FIA) is a disease of domestic cats and is presumed to be caused by the red cell parasite Haemobartonella felis. Definitive proof that H. felis is the causative agent of FIA has not been established, primarily because of the inability to culture the organism and the presence of an asymptomatic carrier state. This disease is characterized by a wide range of clinical signs that include anemia, pyrexia, lethargy, and splenomegaly.4 Historically, the diagnosis of FIA has relied on the visualization of the bacteria on the red cell. The transient parasitemia in acute Haemobartonella infections and the cyclic episodes of parasitemia in carrier animals complicate the diagnosis of this disease. The true nature of this organism and the prevalence of the disease have been disputed because, until recently, the disease could not be reliably diagnosed.2,7,21
The need for a reliable and efficient diagnostic test led to the sequencing of the H. felis 16S rRNA gene and the development of a species-specific polymerase chain reaction (PCR) test.2,7,21,26 With this PCR test, the presence of H. felis DNA was documented to coincide with experimental infection,2 and the amount of DNA could be correlated with the numbers of organisms present in the peripheral blood.6 This is strong circumstantial evidence that H. felis is the causative agent of FIA. However, PCR in solution does not preserve the relationship between the organisms and the tissue or rule out the possibility of contamination by a different Mycoplasma species. With the use of in situ visualization techniques, the DNA sequence can be physically linked to the organisms on the red cell, strengthening the argument that H. felis is the causative agent of FIA.
The in situ hybridization (ISH) of fixed tissue sections uses a labeled nucleic acid probe to identify the location of a cellular nucleic acid sequence of interest. This method is an increasingly popular tool to localize pathogenic organisms to their target tissues and to confirm the identity of those pathogens.11 Specific probes can be designed to locate organisms that cannot be cultured or that fail to react with common immunohistochemical stains. One limitation of conventional ISH is that the threshold for detection is 20 copies of the gene of interest.16 The closest relatives of H. felis, including Haemobartonella muris, Eperythrozoon suis, and Eperythrozoon wenyonii, are members of the order Mycoplasmatales, not Rickettsiales, on the basis of sequence analysis of the 16S rRNA gene.7,21,23,26 Mycoplasmas are known to carry only one or two copies of the 16S rRNA gene;25 therefore, conventional ISH should not detect the genomic 16S rRNA sequence in these species, and additional techniques are needed to enhance the sensitivity of the assay.
In recent years, several methods to improve the sensitivity of conventional ISH have been developed. One technique is to design a probe that binds to the 16S ribosomal RNA sequence as well as the genomic DNA sequence. This method, however, is limited by the secondary structure of the mature ribosomal RNA. The frequent hairpin turns and folding do not allow for adequate denaturation of the RNA and prevent binding of the probe.10 Also, fewer ribosomes are in naturally growing bacteria than in cultured bacteria, making in situ detection more difficult.1 A second technique to increase the sensitivity of ISH is the amplification of the target DNA; this has been achieved by in situ PCR, in situ self-sustained sequence replication, and primed in situ labeling. These techniques have significant limitations. They are time consuming and technically difficult, can produce frequent false-negative and false-positive results, and have relatively low amplification efficiencies.16 The need for maintaining tissue morphology and retaining the amplified product DNA within the permeabilized cell must also be addressed. The most recent advancement in ISH technology is amplification of the signal after the labeled probe has bound to its target. Signal amplification enhances sensitivity of the ISH assay without altering tissue morphology.14 In this study, biotinylated tyramide was used to amplify the ISH signal.
The biotinylated tyramide technique for signal amplification of ISH has been reported to yield a 500–1,000-fold increase in sensitivity for signal detection without production of increased background staining.20 A standard in situ hybridization reaction is performed, and the signal from the labeled probe is amplified by a peroxidase-catalyzed precipitation of tyramide. The free radicals formed by the peroxidase are short lived, thus the biotinylated tyramide is only deposited near the site of the probe. The tyramide forms a strong bond to the electron-rich moieties of adjacent proteins, limiting diffusion of the amplified signal. This technique has been used to detect single copies of incorporated virus,24 eukaryotic rRNA,15 and prokaryotic rRNA,18 as well as in ultrastructural studies of DNA and proteins.27 In this study, signal amplification technology was combined with ISH to visualize the 16S rRNA gene of H. felis within both liver and kidney sections from an experimentally infected cat.
Materials and Methods
Tissues
Haemobartonella felis-positive tissue sections were collected from an experimentally infected cat during a parasitemic episode. Evaluation of the peripheral blood smear revealed that approximately 70% of the red cells contained one or more H. felis organisms. A noninfected cat submitted for routine necropsy served as a source of H. felis-negative tissue. Tissues were fixed in 10% neutral buffered formalin for 48 hours and embedded in paraffin blocks by standard techniques.19 Sections 4–6 mm thick were cut onto charged slides (Surgipath Medical Industries, Richmond, IL) and baked at 60 C for 30 minutes. Care was taken to cut the negative cat tissues first and to change blades between blocks to avoid carryover of H. felis DNA. These sections were stored at room temperature until used for in situ studies.
Bitoin-dUTP probe labeling by PCR
The H. felis 16S rRNA hybridization probe for this study was a 393-base pair (bp) PCR product labeled by the incorporation of biotin-dUTP into the reaction mixture. H. felis-F1 and H. felis-R3 species-specific primers2 were used at a final concentration of 0.2 pmol/μl. The PCR reaction contained 2 mM of each nucleotide: dATP, dGTP, dCTP, 1 mM dTTP, 1 mM biotin-16-2′-dUTP (Boehringer Mannheim Biochemicals, Indianapolis, IN), 0.05 U/μl Taq polymerase (Fisher Scientific, Pittsburgh, PA), 1× PCR reaction buffer A (Fisher Scientific), and H. felis DNA template. The PCR reaction consisted of an initial denaturation of 94 C for 5 minutes followed by 40 three-step cycles of 45 seconds at 94 C, 45 seconds at 54 C, and 1 minute at 72 C. These cycles were followed by a final elongation step of 7 minutes at 72 C. The labeled PCR product was separated from residual primers and nucleotides with a microcolumn according to the manufacturer's instructions (Wizard PCR preps DNA purification system, Promega, Madison, WI).
The nonbinding control probe in this study was designed to be the same length and approximate guanidine and cytosine content of the H. felis probe. This probe was manufactured by the same PCR protocol as described above except DNA extracted from human blood served as the template and the primers were designed to amplify a 450-bp product of the human CYP2D6 (cytochrome P450) gene. This gene was selected because, in eukaryotes, P450 genes are predominately conserved only near the heme-binding region,22 and P450 genes have not been reported in the Mycoplasma species that have been fully sequenced.8,12 The probe sequence was not part of the known conserved eukaryotic regions, and the only two feline P450 sequences that were available did not show homology to this portion of the human CYP2D6 sequence.
Dot blots (probe specificity)
Dot blots were performed to evaluate the specificity of the H. felis probe. A commercial kit was used according to the manufacturer's instructions (Generation, Gentra Systems, Minneapolis, MN) to extract DNA from 200 μl of whole blood from a noninfected cat, a H. felis-infected cat, and a healthy human. This DNA, as well as unlabeled H. felis 16S PCR product and unlabeled P450 PCR product, was applied to a nylon membrane in the following manner. The DNA from each sample was mixed in a 0.4 M NaOH/10 mM ethylenediaminetetraacetic acid (EDTA) solution and was applied to a positively charged nylon membrane with a vacuum manifold. The membrane was incubated in hybridization solution (5× sodium chloride–sodium citrate [SSC] [20× SSC = 3 M NaCl, 0.3 M sodium citrate], 10% dextran sulfate, 2% blocking reagent [Boehringer Mannheim], 0.02% sodium dodecyl sulfate, 0.1% N-laural sarcosine, 50% deionized formamide) for 1 hour at 42 C. Fresh hybridization solution containing either the H. felis 16S probe or the human P450 probe was then added. The membrane was then incubated overnight at 42 C. After hybridization, the membranes were washed in 2× SSC at 60 C for 20 minutes, 0.2× SSC at 42 C for 20 minutes, and 0.1× SSC at room temperature for 5 minutes. The membranes were then rinsed in 2× SSC prior to the color detection steps. Stringency washes and color detection were identical to those used in the in situ hybridization reactions; however, no tyramide amplification was performed.
In situ hybridization
Sections were deparaffinized with xylene for 20 minutes followed by rehydration with 100% ethanol for 10 minutes and 70% ethanol for 5 minutes. The slides were then rinsed in water for 2 minutes and phosphate-buffered saline (PBS) (Sigma-Aldrich Corp., St. Louis, Mo; 120 mM NaCl, 2.7 mM KCl, 10 mM phosphate buffer) for 10 minutes. Slides were overlaid with three drops of endogenous peroxidase blocking reagent (Dako Corporation, Carpinteria, CA), incubated at room temperature for 10 minutes, and then washed twice in PBS for 5 minutes. This wash was followed by a proteinase K (Dako Corp.) digestion for 3 minutes at room temperature. The proteinase reaction was stopped by a 5-minute incubation in 0.1 M glycine in PBS, followed by two 5-minute washes in PBS. Slides were then dehydrated through graded alcohols by washing two times for 5 minutes at each concentration: 50% ethanol, 70% ethanol, and 100% ethanol.
These prepared sections were then overlaid with 65 μl of hybridization solution (50% deionized formamide, 10% dextran sulfate, 0.2% dried milk, 2× SSC) containing 200 ng/ml of probe. A flexible coverslip (Gene Frame, Advanced Biotechnologies Ltd., Epsom, Surrey, UK) was then sealed in place. Slides were incubated for 10 minutes at 95 C followed by an overnight (12–16 hour) hybridization step at 37 C on a dedicated slide thermocycler (Hybaid Limited, Teddington, Middlesex, UK). After hybridization, the slides were washed in 2× SSC at 60 C for 20 minutes, 0.2× SSC at 42 C for 20 minutes, and 0.1× SSC at room temperature for 5 minutes. Slides were rinsed in 2× SSC prior to the tyramide amplification steps.
Biotin tyramide amplification
Amplification of the probe's biotin signal was achieved with the tyramide signal amplification (TSA)-indirect (ISH) kit (NEH Life Science Products, Boston, MA) according to the manufacturer's instructions. Minor modifications, outlined below, were made to optimize the signal intensity. The following steps were all performed at room temperature. After the stringency washes, the slides were overlaid with 100 μl of blocking solution and incubated in a humid chamber for 30 minutes. The slides were then incubated for 45 minutes in a streptavidin–horseradish peroxidase solution diluted 1:75 in blocking buffer. This incubation was followed by three 5-minute washes in PBS containing 0.05% Tween 20 (Sigma). The slides were then overlaid with a 1:40 dilution of the biotinyl tyramide for 7 minutes. This step was followed by three 5-minute washes in the PBS/Tween solution. The slides were then treated to detect the signal as described below.
Colorimetric detection
The slides were incubated in a 0.375-U/ml (1:2,000 dilution) strepavidin–alkaline phosphatase (Boehringer Mannheim) solution for 30 minutes. This incubation was followed by two 5-minute washes in buffer 1 (0.1 M Tris-HCl, 0.1 M NaCl, 2 mM MgCl2, pH 7.4) and a 10-minute wash in buffer 2 (0.1 M Tris base, 0.1 M NaCl, 50 mM MgCl2, pH 9.5). This step was followed by the color substrate; NBT/BCIP (nitro blue tetrazolium chloride 18.75 mg/ml and 5-bromo-4-chloro-3-indolyl phosphate 9.4 mg/ml [Boehringer Mannheim]) diluted 1:50 in buffer 2 containing 0.2 mM levamisole (Dako Corp.). The slides were monitored for color change and the reaction was stopped after 5–20 minutes by immersion in buffer 3 (20 mM Tris-HCl, 5 mM EDTA, pH 7.5). Slides were counterstained with an aqueous xanthene dye (Diff-Quik, Dade Diagnostics, Aquada, PR), and the color was preserved with a commercial aqueous mounting media (Crystal/Mount, Biomeda Corporation, Foster City, CA).
Controls consisted of 1) H. felis-negative feline tissue probed with the H. felis 16S probe, 2) H. felis-infected tissue with no probe or the human P450 probe, and 3) H. felis-infected tissue with the H. felis 16S probe but without tyramide amplification.
Results
Bitoin-dUTP probe labeling by PCR
Incorporation of the biotinylated nucleotide was verified by comparing the mobility of the PCR products amplified with and without the biotin-dUTP. The labeled 393-bp DNA probe had a decreased electrophoretic mobility when compared with an unlabeled PCR product generated with the same primers, confirming adequate biotin-dUTP incorporation17 (data not shown).
Dot blots (probe specificity)
The H. felis 16S probe bound only to the DNA from the H. felis-infected cat blood and the unlabeled H. felis PCR product. No binding occurred to the DNA from noninfected cat blood, the human blood, or the unlabeled P450 PCR product (Fig. 1).
The P450 probe showed a strong affinity for the human blood DNA and the unlabeled P450 PCR product. The P450 probe also showed a weak affinity for the DNA from both the noninfected and H. felis-infected feline blood. The probe did not bind to the H. felis unlabeled 16S PCR product (Fig. 2).
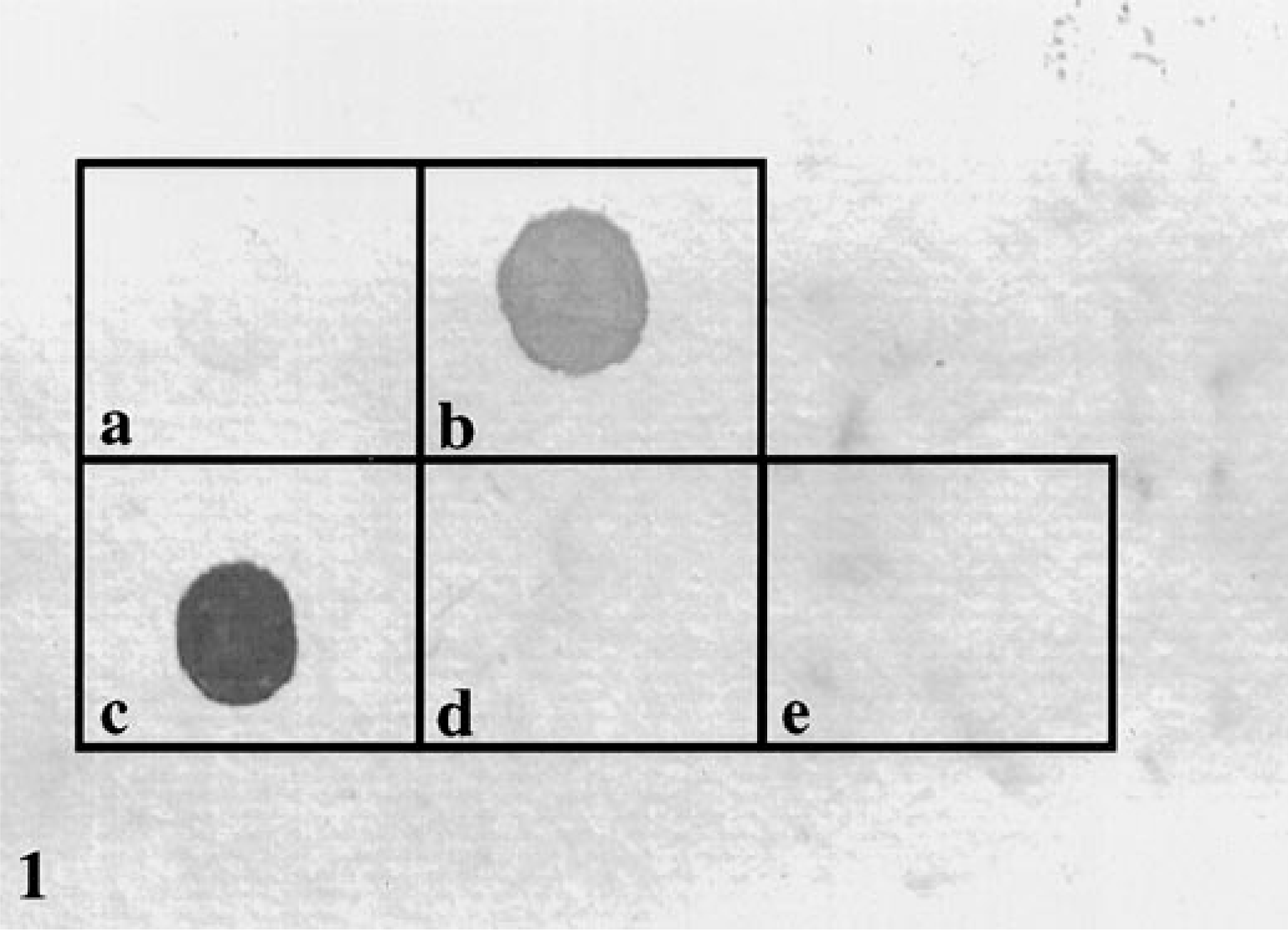
Dot blots probed with the H. felis 16S probe.
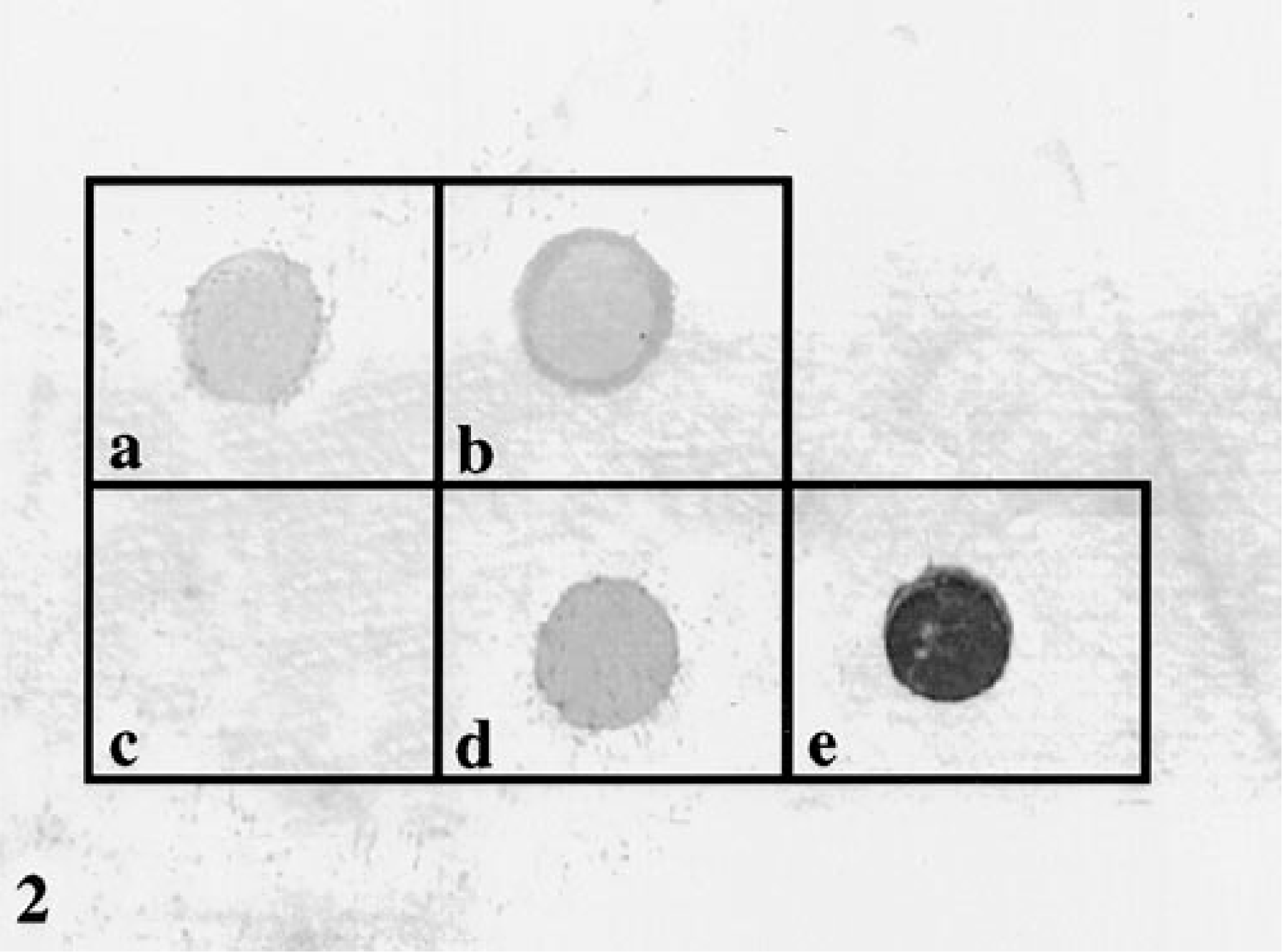
Dot blots probed with the human P450 probe.
In situ hybridization and tyramide amplification
The H. felis 16S probe failed to hybridize to the noninfected feline liver and kidney (Figs. 3, 4). Also, no binding was seen when the H. felis-infected tissues were probed with the human P450 probe or when the probe was deleted from the hybridization solution (data not shown).
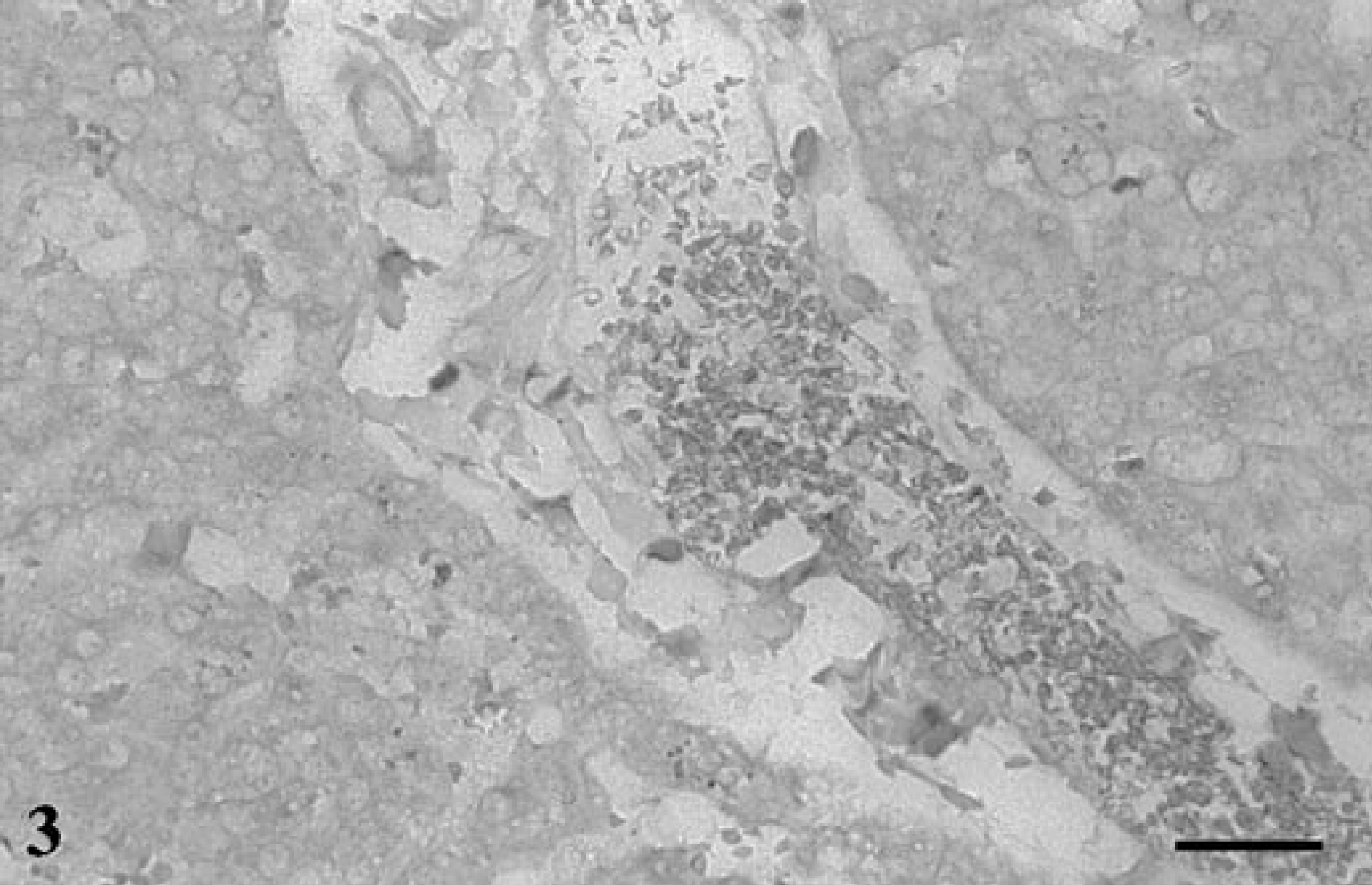
Liver; noninfected cat. No hybridization signal is seen in the negative control tissues. In situ hybridization, H. felis 16S probe. Biotin-TSA method, NBT/BCIP, xanthene counterstain. Bar = 30 μm.
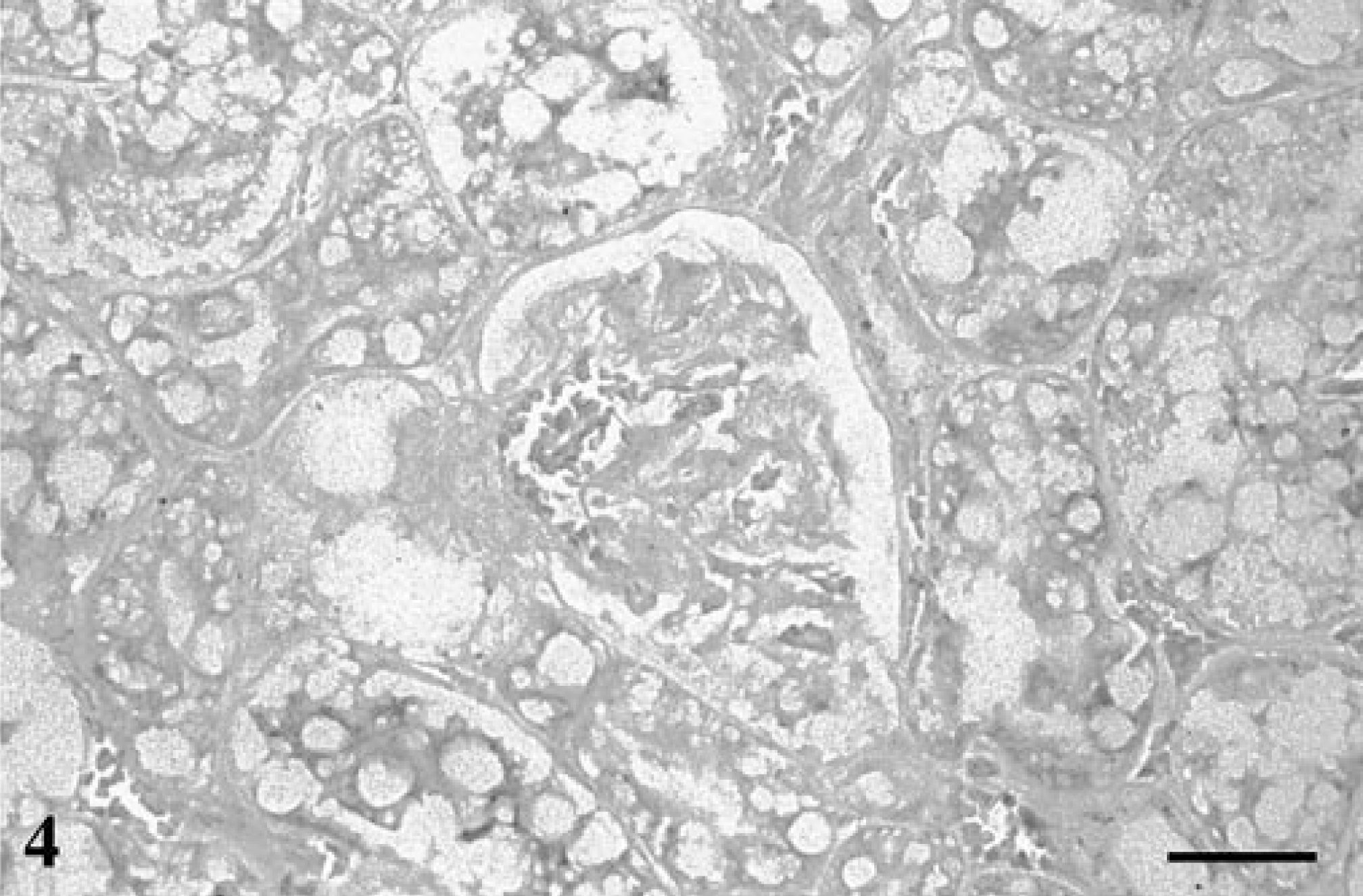
Kidney; noninfected cat. No hybridization signal is seen in the negative control tissues. In situ hybridization, H. felis 16S probe. Biotin-TSA method, NBT/BCIP, xanthene counterstain. Bar = 30 μm.
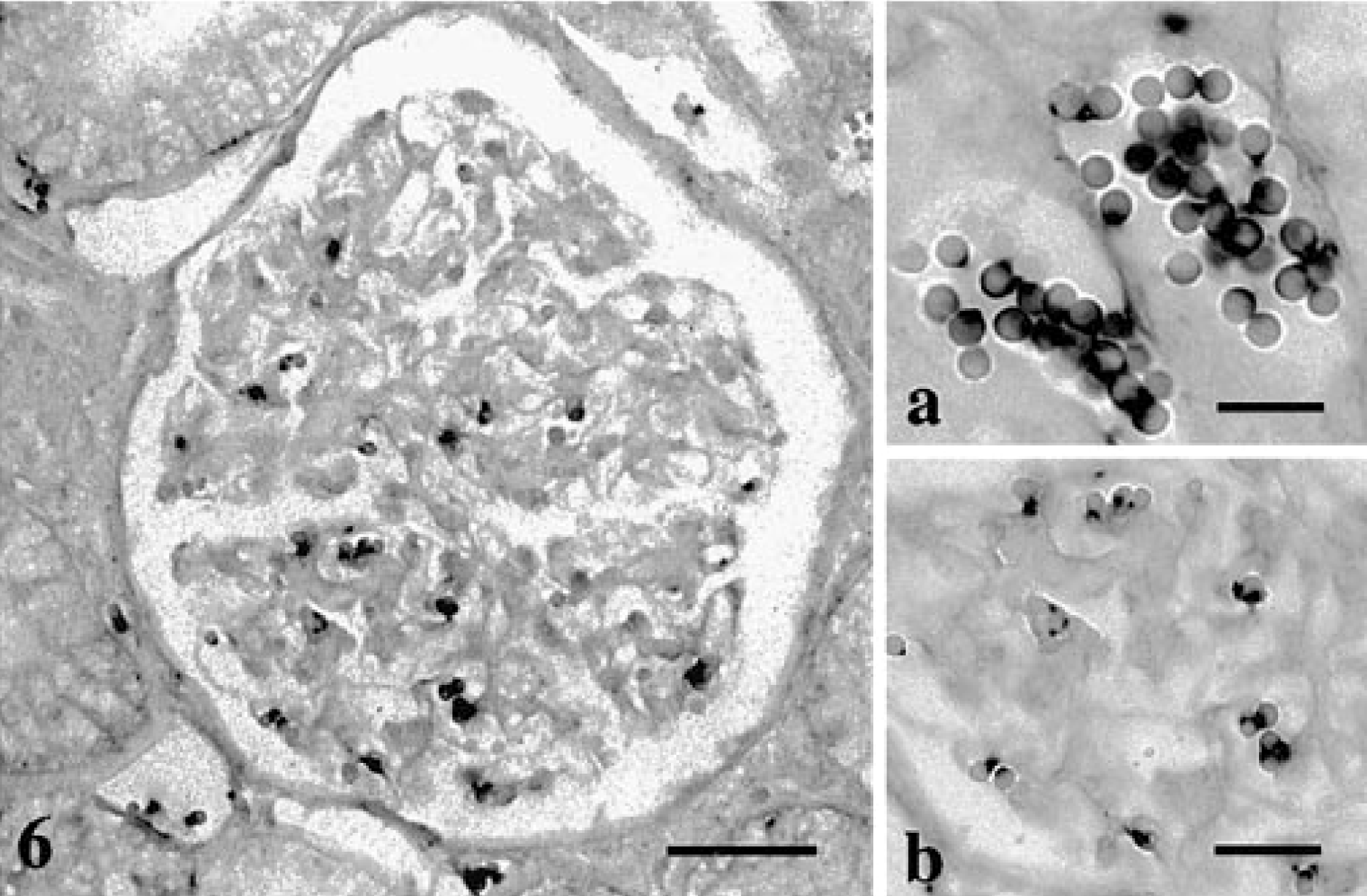
The H. felis 16S probe hybridized to the liver and kidney sections from the H. felis-infected cat and showed a strong dark purple signal on the edges of the red cells. This positive signal was seen within major blood vessels, hepatic sinusoids, and renal glomeruli (Figs. 5, 6). The signals seen in these tissue sections were at a site consistent with the location of the H. felis bacteria as seen in the peripheral blood (Fig. 7). The amplified signal was focal, restricted to red blood cells, and without any visual diffusion. When infected tissues were probed with the H. felis 16S probe but without the tyramide amplification step, faint and inconsistent positive signals were seen on the red cells (data not shown).
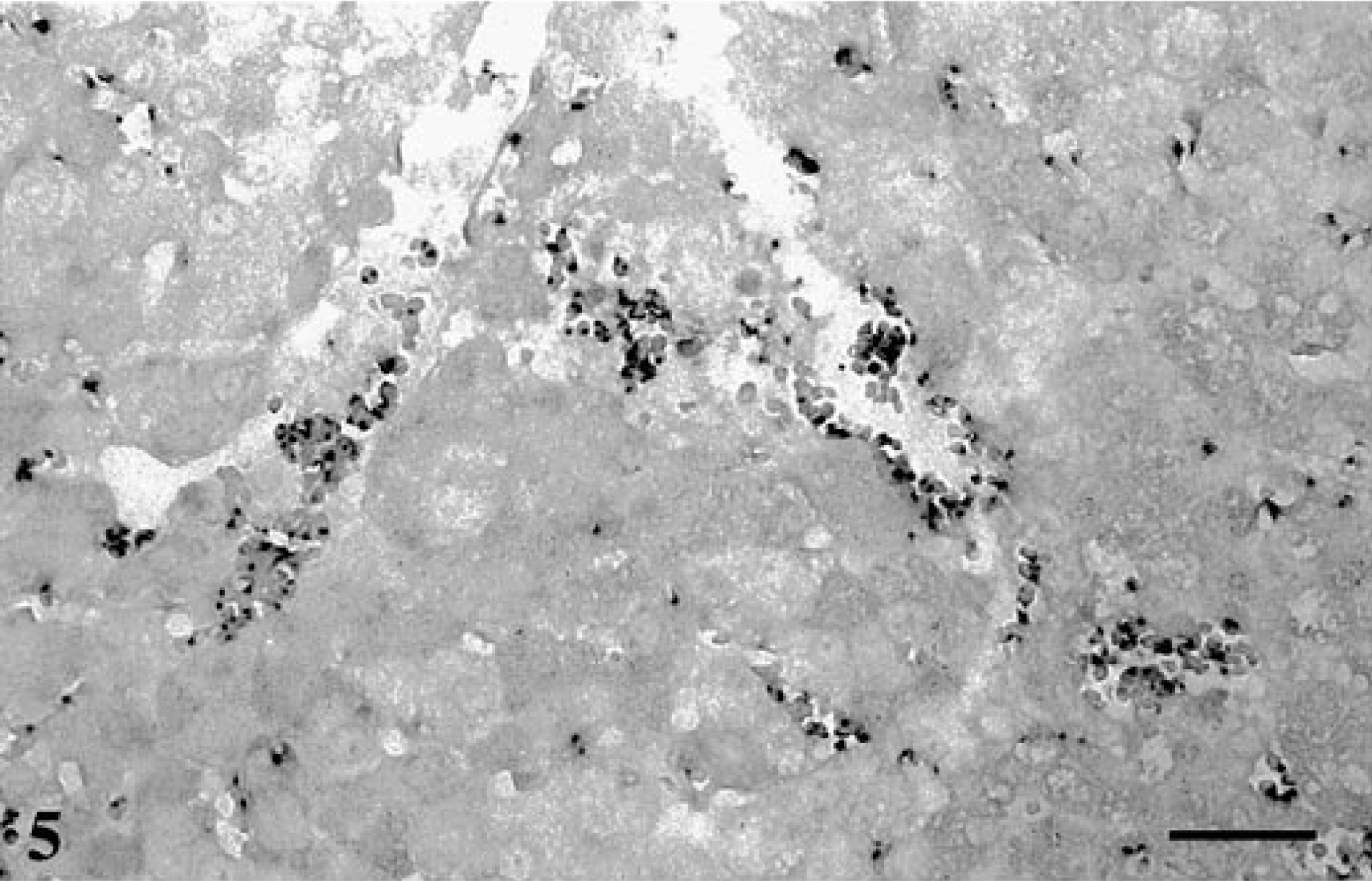
Liver; H. felis-infected cat. A dark hybridization signal is seen on the edge of the red cells indicating the position of H. felis. In situ hybridization, H. felis 16S probe. Biotin-TSA method, NBT/BCIP, xanthene counterstain. Bar = 30 μm.
Kidney; H. felis-infected cat. A dark hybridization signal is seen on the edge of the red cells indicating the position of H. felis. In situ hybridization, H. felis 16S probe. Biotin-TSA method, NBT/BCIP, xanthene counterstain. Bar = 30 μm.

Peripheral blood; H. felis-infected cat. H. felis organisms are seen as small dark dots overlaying and on the periphery of the red cells. Wright's stain. Bar = 10 μm.
Discussion
The ISH of fixed tissue sections has become an increasingly popular tool for localizing pathogenic organisms to their target tissues. This study was the first demonstration of H. felis nucleic acid by ISH in formalin-fixed, paraffin-embedded tissue specimens. Results of this in situ hybridization assay, with a biotin-labeled, double-stranded DNA probe with tyramide signal amplification, localized H. felis to the edge of feline red cells.
The results of an in situ study are reliable only if the proper controls are conducted to rule out false-positive signals. With a biotin-labeled probe, one potential source of false-positive signal is amplification of endogenous biotin.5 As a negative control, H. felis-infected tissues were processed in the absence of the biotinylated probe to determine if the naturally occurring biotin would interfere with the assay. Signal amplification was not seen in these control tissues with the exception of early attempts when the tissues were overdigested (data not shown). This finding is consistent with other studies that demonstrated the endogenous biotin will remain masked unless revealed by antigen retrieval techniques such as microwave heating, pressure cooking, or excessive protein digestion.3,13 Endogenous biotin reactions can also be circumvented by pretreatment with an avidin compound;5 however, this procedure was not needed in this study.
A second potential pitfall in this study is the loss of cellular localization of the amplified product. No diffusion of the signal was visible in this study because the tyramide signal amplification system deposits the biotinylated tyramide only near the site of the probe, and the biotinylated tyramide does not diffuse after forming a strong bond to the proteins adjacent to the probe.16
Nonspecific binding of the DNA probe to the H. felis organism or to the feline tissues is a third potential source of false-positive signals. This nonspecific binding can be avoided by using high stringency washes and assayed for by using an irrelevant or nonbinding probe. The P450 probe used in this study bound weakly to both the infected and noninfected feline blood in the dot blot hybridization experiments. However, no binding of the P450 probe occurred in the in situ experiments. This finding suggests the presence of a sequence in the feline genome to which the probe has some affinity. This affinity was not enough to create a signal in the in situ hybridization experiments. The H. felis 16S probe did not bind to the noninfected feline blood in the dot blot or the in situ experiments, indicating no homology between parts of the feline genome and the H. felis probe. Thus, the signals seen with the H. felis probe were a result of binding to the organism and were not due to nonspecific binding of the probe to parts of the feline genome.
The manufacturer of the TSA kit suggested the inclusion of a control in which the signal amplification step was eliminated and infected tissues were processed as if for conventional ISH. The degree of signal amplification achieved was assessed by comparing the results of the straight ISH with the amplified slides. No signal was expected on the straight ISH slides because the copy number of the 16S rRNA gene of H. felis was below the threshold of detection by this method. However, a faint signal was detected on the edges of some of the red cells. Possibly this signal is the result of binding of the DNA probe to the ribosomal RNA as well as the genomic DNA. Another explanation is that several organisms are in close proximity on the red cell. Regardless, the signal was found in the same location on both the straight in situ slides and the slides processed with the signal amplification and was consistent with the expected location of the organism. The TSA method, however, provided a clearer and more consistent signal than that seen on the nonamplified slides. The large numbers of controls used in this study were needed to ensure that the H. felis organisms were the only structures to give a specific positive signal on the slides.
The specificity of the in situ signal provides the final evidence needed to prove that H. felis is the causative agent of FIA. Previous experiments in this laboratory have shown that H. felis was likely the causative agent of FIA by correlating the presence of a species-specific PCR product to the presence of organisms in the cat.2,6,21 Those experiments fulfilled six of the seven molecular guidelines of establishing microbial disease causation as outlined by Fredricks and Relman.9 However, the sequencing and PCR studies were done with DNA in solution and did not rule out the possibility of infection or contamination with a Mycoplasma species other than H. felis. The final proof of molecular disease causation requires the demonstration of specific in situ hybridization of the microbial sequence to an area where the microorganisms are located. In this study, the chromagenic signal was found only on the red blood cell; this signal was consistent with the location of the bacteria as seen on peripheral blood smears. Thus, these in situ hybridization studies physically linked the 16S rRNA sequence to H. felis in an area of known cellular pathology. This final step proves that FIA is caused by the mycoplasmal organism H. felis.
Footnotes
Acknowledgements
We thank Abbott Laboratories, Division of Drug Safety Evaluation, for financial support for this project. We also thank Drs. James Zachary and Dan Morton, who were of great help in capturing the images for this paper, and Ms. Maria Davey and Mr. Steve Postl of Abbott Laboratories, who provided invaluable technical assistance.