
Abstract
Polysaccharide storage myopathy (PSSM) has been found in more than 35 different horse breeds through identification of abnormal storage of polysaccharide in muscle biopsies. A dominant mutation in the glycogen synthase 1 gene (GYS1) accounts for a substantial proportion of PSSM cases in at least 17 breeds, including Quarter Horses, but some horses diagnosed with PSSM by muscle histopathologic analysis are negative for the mutation. We hypothesized that a second distinct form of glycogen storage disease exists in GYS1 -negative horses with PSSM. The objectives of this study were to compare the histopathologic features, ultrastructure of polysaccharide, signalment, history, and presenting complaints of GYS1 -negative Quarter Horses and related breeds with PSSM to those of GYS1 -positive horses with PSSM. The total histopathologic score in frozen sections of skeletal muscle stained with hematoxylin and eosin, periodic acid Schiff (PAS) and amylase-PAS stains from 53 GYS1-negative horses did not differ from that of 52 GYS1 -positive horses. Abnormal polysaccharide was fine granular or homogenous in appearance (49/53; 92%), often amylase-sensitive (28/53; 53%), more commonly located under the sarcolemma, and consisting of β glycogen particles in GYS1 -negative horses. However, in GYS1 -positive horses, abnormal polysaccharide was usually coarse granular (50/52; 96%), amylase-resistant (51/52; 98%), more commonly cytoplasmic, and consisting of β glycogen particles or, in some myofibers, filamentous material surrounded by β glycogen particles. Retrospective analysis found that GYS1 -negative horses (n = 43) were younger at presentation (4.9 ± 0.6 years vs. 6.7 ± 0.3 years for GYS1 -positive horses) and were more likely to be intact males than GYS1 -positive horses (n = 160). We concluded that 2 forms of PSSM exist and often have distinctive abnormal polysaccharide. However, because evaluation of the histologic appearance of polysaccharide can be subjective and affected by age, the gold standard for diagnosis of PSSM at present would appear to be testing for the GYS1 mutation followed by evaluating muscle biopsy for characteristic abnormal polysaccharide in those horses that are negative for the mutation.
Introduction
Polysaccharide storage myopathy (PSSM) is a prevalent, clinically significant cause of neuromuscular disease in horses.12–14, 18 It has been diagnosed in 35 different horse breeds based on muscle pathology in frozen sections.13 In addition, breeds ranging from mules to thoroughbred racehorses have been diagnosed with equine polysaccharide storage myopathy (EPSM or EPSSM) based largely on pathology in formalin-fixed muscle tissue.13, 25, 26 Our laboratory has recently identified a dominantly inherited mutation in the skeletal muscle glycogen synthase (GYS1) gene that is responsible for a form of PSSM in at least 17 different horse breeds.16 The GYS1 mutation results in a constitutively active glycogen synthase enzyme when assayed on muscle homogenates in vitro. This mutation, if also occurring in muscle in vivo, could explain the accumulation of normal glycogen and a less highly branched amylase-resistant polysaccharide.3 The mutation is present in a large number of breeds of horses with PSSM; however, it clearly does not account for all cases of PSSM. For example, although the prevalence of the GYS1 mutation is particularly high in draft (87%) and Quarter Horse–related breeds (72%) with histopathologic evidence of PSSM, it is lower in warmbloods (18%) and other light horse breeds with histopathologic evidence of PSSM.16
Three possibilities exist to explain the absence of the GYS1 mutation in a proportion of horses with PSSM. The first possibility is that the Arg309His glycogen synthase mutation is not the actual causative mutation for PSSM. However, the region on equine chromosome 10 containing the GYS1 gene was very strongly associated with PSSM in a whole genome scan; it is inherited with PSSM in a very large Quarter Horse kindred; and sequencing of the entire coding region of the GYS1 gene, as well as the 5′ and 3′ untranslated regions, did not identify any other sequence differences between control horses and horses with PSSM.17 Further, genetic association analysis in Quarter Horses with PSSM that do not have the GYS1 mutation has excluded the entire GYS1 chromosomal locus as the cause of abnormal polysaccharide accumulation in this subset of horses with PSSM.17
The second possibility is that many of the horses without the GYS1 mutation in our studies received a false-positive diagnosis of PSSM. Interpretation of the amount and character of polysaccharide in skeletal muscle is somewhat subjective, and at least 2 different diagnostic criteria have been established by different laboratories to diagnose PSSM, indicating a lack of agreement as to what does and does not constitute a histopathologic diagnosis of PSSM.21, 25 The gold standard for diagnosis of PSSM in our laboratory is the presence of periodic acid Schiff (PAS) positive inclusions in type 2A and type 2B muscle fibers, which are resistant to amylase digestion.21 However, alternative diagnostic criteria for the histopathologic diagnosis of PSSM have also been suggested, including increased amylase-sensitive glycogen and sarcoplasmic masses.10, 14, 22, 25 The specificity of these criteria for PSSM has been questioned.1, 11, 13 Adherence to the strict diagnostic criteria of the presence of abnormal polysaccharide that is typically amylase-resistant, compared with alternative criteria of abnormal polysaccharide that is typically amylase-sensitive, results in a higher proportion of GYS1-positive horses with PSSM.13 However, a significant proportion of horses have amylase-resistant PAS-positive inclusions but do not have the GYS1 mutation, so it is unlikely that all histopathologic diagnoses of PSSM are attributable to a false-positive diagnosis.16
The third explanation for the absence of the GYS1 mutation in horses diagnosed with PSSM by histopathologic means is that a population of horses with PSSM have separate, distinct glycogenosis(es) resulting from different molecular and cellular causes.16, 17 If so, further evaluation of PSSM cases in our Neuromuscular Diagnostic Laboratory (NDL) database might allow us to identify unique features associated with the forms of PSSM attributable and not attributable to the GYS1 mutation. The objectives of this study were to contrast the histopathologic and ultrastructural features of muscle biopsies from GYS1-positive horses with PSSM with biopsies from GYS1-negative Quarter Horses and related breeds (Paint and Appaloosa horses; collectively termed QHR) with PSSM and to compare the signalment, history, and presenting complaints in QHR with the 2 forms of PSSM.
Materials and Methods
Selection of cases
Records from the NDL were searched to identify 285 QHR horses diagnosed with PSSM with a severity of grade 2 for which biopsies were available for reevaluation. A diagnosis of grade-2 PSSM was based on the presence of numerous muscle fibers containing large amounts of fine granular cytoplasmic or homogeneous subsarcolemmal glycogen or the presence of fibers with abnormal coarse granular PAS-positive inclusions, which are reported to be typically amylase-resistant.11, 13 Amylase staining was not initially performed on all of the 285 samples. Horses diagnosed with grade-1 PSSM by muscle biopsy were excluded from this study to reduce possibilities of a false-positive histopathologic diagnosis of PSSM. Grade-1 PSSM was defined as muscle biopsies that contained a number of fibers with increased amounts of normal-appearing glycogen, which is typically amylase-sensitive.13
All horses with grade-2 PSSM were genotyped for the Arg309His allele of the GYS1 gene using a restriction fragment length polymorphism assay described previously,17 and genotypes were recorded as homozygous affected P/P, heterozygous affected P/N, or homozygous normal N/N at the GYS1 locus.16 Each horse was genotyped on at least 2 separate occasions to ensure accuracy of genotyping results. Through the genotyping, 75 N/N horses (non-GYS1 PSSM), 197 GYS1 P/N horses, and 13 GYS1 P/P horses were identified.
Histopathologic evaluation
The 75 available muscle biopsies from GYS1-negative (N/N) horses with PSSM were re-sectioned and reevaluated at one time by a single investigator (SJV) blinded to the horse's genotype or previous biopsy grade. Frozen (n = 70) or formalin (n = 5) 10-μm sections from each biopsy were stained with amylase-PAS, PAS, and hematoxylin eosin (HE) stain. For amylase-PAS stains, sections were placed in Carnoy's fixative, digested with 0.35% amylase for 15 minutes at 37°C and then stained with PAS.
In the reevaluation of GYS1-negative muscle biopsies, 22 biopsies were rescored as grade 1 and were excluded from further analysis. Histopathologic features were compared between the 53 remaining GYS1-negative grade-2 horses with PSSM and 52 GYS1-positive P/N (n = 47) or GYS1-positive P/P (n = 5) horses that were selected at random from the total of 210 grade-2 GYS1-positive horses (51 frozen, 1 formalin sections). The scoring system used for 20× magnification was 0 = not present, 1 = present in at least 1 fiber in 1 of 3 random microscopic fields (rmfs), 2 = present in at least 2 fibers in 2 of 3 rmfs, 3 = present in at least 2 fibers in 3 of 3 rmfs. The HE stains were scored for the presence of anguloid atrophy (0–3), necrosis/macrophages (0–3), central nuclei (0–3), subsarcolemmal vacuoles (0–3), and rimmed vacuoles (0–3). In the PAS stain, the amount of abnormal polysaccharide was also scored (0–3). The sum of each of these features was totaled for each biopsy (0–18). The appearance of abnormal polysaccharide was evaluated as coarse granular or fine granular/homogeneous in PAS stains. The amount of abnormal polysaccharide in subsarcolemmal (0–3), and cytoplasmic (0–3) locations was also evaluated but not included in the total score. Amylase-PAS stains were evaluated to determine if the abnormal polysaccharide was amylase-sensitive or amylase-resistant.
To further characterize the histopathologic features of horses with PSSM, 6 biopsies from GYS1-positive and GYS1-negative were selected and stained with Gomori's trichrome, nicotinamide adenine dinucleotide tetrazolium reductase (NADH), oil red O (ORO), adenosine triphosphatase (ATPase, pH 4.6), and acid phosphatase stains.
Electron microscopy
Immediately after obtaining a percutaneous needle biopsy from 2 GYS1-positive horses and 1 GYS1-negative horse, gluteus muscle tissue was trimmed in chilled lactated Ringer's. Muscle fragments (1 mm3) were fixed in 2.5% glutaraldehyde in 0.166 M sodium cacodylate buffer (pH = 7.4) at 4°C and postfixed in 1% osmium tetroxide (OsO4) in 0.166 M sodium cacodylate buffer (pH = 7.4, 300 mOsm) with 2% potassium ferrocyanide (K4Fe[CN]6). Muscles were sequentially dehydrated in increasing concentrations of acetone and infiltrate by resin Epon. Sample were embedded in resin and polymerized at 60°C. Selected blocks were sectioned at 70–80 nm, stained with uranyl acetate and lead citrate, and examined with a GEOL 1200 EXII transmission electron microscope (Japan). Because most thin sections of these samples did not contain fibers with coarse granular polysaccharide, we elected to use samples from paraffin-embedded blocks. A specific area containing numerous PAS-positive course granular inclusions was removed from a paraffin-embedded formalin-fixed tissue block from 4 GYS1-positive horses, 4 GYS1-negative horses and 3 control horses. These samples were sequentially deparaffinated in decreasing concentrations xylol/ethanol and processed for transmission electron microscopy as described previously.
Immunohistochemistry
To further characterize the abnormal filamentous material identified by electron microscopy, formalin-fixed sections from the control and GYS1-positive horses used for electron microscopy as well as an additional formalin-fixed sample from a GYS1-negative horse were stained for desmin and myoglobin. Briefly, paraffin-embedded tissues were deparaffinized. For desmin, antigens were unmasked using the heat-induced epitope retrieval method. Tissues were subsequently incubated in 3% H2O2 to block endogenous peroxidase. Nonspecific binding sites were blocked with normal goat serum. Samples were incubated with either mouse anti-human desmin (DakoCytomation) or rabbit anti-human myoglobin (DakoCytomation) antibodies. For the detection of primary antibodies, tissues were incubated with EnVision+/HRP Goat anti-Mouse IgG (DakoCytomation) and EnVision+/HRP goat anti-rabbit IgG (DakoCytomation). Chromogen substrate was then used for detection (Dako). All tissues were counterstained with Mayer's hematoxylin (Dako).
Retrospective analysis of phenotype
History and physical examination findings provided on the submission forms were available for 48/53 GYS1-negative horses with PSSM and 182/210 GYS1-positive horses with PSSM. Age, breed, gender, serum creatine kinase (CK) and aspartate aminotransferase (AST) activities were recorded, and the occurrences of exertional rhabdomyolysis (tying-up), muscle atrophy, gait abnormality, and muscle fasciculations were tabulated. Horses with concurrent GYS1 and ryanodine receptor (RYR1) mutations2 were excluded from analysis because the RYR1 mutation affects severity of PSSM clinical signs.15
Statistical analysis
Distribution of age, histopathologic criteria scores, and total biopsy scores were tested for normality using the Kolmogorov-Smirnov normality test with the Dallal-Wilkinson-Lillefor P value. These distributions were non-normally distributed, and significant differences were determined using the Mann-Whitney U-test. Logarithmically transformed CK and AST values and mean age between groups were compared using an unpaired t-test. Contingency tables of clinical history and physical examination findings were compared using a Fischer's exact test. Contingency tables of gender and breed distribution were compared using a chi-square test of independence. For all analyses P < .05 was considered significant.
Results
Histopathology
The total histopathologic score for all the GYS1-negative grade-2 PSSM muscle biopsies and individual scores for atrophy, myonecrosis, and centrally located nuclei were not significantly different from all the GYS1-positive grade-2 PSSM muscle biopsies (Table 1). Subsarcolemmal vacuoles were either devoid of stain or contained lightly eosinophilic material in HE stains (horizontal arrow, Fig. 1). Rimmed vacuoles had a basophilic rim and contained eosinophilic material in HE (vertical arrows, Fig. 1). The rim of these vacuoles was red in Gomori trichrome stain and the contents were intensely PAS-positive in amylase-PAS stains. Subsarcolemmal and rimmed vacuoles were less prevalent in GYS1-negative PSSM compared to GYS1-positive PSSM horse muscle (Table 1). There were no apparent abnormalities in muscle fiber type distribution, NADH staining, lipid storage, and lysosomal acid phosphatase staining in special stains performed on GYS1-negative horses.
Mean and total scores for individual histopathologic criteria and mean scores for glycogen cellular location in GYS1-positive compared to GYS1-negative cases.
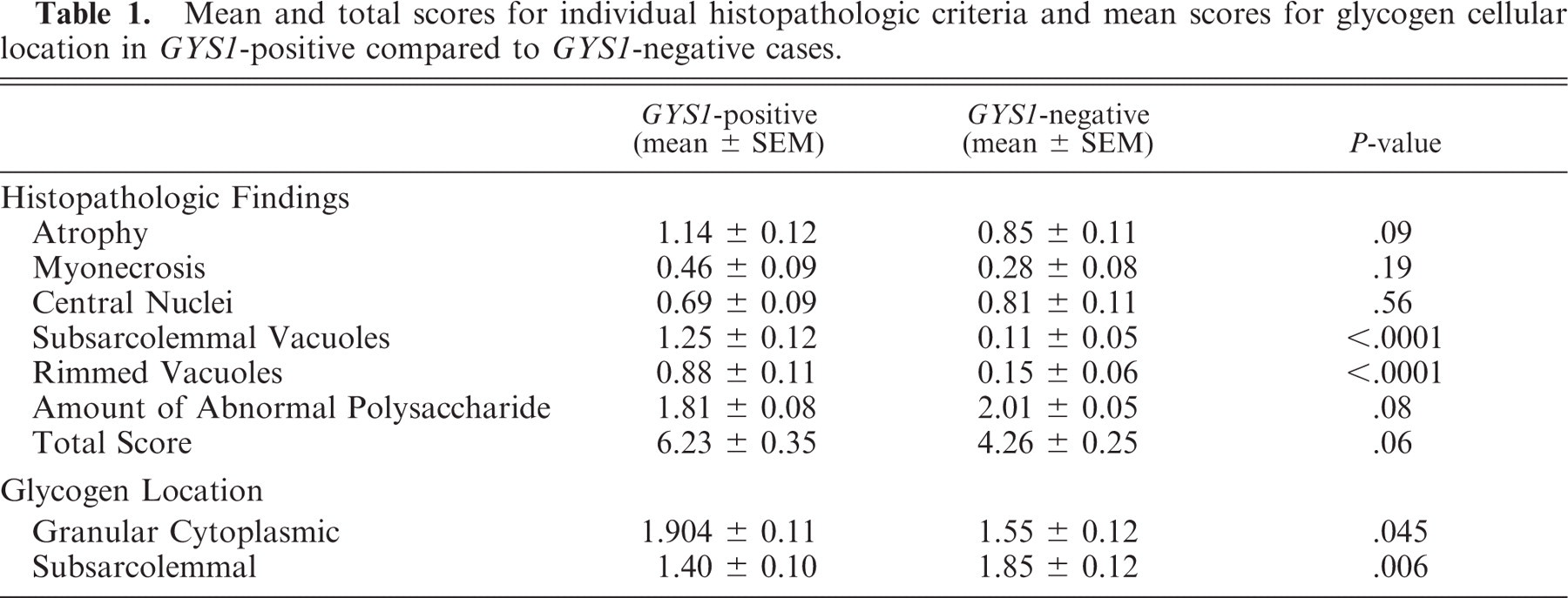
A cross-section of frozen semimembranosus muscle from a GYS1-positive horse stained with HE. Horizontal arrow shows a subsarcolemmal vacuole that likely represents a lake of glycogen accumulation. Vertical arrows indicate rimmed vacuoles. These usually contain amylase-resistant polysaccharide and may represent activated lysosomes. Bar = 50 μm.
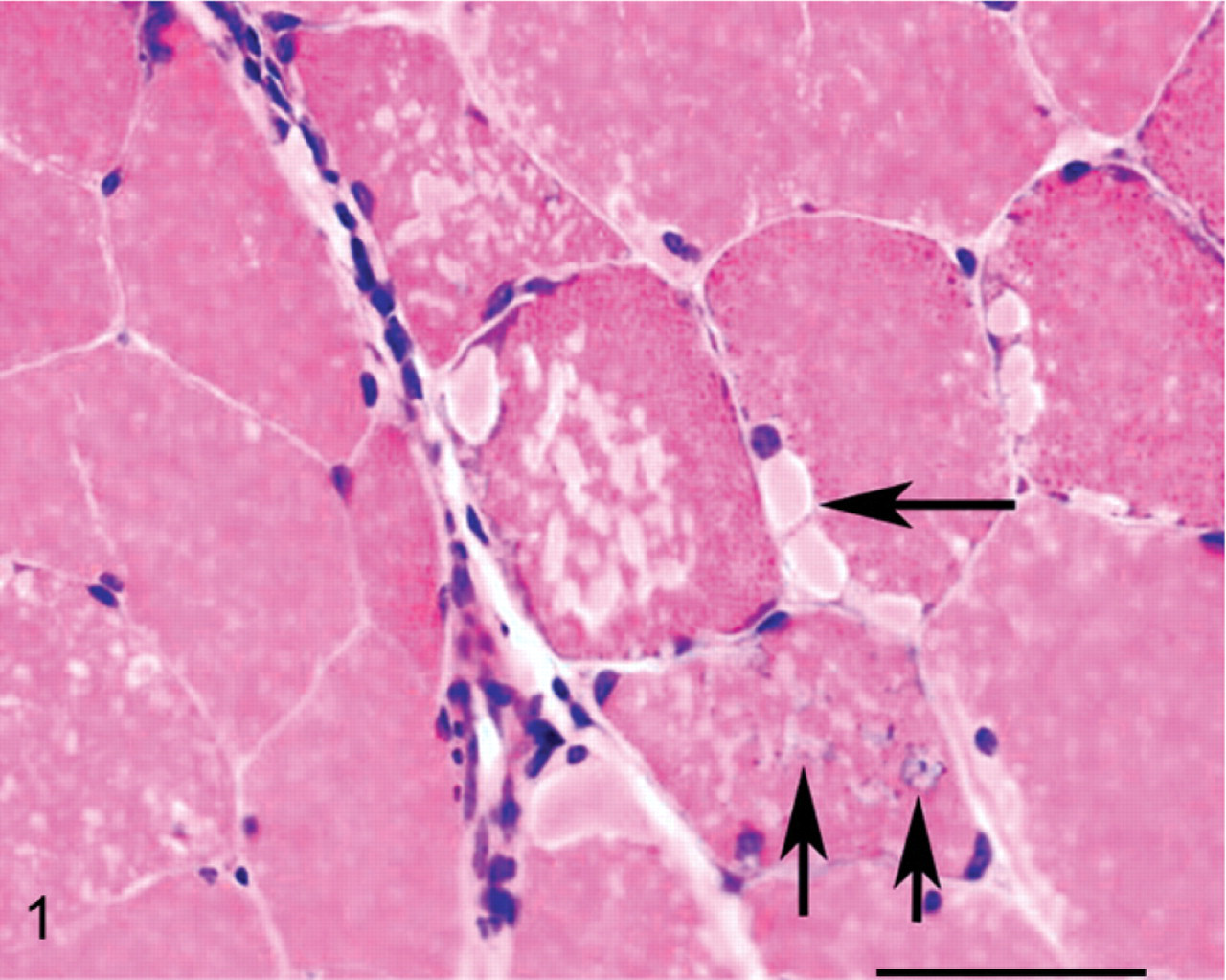
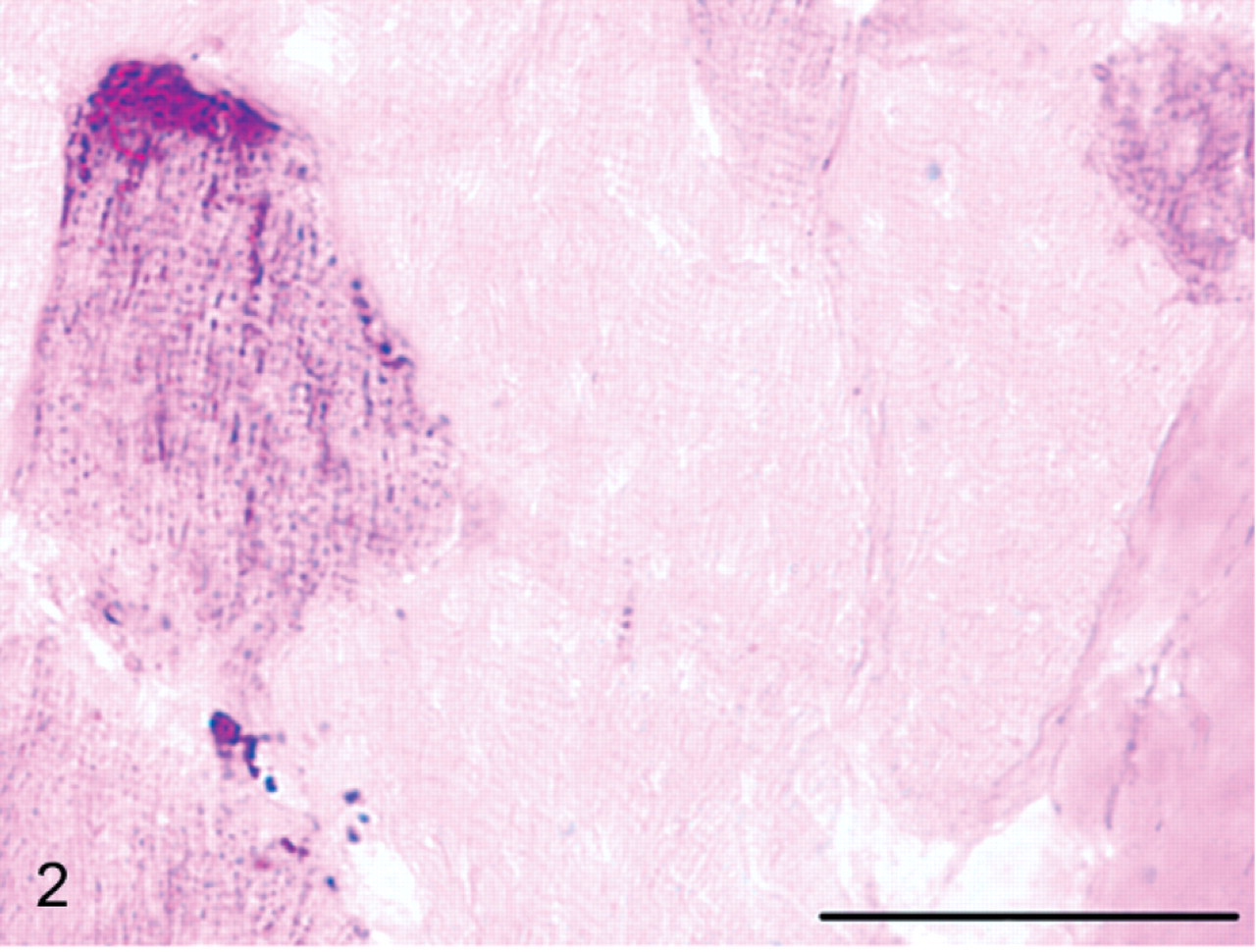
The histopathologic features of grade-2 muscle biopsies that appeared to most clearly differentiate the 2 types of PSSM were the appearance and location of abnormal polysaccharide (Tables 1, 2). Abnormal polysaccharide in GYS1-negative horses with PSSM was often characterized as having a fine granular/homogeneous appearance (92%) (Fig. 2) rather than containing coarse granular abnormal polysaccharide (8%) (Fig. 3). There were significantly greater numbers of muscle biopsies from GYS1-positive horses with PSSM that contained the coarse granular type of abnormal polysaccharide (96%) compared to GYS1-negative horses with PSSM (Table 2). Of the 2 GYS1-positive PSSM biopsies not exclusively in the coarse granular category, one had both coarse granular and fine granular/homogeneous polysaccharide whereas the remaining biopsy was classified as having fine granular/homogeneous-appearing abnormal polysaccharide. The score for the amount of abnormal polysaccharide did not differ significantly between GYS1-positive horses with PSSM and GYS1-negative horses with PSSM (Table 1).
Appearance and amylase resistance of glycogen inclusions in GYS1-positive compared to GYS1-negative cases.

A cross-section of frozen semimembranosus muscle fibers from a horse with GYS1-negative PSSM stained with PAS showing fine granular abnormal polysaccharide in the cytoplasm and accumulating under the sarcolemma. Bar = 50 μm.
A cross-section of frozen semimembranosus muscle from a GYS1-positive PSSM horse stained with PAS showing the coarse granular appearance of abnormal polysaccharide in the cytoplasm. Bar = 50 μm.
The location of the polysaccharide within the cell also differed between GYS1-negative PSSM horse muscle and GYS1-positive PSSM horse muscle. Large amounts of abnormal polysaccharide were more commonly found under the subsarcolemmal of muscle fibers in GYS1-negative PSSM horse muscle (Fig. 4a) than in GYS1-positive PSSM muscle (Fig. 5a). Aggregates of granular cytoplasmic polysaccharide were more common in muscle biopsies of GYS1-positive horses with PSSM compared to GYS1-negative PSSM muscle biopsies (Table 1).
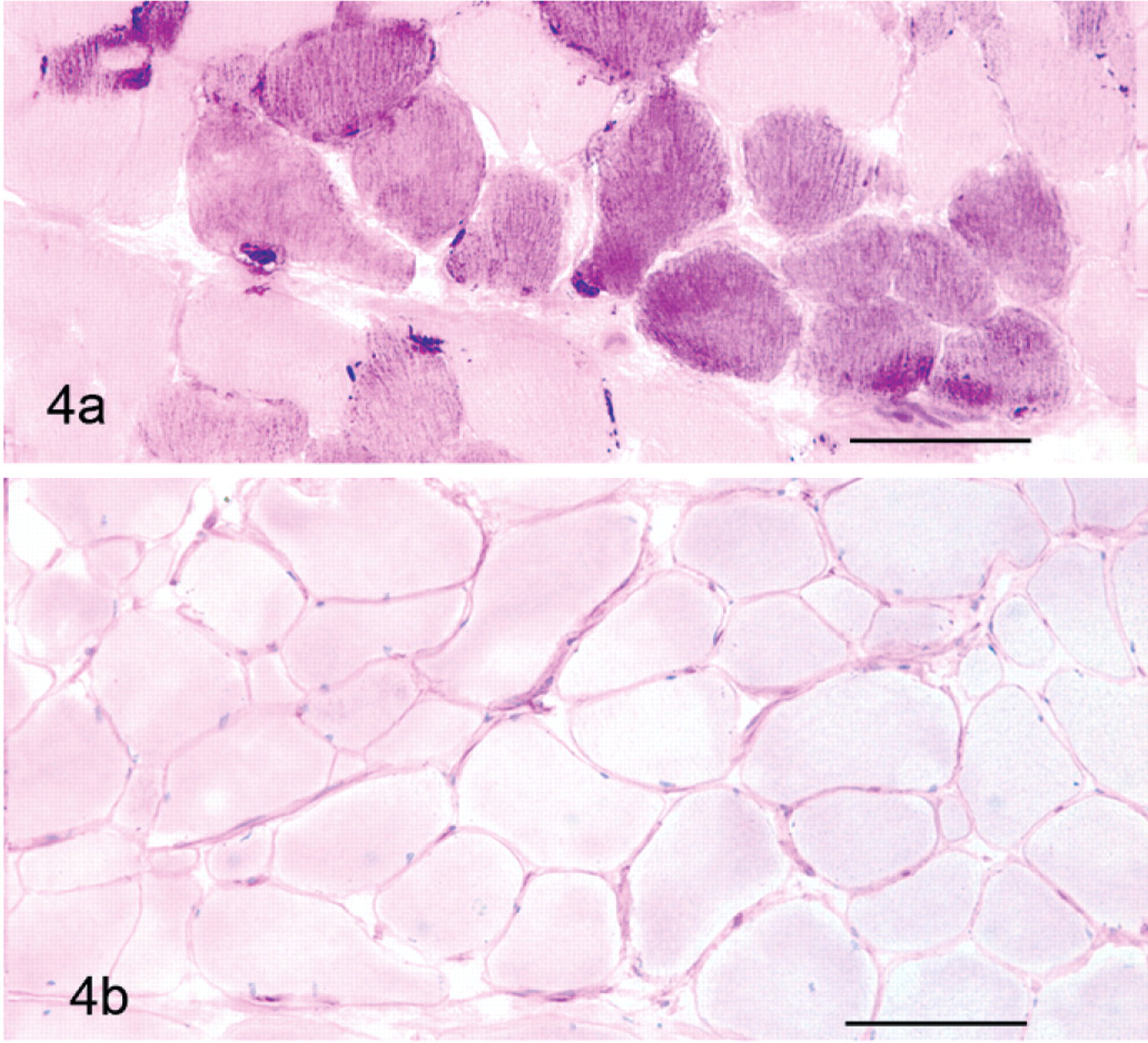
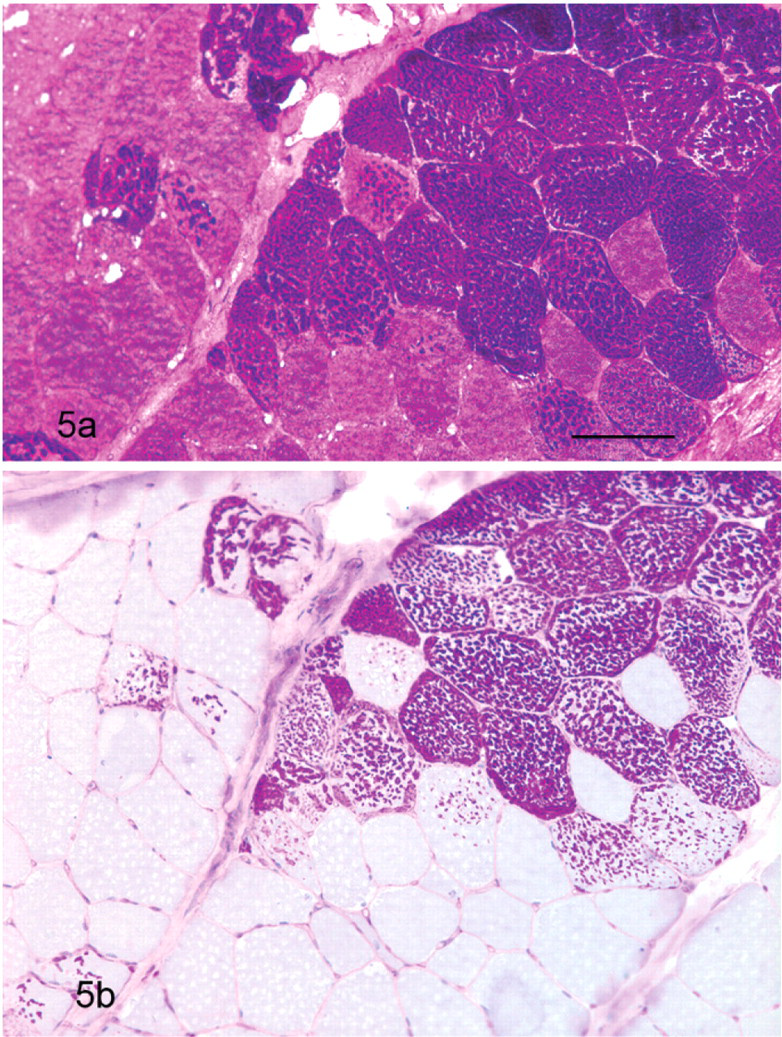
The fine granular/homogeneous abnormal polysaccharide in GYS1-negative PSSM horse muscle biopsies was more likely to be amylase-sensitive (Fig. 4b) than the abnormal polysaccharide found in GYS1-positive horses (Table 2, Fig. 5b). In contrast, the abnormal polysaccharide in GYS1-positive PSSM muscle was largely amylase-resistant (Table 2). The single GYS1-positive PSSM biopsy with non–amylase-resistant polysaccharide was from a 5-month-old foal that may have been too young to begin to accumulate amylase resistant polysaccharide.7
Electron microscopy
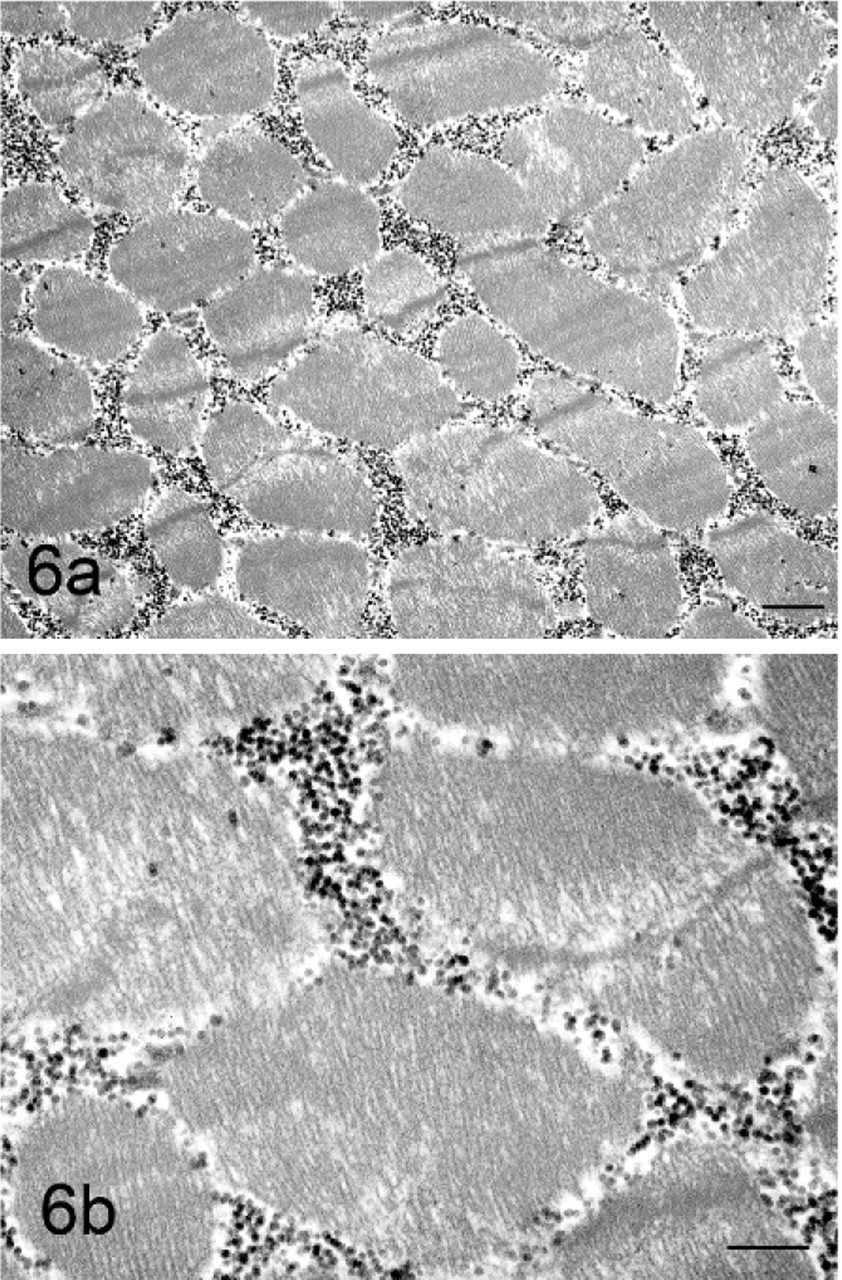
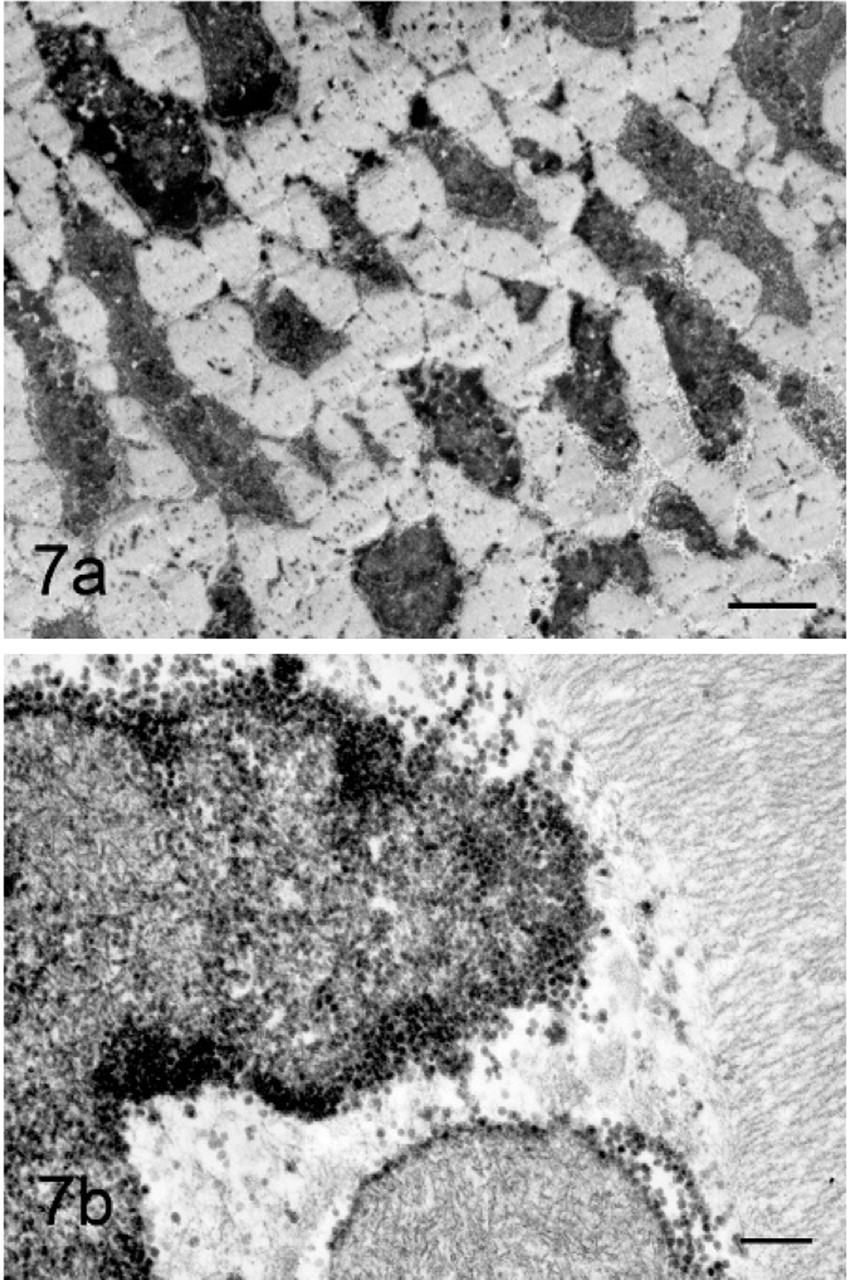
Myofibers from both GYS1-negative and GYS1-positive horses contained large amounts of β glycogen particles in both cytoplasmic and subsarcolemmal locations (Figs. 6a, 7a, 8a, b). Based on distribution or structure of β glycogen particles, it was not possible to distinguish between the 2 forms of PSSM in the initial freshly obtained muscle samples. When areas known to contain course granular amylase-resistant polysaccharide were evaluated in a GYS1-positive horse, distinctive polyglucosan bodies were identified (Fig. 7a). These bodies consisted of a filamentous material surrounded by β glycogen particles (Figs. 7b, 9).

Immunohistochemistry
Desmin staining was minimal in control horses (Fig. 10b) and GYS1-negative horses (Fig. 12b), but strong staining for desmin was apparent in regions containing the abnormal coarse granular polysaccharide (Figs. 11a, b). Myoglobin staining of control horses (Fig. 10c) and GYS1-negative horses (Fig. 12c) showed a mosaic pattern likely attributable to the variable myoglobin content of slow or fast twitch muscle fibers. Myofibers from GYS1-positive horses that contained coarse granular polysaccharide showed colocalization of myoglobin with the abnormal polysaccharide (Figs. 11a, c).
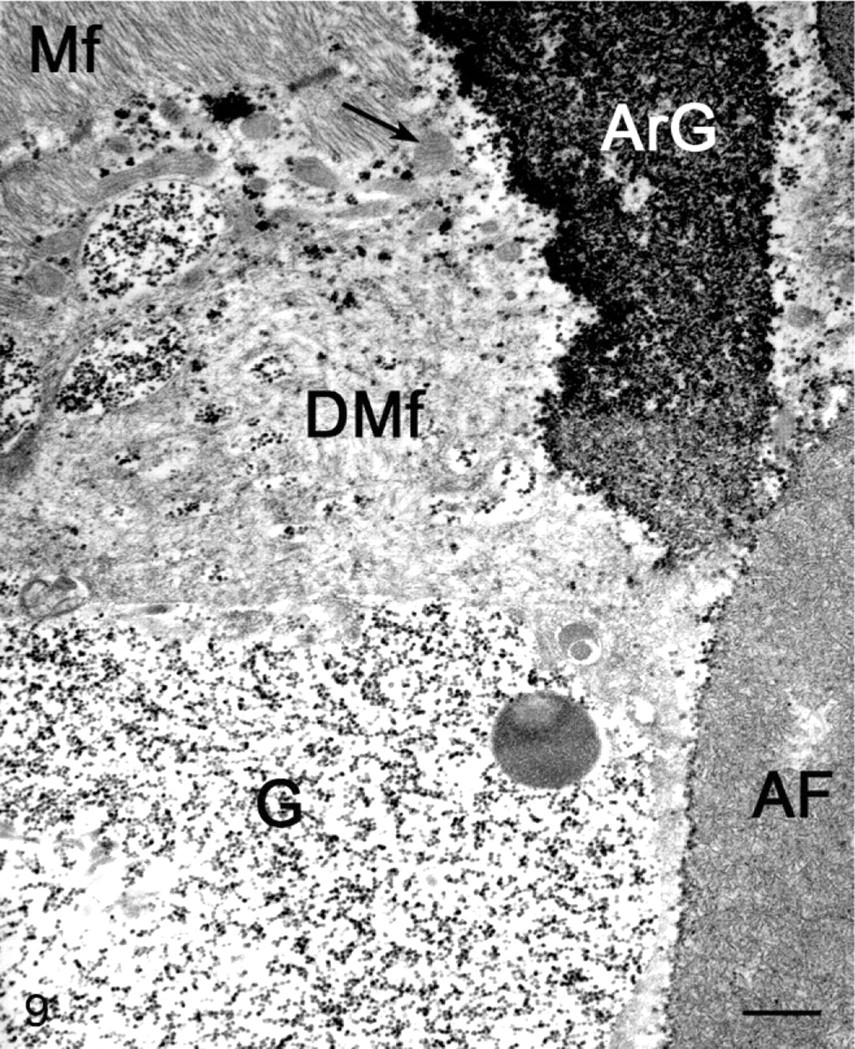
Electron micrograph of polyglucosan body from a GYS1-positive horse. Note normal myofibrils (Mf), degenerated myofibrils (DMf), free loose β glycogen particles (G), polyglucosan-like bodies that contain filamentous material associated with amylase resistant glycogen (ArG), and dense areas of filamentous material (Af). Bar = 200 nm.
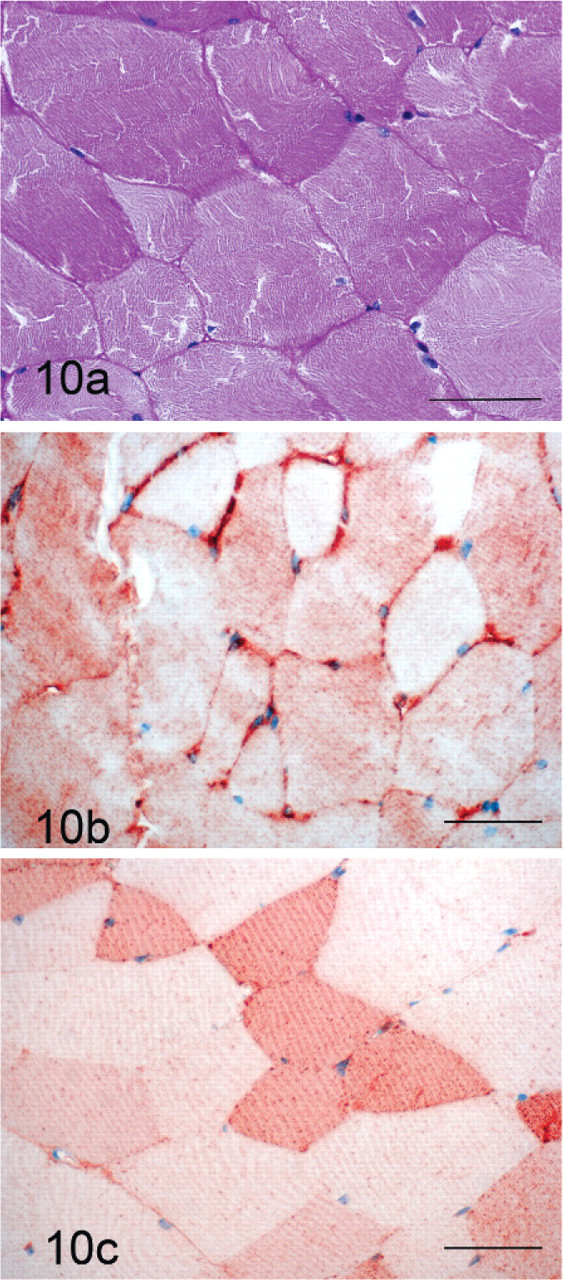
Control horse formalin-fixed muscle in cross-section stained with PAS (a), immunohistochemical staining for desmin (b) and myoglobin (c). Bar = 50 μm.
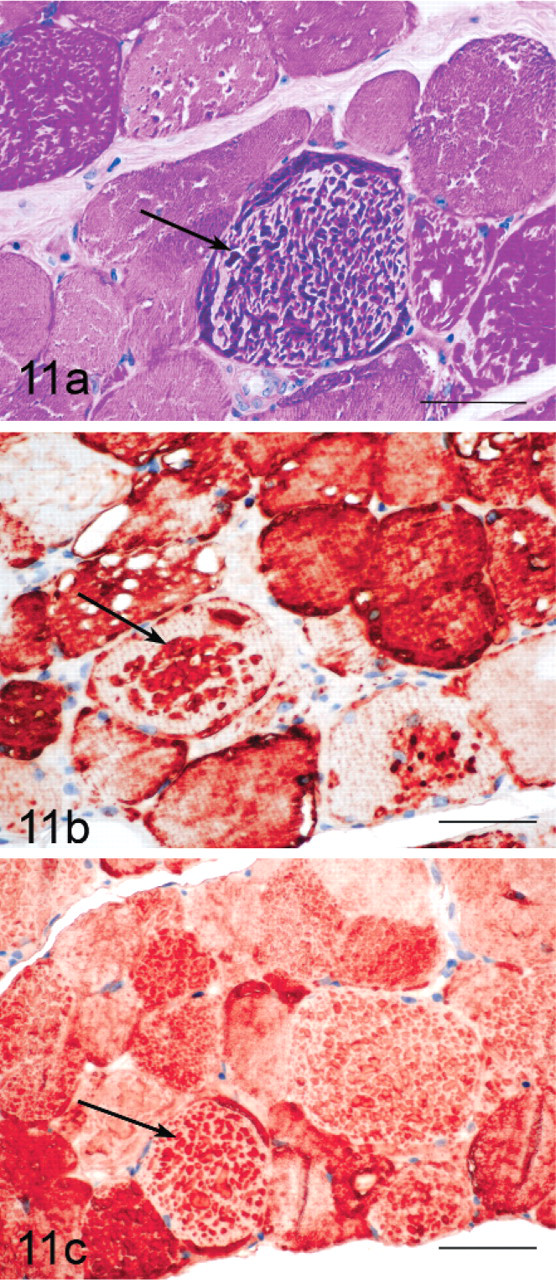
GYS1-positive horse formalin-fixed muscle in cross-section stained with PAS (a), showing abnormal course granular cytoplasmic inclusions that stain intensely for desmin (b) and myoglobin (c) in immunohistochemical stains. Bar = 50 μm.
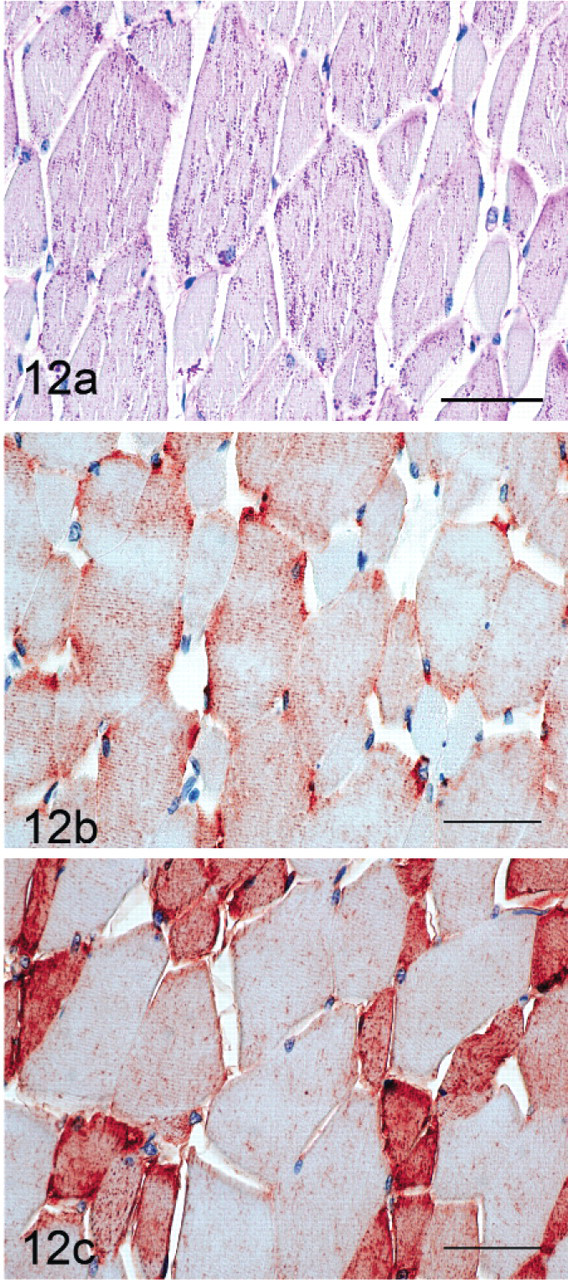
GYS1-negative formalin-fixed muscle in cross-section stained with PAS (a) showing fine granular cytoplasmic inclusions that do not stain for desmin (b) or myoglobin (c). Bar = 50 μm.
Retrospective analysis
Breed distribution was not significantly different between GYS1-negative horses with PSSM (43 Quarter Horses, 9 Paint horses, and 1 Appaloosa) and GYS1-positive horses with PSSM (145 Quarter Horses, 25 Paint horses, and 12 Appaloosas). Mean age at time of biopsy was significantly lower in GYS1-negative horses with PSSM (4.9 ± 0.6 years, range = <1–17 years) compared to GYS1-positive horses with PSSM (6.7 ± 0.3 years, range = <1–22 years, P = .005). In particular, there was a high proportion of GYS1-negative horses with PSSM that were younger than 1 year old (Fig. 13). There was a significantly higher proportion of intact male GYS1-negative horses with PSSM (24 females, 21 geldings, and 7 intact males, P = 0.02) compared to GYS1-positive horses with PSSM (94 females, 76 geldings, and 6 intact males).
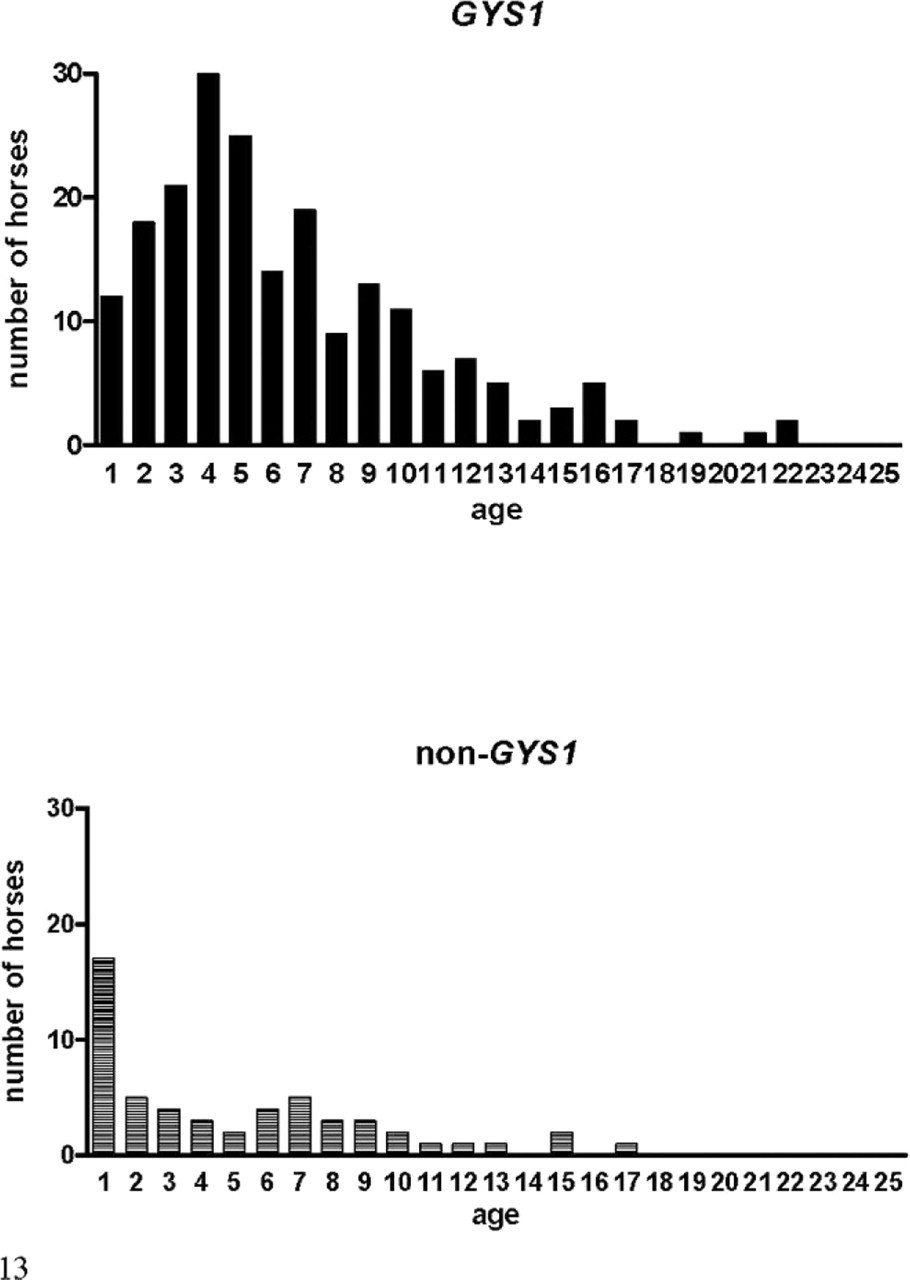
Histograms of the age distribution at time of biopsy for GYS1-positive and GYS1-negative horses. Mean age was significantly younger for GYS1-negative horses.
Clinical history was provided for 160 GYS1-positive horses with PSSM and 43 of the GYS1-negative horses. There was no difference in the reported incidence of exertional rhabdomyolysis, muscle atrophy, or muscle fasciculations between GYS1-negative horses with PSSM and GYS1-positive horses with PSSM. GYS1-negative horses with PSSM were significantly more likely to have reported a history of gait abnormality compared to GYS1-positive horses with PSSM (Table 3). However, the type of gait abnormality reported did not differ between groups (Table 3). Serum CK and AST activities were not significantly different between groups (CK: 37,738 ± 11,115 U/L and 42,085 ± 18,669 U/L, P = .66; and AST: 4,207 ± 668 U/L and 7,439 ± 2,054 U/L, P = .98, for GYS1-positive and GYS1-negative horses, respectively).
Reported clinical findings in GYS1-positive compared to GYS1-negative cases.
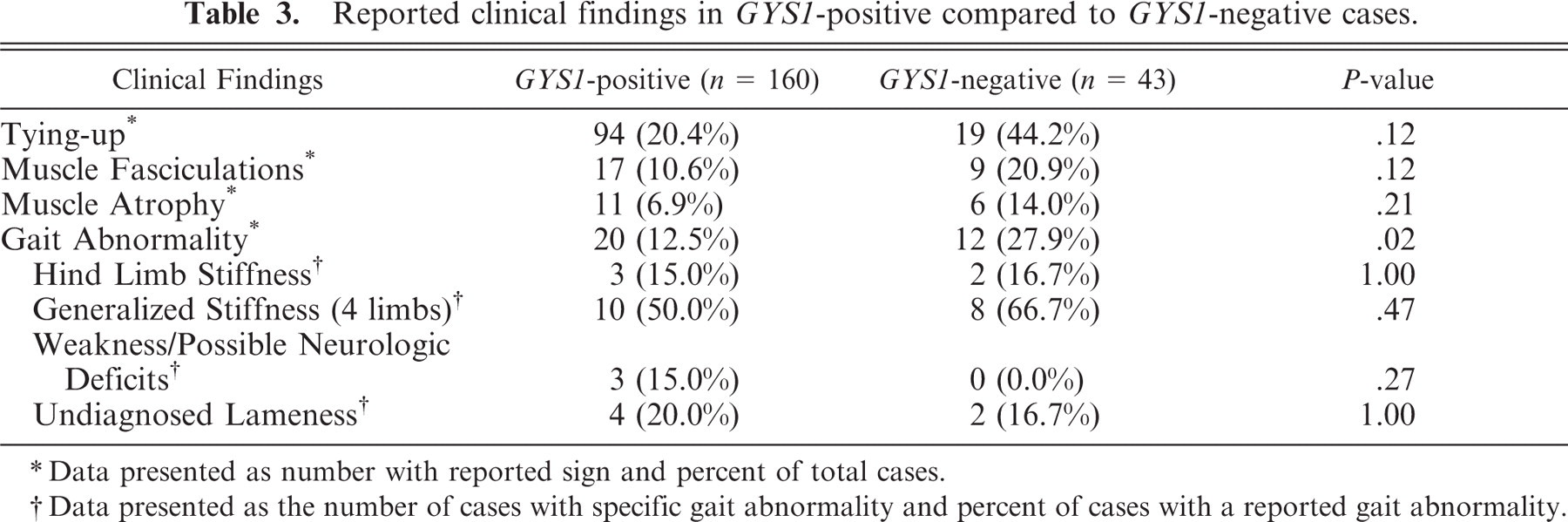
Data presented as number with reported sign and percent of total cases.
Data presented as the number of cases with specific gait abnormality and percent of cases with a reported gait abnormality.
Discussion
The results of this study indicate that at least 2 forms of PSSM exist in Quarter Horses and related breeds that have some characteristic features evident in PAS and amylase-PAS stains of muscle biopsies. While the amount of abnormal polysaccharide did not differ between GYS1-positive and GYS1-negative horses, the type of abnormal polysaccharide differed. The abnormal polysaccharide in GYS1-positive PSSM biopsies typically had an amylase-resistant coarse granular appearance, with large aggregates most often found in the cytoplasm. Ultrastructural evaluation showed that in GYS1-positive horses, many muscle fibers had increased β glycogen particles and, in a lesser number of fibers, course granular PAS-positive material contained polyglucosan bodies. Polyglucosan bodies consist of β glycogen particles, less highly branched glycogen, and a variety of protein filaments, including myoglobin, desmin, and ubiquitin.19, 23, 24 Polyglucosan bodies are found in specific glycogenoses, such as phosphofructokinase (PFK) and AMP-kinase enzyme and glycogen branching enzyme deficiency, which are characterized by glycogen with a less highly branched structure.4, 6 In contrast, the abnormal polysaccharide in GYS1-negative PSSM biopsies often had a fine granular or homogeneous amylase-sensitive appearance, commonly accumulated under the sarcolemma, and appeared to consist of β glycogen particles that were not membrane bound. Increased non–membrane-bound glycogen without polyglucosan body accumulation is characteristic of glycogenoses such as myophosphorylase deficiency, early stages of PFK deficiency, and phosphoglycerate mutase and kinase deficiencies,9 which do not affect the ratio of branched-to-unbranched glycogen.
A diagnosis of PSSM could not be established from HE stains,11 although subsarcolemmal and rimmed vacuoles were more common in HE stains of GYS1-positive muscle biopsies. Subsarcolemmal vacuoles likely represented large lakes of glycogen accumulation, which were visualized in electron micrographs. Serial sections showed that rimmed vacuoles, which are apparent in frozen but not formalin-fixed11 sections, contained amylase-resistant polysaccharide. Rimmed vacuoles may represent a complex of abnormal (degenerated) fibrils and proteins, such as desmin- and ubiquitin-containing amylase-resistant polysaccharide. Accumulation of ubiquitin may tag these bodies as abnormal material ready for lysosomal degradation.23
In the present study, only horses with PSSM diagnosed with grade-2 accumulation of polysaccharide were used. There are, however, numerous horses diagnosed with PSSM based on grade-1 criteria of increased amylase-sensitive glycogen.13, 22 It is important to recognize that these diagnostic criteria are subjective, and their use decreases the specificity and increases the sensitivity of a diagnosis of GYS1-positive (type 1)11 and likely GYS1-negative (type 2) PSSM. Subjectivity is due in part to the fact that PAS stains for polysaccharide are affected by tissue handling before freezing,5 trauma to specimen,5 formalin fixation,5 thickness of sections,5 low specificity of sarcoplasmic masses for PSSM,1, 11 and higher glycogen content in athletic horse muscle.20 Thus, to ensure an accurate diagnosis, horses with a gait abnormality and grade-1 PSSM should also be thoroughly evaluated for other causes of lameness or muscle disease.
The most accurate means of diagnosing glycogenoses is to identify the molecular basis for the myopathy whenever possible. The discovery of the genetic mutation in the GYS1 gene now provides a simple means to diagnose GYS1 PSSM by DNA analysis of blood, hair root samples, or fresh or frozen tissue samples as well as on portions of paraffinized tissue (http://www.cvm.umn.edu/umec/lab/home.html). Genetic testing for PSSM is recommended, particularly in those 17 breeds where the genetic mutation has been identified.16 However, for horses with clinical signs of a muscle disorder that are negative for the GYS1 mutation, muscle biopsy diagnosis of PSSM will still be necessary. To readily distinguish the 2 forms of PSSM we propose that the term “type-1 PSSM” be used for horses that possess the GYS1 mutation and “type-2 PSSM” be used to distinguish the group of horses with abnormal polysaccharide in muscle biopsies that do not have the GYS1 mutation.16
Both forms of PSSM occurred in young horses, but it is of particular note that a high percentage of horses with type-2 PSSM, 32% (17/53), were younger than 1 year old. In contrast, the mean age of GYS1-positive horses with PSSM was greater than GYS1-negative PSSM, and only 6.6% (12/182) of type-1 horses with PSSM were younger than 1 year old. Age is an important consideration in attempting to diagnose PSSM by muscle biopsy because horses younger than 2 years old may not yet have any accumulation of abnormal polysaccharide within myofibers, resulting in a false-negative diagnosis.7 Therefore, to avoid false-negative histopathologic diagnoses of PSSM in foals with rhabdomyolysis, genetic testing should be done whenever possible.
The overrepresentation of intact males with GYS1 PSSM compared to type-1 PSSM may also have been affected by age in that young horses would likely not yet be castrated and therefore would be recorded as stallions rather than geldings. Of the 7 intact males in the GYS1-negative group, 6 were younger than 2 years old, 4 were younger than 1 year old (mean age of GYS1-negative intact males was 1.5 ± 0.6 years), and it is likely that most of these males would eventually be castrated. In contrast, all of the type-1 intact males were 2 years old or older, an age where most horses have been gelded (mean age was 4.6 ± 1.5 years).
The only significant difference in clinical signs between the 2 forms of PSSM in our Quarter Horse populations was that GYS1-negative horses with PSSM more commonly presented with an obscure or undiagnosed gait abnormality compared to GYS1-positive horses with PSSM. The similarity in clinical presentation between the two forms of PSSM is consistent with findings in other species. There are 11 described inherited glycogenoses that affect skeletal muscle in humans8, 9, 27 and cause rhabdomyolysis, pain with exertion, muscle cramping, and myoglobinuria or fixed, progressive weakness.8, 9 These clinical characteristics are shared by GYS1-positive and GYS1-negative horses with PSSM. Although glycogenoses described in other species are inherited, a more thorough investigation of pedigrees, clinical history, clinical findings and muscle biochemistry should help to determine if there is a familial basis for GYS1-negative PSSM and what the major distinguishing clinical and biochemical characteristics are for this disorder.
In conclusion, there appears to be 2 forms of PSSM in Quarter Horse–related breeds that have some characteristic features in muscle histopathology. Abnormal polysaccharide in GYS1 (type 1) PSSM is often amylase-resistant and coarse granular in appearance and is commonly located in the cytoplasm whereas in GYS1-negative (type 2) PSSM it is often amylase-sensitive, fine granular/homogeneous in appearance, and commonly located under the sarcolemma. Due to some subjectivity in evaluating the appearance of polysaccharide in muscle sections and the absence of abnormal polysaccharide in young horses with PSSM, the gold standard for diagnosis of PSSM at present would appear to be testing for the GYS1 mutation (type 1) followed by evaluation of muscle biopsy for abnormal polysaccharide in those horses that are negative for the mutation (type 2).
Footnotes
Acknowledgements
The authors would like to thank Don Ariyakumar for electron microscopy preparations. Funding was provided by the Morris Animal Foundation grants D07EQ-041 and D07EQ-402 (ME McCue, salary support) and the American Quarter Horse Association grant “Genetic Analysis of Glycogen Storage Disorders in Quarter Horses.” All work was done at the University Of Minnesota College Of Veterinary Medicine.
Conflict of interest: Drs. McCue, Mickelson, and Valberg own the patent for PSSM genetic testing. A portion of the profit from testing goes to their ongoing research and patent royalties.