
Abstract
Abstract
Purpose
Chondrosarcomas typically present in adults during the fifth to seventh decades and are rare in young patients. The biological behaviour and oncological outcomes may be different in children and adolescents.
Methods
We retrospectively evaluated the outcomes of all patients with chondrosarcoma of bone who were younger than 18 years of age at the time of diagnosis and were treated at our centre between 1995 and 2018.
Results
The 15 consecutive patients studied included nine male and six female cases, with a mean age at diagnosis of 13 years (7 to 17). The median follow-up was 117 months (30 to 277). The tumours were primary and secondary in ten and five patients, respectively. The tumours were central in 13 and surface in two patients. The tumour locations were the humerus in five, digits in five, femur in three, radius in one and pelvis in one patient. The histological grades were grade I in seven, grade II in seven and grade III in one patient. The surgical treatments were limb salvage in ten patients and ray amputation in five patients. The surgical margins were wide in eight, marginal in two and intralesional in five patients. All the patients were alive and continuously free of disease at the time of the last follow-up. No patient developed metastases or local recurrence.
Conclusion
Chondrosarcoma of bone in children and adolescent patients has a very good prognosis and is less aggressive compared with published outcomes in older patients.
Level of evidence
IV
Introduction
Chondrosarcoma is a cartilage-forming malignant bone tumour. It is the second most common primary malignant bone tumour following osteosarcoma. It often occurs in middle to old age, with a peak incidence in the fifth to seventh decades of life.1,2
Due to the rarity of chondrosarcoma in children and adolescents, there is a scarcity of studies that have described the outcome of this tumour in children.3–8 Thus, the biological behaviour of chondrosarcomas in young patients is unclear. In some of these studies, the survival rates of paediatric chondrosarcoma were reportedly worse than in adults; 3 in contrast, other studies have reported equivalent survival4,5 and improved survival in young patients compared with adults. 6 However, none of these studies have concentrated on the outcome of chondrosarcoma in young patients with open and growing physis.
We hypothesized that chondrosarcomas occurring in young patients with open and growing physis may have different characteristics and may have a less biologically aggressive course compared with those occurring after closure of growth plates, i.e. skeletal maturity. The present study was, therefore, aimed at reporting the clinical outcome of conventional chondrosarcoma of bone in patients younger than 18 years old at the time of diagnosis.
Patients and methods
A retrospective review was conducted of all patients who had been diagnosed with chondrosarcoma between 1st January 1995 and 31st December 2018 at a single tertiary sarcoma centre. We identified 15 patients younger than 18 years of age at the time of diagnosis with a histologically confirmed diagnosis of conventional chondrosarcoma. We excluded patients with variants of chondrosarcoma such as mesenchymal chondrosarcoma and extraskeletal lesions and also excluded patients with chondrosarcomas of the skull, facial bones, and head and neck regions. The patients presented either with pain or enlargement of a previously observed incidental lesion. Surgeries were conducted only when the lesions showed high-grade malignancy by biopsy or low-grade malignancy with rapid progression with or without pain in a few months. In contrast, the unchanged lesions in terms of size which showed some cellular atypia were considered as a non-malignant lesion such as enchondroma and the lesions were carefully followed up and not considered chondrosarcoma. This study was approved by our institutional review board and all data were collected from the clinical records and imaging systems as part of routine patient follow-up.
The demographic information reviewed included age, gender, predisposition to secondary chondrosarcoma, tumour locations, histological grades, biopsy types, surgical treatment types, surgical margins and tumour stages. The diagnosis of chondrosarcoma in this study was made based on comprehensive review of the clinical, radiological and histopathological findings, not histology alone, by a specialist supra-regional sarcoma multidisciplinary board. The biopsy results, treatment plans and definite diagnosis of surgical specimens were made at the sarcoma multidisciplinary meetings with participating paediatric oncologists, radiologists, bone sarcoma pathologists and oncological orthopaedic surgeons. Tumour staging was determined with the use of the Enneking surgical staging system for bone sarcomas. 9
Histological grades were divided into three groups (grades I, II and III). 2 The histological grades assigned to the tumours were the highest grades seen on the histology, even when the higher-grade cells comprised a focal region or a small number of cells.
The survival analyses were performed according to the Kaplan-Meier method and log-rank test using SPSS version 26.0 (IBM, Armonk, New York). Event-free survival was defined as lasting from the date of the initial surgery to the date of the surgery for a subsequent chondrosarcoma. Overall survival was defined as lasting from the date of the surgery to the date of the last follow-up. The difference between the survival curves was evaluated by Cox proportional hazard modelling.
Results
In all, 15 consecutive patients with conventional chondrosarcomas were studied, including nine male and six female cases (Table 1). The mean age at the time of diagnosis was 13 years (7 to 17). Primary and secondary chondrosarcomas occurred in ten (67%) and five (33%) patients, respectively. Secondary chondrosarcomas arose from Ollier's disease in four patients and Maffucci's syndrome in one patient. The tumours were central in 13 (87%) and surface in two patients (13%). The tumour location was in the humerus in five (33%), digits in five (one metacarpal and four phalangeal bone; 33%), femur in three (20%), radius in one (7%) and pelvis in one patient (7%).
Patient details
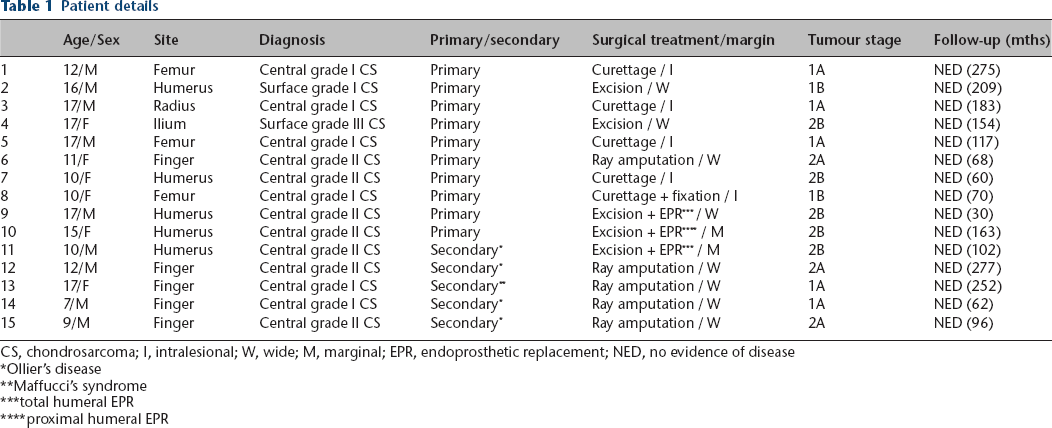
CS, chondrosarcoma; I, intralesional; W, wide; M, marginal; EPR, endoprosthetic replacement; NED, no evidence of disease
Ollier's disease
Maffucci's syndrome
total humeral EPR
proximal humeral EPR
All the patients had histological diagnoses of conventional chondrosarcomas. Preoperative biopsy was performed in ten patients, including open biopsy in four and needle biopsy in six. Histological diagnoses were made in three patients after curettage and digital amputations in two patients with radiologically suspected chondrosarcomas (one had Ollier's and the other had Maffucci's syndrome).
The surgical treatment involved limb salvage in ten patients and ray amputation in five patients. In patients who underwent limb salvage, tumours were treated with curettage in five patients, including four patients with grade I and one patient with grade II chondrosarcoma; excision alone in two patients with grade I and III chondrosarcoma; and excision followed by endoprosthetic reconstruction in three patients with grade II chondrosarcoma. In the ten patients who underwent
Event-free survival at five years was 100% in primary chondrosarcoma and 60% in secondary chondrosarcoma due to the development of further chondrosarcoma at other sites in patients with Ollier's disease or Mafucci's syndrome (Fig. 1). Patient with Ollier's disease encountered a subsequent chondrosarcoma at another finger ten months after the initial operation, and another patient with Maffucci's syndrome developed a subsequent chondrosarcoma at distal radius and below elbow amputation was performed at 19 months after the initial surgery.
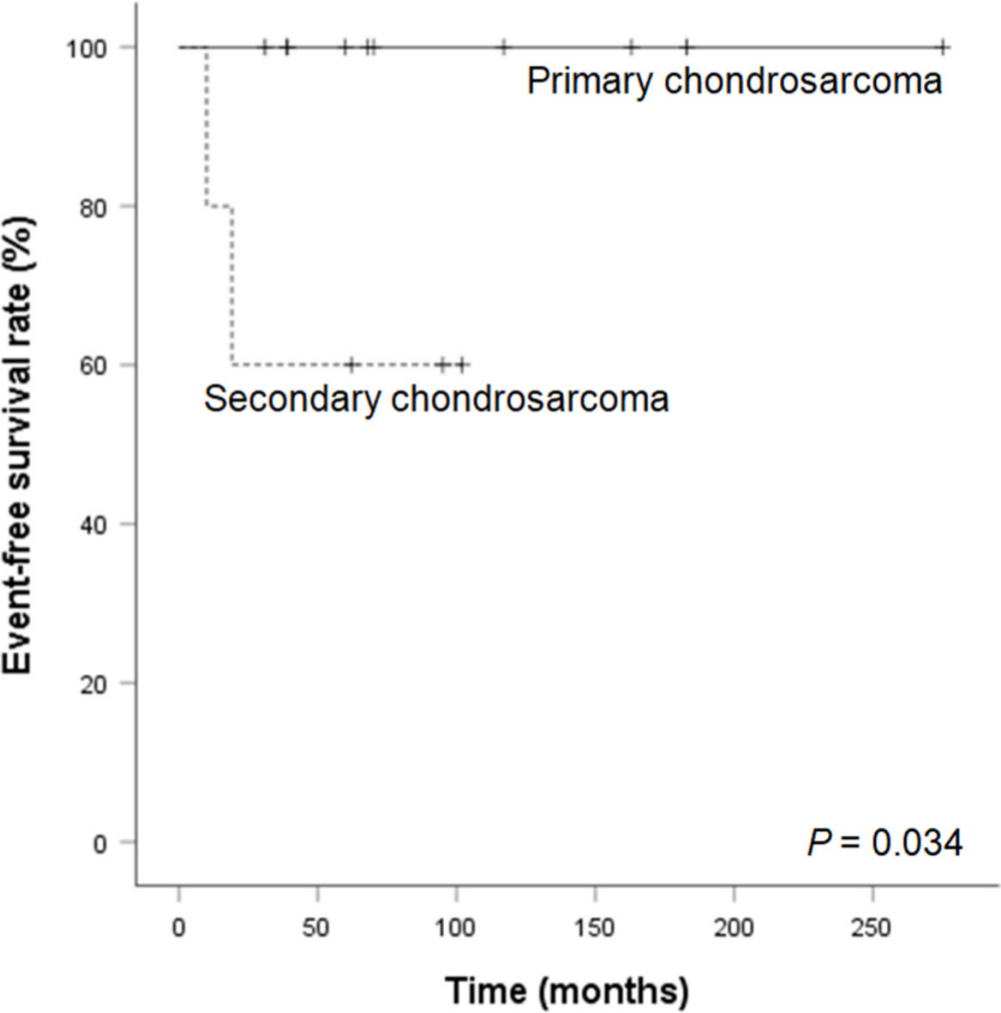
Kaplan-Meier curve for event-free survival.
Although one patient was lost to follow-up after 30 months, 14 patients were followed up for more than five years with a median follow-up of 117 months (30 to 277). All the patients were alive and continuously free of disease at the time of the last follow-up. No patient developed metastases or local recurrence.
Discussion
Chondrosarcoma is a disease often associated with middle-aged and older patients.1,10 It is extremely rare in young patients, with < 7% of all chondrosarcomas occurring in patients younger than 21 years of age at the time of diagnosis.1,4,5,11 In addition, chondrosarcomas are even more rare in children, with a reported incidence of only 0.9% to 1.6% of chondrosarcomas occurring in patients younger than 18 years of age.4,5 The 15 patients studied in this series represent only 2.5% of 598 patients treated for chondrosarcoma at our centre over the period of the study. Therefore, there is a paucity of literature on the biological behaviour and prognosis of this tumour in children, with only a few papers having been published.3–8
Several of the published papers extend over many decades, over which the diagnosis of bone tumours has significantly evolved, or have included several variants of chondrosarcomas and/or have also included extra-skeletal or head and neck chondrosarcomas, which may have a different pattern of behaviour and prognosis. For example, Aprin et al 7 retrospectively reviewed 12 paediatric and adolescent chondrosarcoma patients (aged from six to 20 years) who were treated at the Children's Hospital Medical Center in Boston from 1957 to 1980, while Huvos and Marcove 3 described 79 patients with chondrosarcoma under 21 years of age who were diagnosed and treated at the Memorial Hospital for Cancer and Allied Diseases in New York over a period of 54 years, but Huvos's study was not directly comparable as it included several histological variants (20 mesenchymal chondrosarcomas, eight myxoid chondrosarcomas, five spindle cell chondrosarcomas and one clear cell chondrosarcoma) and two extra-skeletal lesions. Similarly, Wu et al 6 reported a large series of patients with chondrosarcomas who were aged from 0 to 18 years, but most of the tumours were extra-skeletal or head and neck chondrosarcomas.
There are three recent and comparable studies (Table 2).4,5,8 In the series of the Mayo clinic and their consultation files, Young et al 8 retrospectively reviewed 47 patients with conventional chondrosarcoma less than 17 years of age. In a report from the Rizzoli Institute, Gambarotti et al 4 retrospectively reviewed 17 patients with chondrosarcoma under 18 years of age from 1981 to 2014, while Puri et al 5 reported 11 adolescent chondrosarcoma cases (aged from 14 to 21 years) who were operated on at the Tata Memorial Hospital from January 2006 to December 2016.
Previous reports of children and adolescent chondrosarcoma
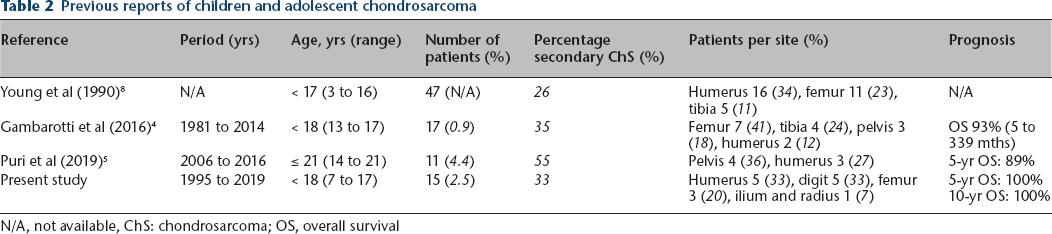
N/A, not available, ChS: chondrosarcoma; OS, overall survival
Secondary chondrosarcomas make up a larger proportion of all chondrosarcomas in children and adolescents compared with adults.3–5,7,8 Patients with hereditary multiple exostosis, Ollier's disease and Maffucci's syndrome have a higher incidence of secondary chondrosarcomas, and these secondary chondrosarcomas generally present at a younger age than do those in the general population, as confirmed in our series. 12 In our series, five of 15 cases (33%) arose from pre-existing enchondroma lesions (Ollier's disease in four and Maffucci's syndrome in one case).
In general, the pelvis is the most common site of chondrosarcoma, followed by the femur and shoulder girdle.11–15 The pattern of skeletal distribution in paediatric chondrosarcoma patients, however, might be different that in adults.4,6,8 In our series, only one case (7%) occurred in the pelvis, and the most common sites were the humerus (33%) and digits (33%). This is similar to the series of Young et al 8 (humerus (34%), femur (23%) and tibia (11%)) and that of Gambarotti et al 4 (pelvic lesion (18%), femur (41%) and tibia (24%)). This discrepancy in tumour location might be one of the reasons for better oncological outcomes in paediatrics than those of adults.
Histological grade has been shown to be a significant predictor of both patient survival and the risk of local recurrence.16,17 Accurate pathological evaluation in grading is, therefore, crucial. Histopathological grading, however, remains difficult, with the possibility of misdiagnosis.14,18 Previous studies have described a relatively high discrepancy in grading (43% to 59%) between biopsy specimens and surgically excised specimens.19,20 This is similar in our series, with a discrepancy between grades noted on biopsy and excised specimens in 42% of patients.
Since chondrosarcomas are generally resistant to both chemotherapy and radiotherapy, surgery remains the mainstay for treatment.10,17 The recommended treatment for intermediate- and high-grade chondrosarcomas is
Huvos and Marcove 3 reported worse survival in paediatric chondrosarcoma than in adult chondrosarcoma. This could be attributed to the study design, which included high-grade chondrosarcoma variants such as mesenchymal chondrosarcoma. Mesenchymal chondrosarcoma is a highly malignant lesion with a poorer prognosis than conventional chondrosarcoma.22,23 In addition, Young et al 8 found that chondroblastic osteosarcoma might have been included as chondrosarcoma in Huvos and Marcove's series. Several authors, therefore, disagreed with Huvos and Marcove's conclusion.4–6,8 In a series of 27 paediatric patients with chondrosarcoma, Young et al 8 reported that five of the eight patients (63%) who underwent curettage and two of the seven patients (29%) who received local excision experienced recurrence but 12 chondrosarcoma patients resected with adequate margins showed no recurrence. Gambarotti et al 4 and Puri et al 5 also showed that grade II chondrosarcoma cases with involved margins had no recurrence more than six years after the surgery. None of the patients in our series with intralesional, marginal or wide margins have developed local recurrence. These data may indicate that paediatric chondrosarcoma has better outcomes than adult chondrosarcoma.
Puri et al 5 reported 89% overall survival and disease-free survival at five years for all patients with chondrosarcomas who were 21 years of age or younger. In our series, both the five-year and ten-year overall survival rates for all patients were 100%; event-free survival at five years was 100% in primary chondrosarcoma and 60% in secondary chondrosarcoma.
Despite the limited number of patients in this and other studies, our study indicates that the prognosis of young patients who develop chondrosarcoma is far better than that of adults.
There are some limitations to this study. First, this is a retrospective and single institutional study. Second, the sample size is small for the purpose of statistical analysis. We did not include other chondrosarcoma variants, such as mesenchymal and dedifferentiated chondrosarcoma or adult patients older than 18 years of age in this study. This study aimed purely to investigate chondrosarcoma in children and adolescent patients.
In conclusion, chondrosarcoma in young patients is rare and has different tumour location characteristics compared with chondrosarcoma in adult patients. Also, chondrosarcomas of the bone in children and adolescent patients has a good prognosis compared with adult chondrosarcomas.
Footnotes
TF: Conception and design, Acquisition of the data, Analysis and interpretation of the data, Drafting of the article, Critical revision of the article for important intellectual content, Final approval of the article.
YT: Conception and design, Acquisition of the data, Analysis and interpretation of the data, Drafting of the article, Final approval of the article.
SY: Conception and design, Acquisition of the data, Analysis and interpretation of the data, Drafting of the article, Final approval of the article.
JDS: Conception and design, Drafting of the article, Critical revision of the article for important intellectual content, Final approval of the article.
AA: Conception and design, Analysis and interpretation of the data, Drafting of the article, Critical revision of the article for important intellectual content, Final approval of the article.