
Abstract
KIDAR syndrome (AP1B1-deficiency syndrome) is an ultra-rare autosomal recessive disorder characterized by ichthyosis, sensorineural deafness, and developmental delay, with fewer than 15 molecularly confirmed cases reported worldwide. We describe a 2.5-year-old Palestinian boy with a homozygous frameshift variant in AP1B1 (p.Leu166TrpfsTer38) who presented with the classic phenotype in addition to persistent vomiting and severe enteropathy, hyperparathyroidism, subclinical hypothyroidism, and progressive elevations in hepatic transaminases. His presentation overlaps with previously reported cases while further illustrating the persistence and complexity of gastrointestinal and endocrine involvement in KIDAR syndrome. This case underscores the importance of early genetic confirmation, coordinated multidisciplinary care, and continued case aggregation to refine the phenotype and guide management of this ultra-rare disorder.
Introduction
KIDAR syndrome (Keratitis–Ichthyosis–Deafness with Annular Erythroderma and Reticulated Hyperkeratosis), also known as AP1B1-deficiency syndrome or MEDNIK-like syndrome, is an ultra-rare autosomal recessive genodermatosis caused by biallelic loss-of-function mutations in the AP1B1 gene.1,2 This gene encodes the β1 subunit of the adaptor protein complex AP-1, which is essential for intracellular trafficking, including the localization of copper-transporting ATPases.1,3 Both homozygous and compound heterozygous AP1B1 variants have been reported.2,4,5 To date, fewer than a dozen molecularly confirmed cases have been reported worldwide.3,6
The syndrome presents with a core triad of ichthyosis, sensorineural hearing loss, and developmental delay, frequently accompanied by enteropathy, elevated liver enzymes, and variable endocrinopathies. Due to its extreme rarity, the full phenotypic spectrum is still being defined.
Diagnosis is confirmed by molecular genetic testing demonstrating biallelic pathogenic AP1B1 variants, often via exome sequencing or targeted gene panels in the context of the characteristic phenotype.1,2,4,6 Management is supportive and multidisciplinary, as no disease-specific targeted therapy is currently established, focusing on skin care (emollients/keratolytics), nutritional rehabilitation and treatment of enteropathy, audiologic interventions, and surveillance/management of hepatic, hematologic, and endocrine complications.1,3,6 Prognosis appears variable but frequently involves persistent developmental delay and chronic multisystem morbidity; early genetic confirmation can streamline evaluation, guide anticipatory monitoring, and inform genetic counseling.1,3,6
Here, we report a Palestinian male with a homozygous frameshift variant in AP1B1 who exhibits multisystem involvement consistent with, and expanding upon, previous reports. Notably, his persistent enteropathy, progressive dermatologic findings, and multiple endocrine abnormalities underscore the syndrome’s systemic nature and provide new insights into its clinical course.
Case Presentation
The patient is a 2.5-year-old boy born to first-cousin parents after a pregnancy complicated by suspected partial placental abruption. He was delivered prematurely at 32 weeks and 2 days of gestation (birth weight 2.3 kg). In the neonatal period, he required intubation and mechanical ventilation for respiratory distress syndrome and received surfactant and caffeine. His early course was complicated by 2 episodes of neonatal seizures, and neuroimaging demonstrated bilateral grade 1 intraventricular hemorrhage. Echocardiography showed a mild-to-moderate patent foramen ovale. Due to poor oral intake and failure to thrive, nasogastric tube feeding was initiated at 4 months of age.
Key clinical events and investigations are summarized in Table 1.
Timeline of Key Clinical Events, Investigations, and Management.
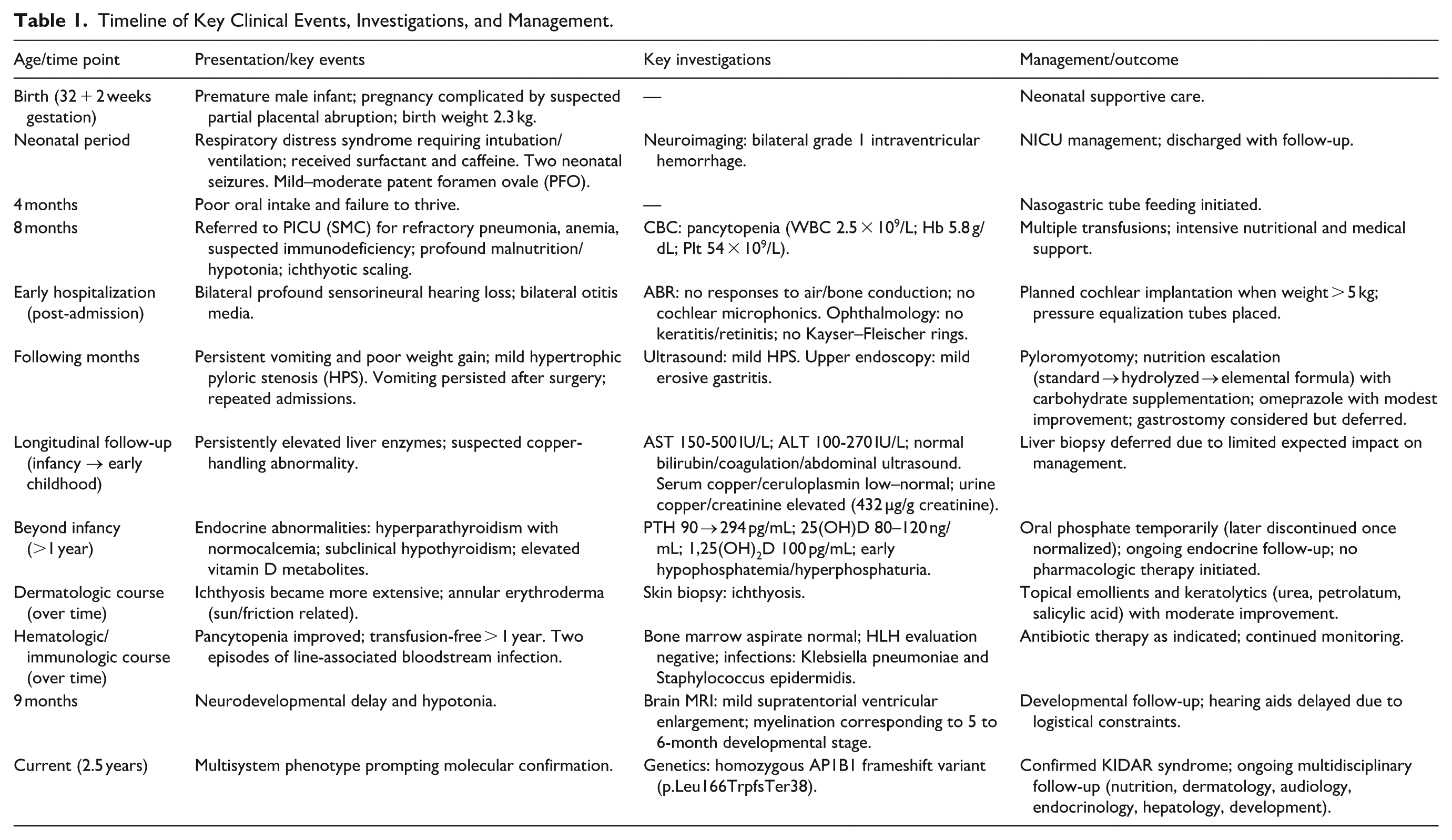
At 8 weeks of age, the patient was referred from a secondary hospital in Nablus to the pediatric intensive care unit (PICU) at Sheba Medical Center (SMC) due to refractory pneumonia, anemia, and suspected immune deficiency. His weight at that time was below his birth weight. At the referring hospital, he had been treated for infections with Klebsiella pneumoniae and Pseudomonas aeruginosa, and hypertrophic pyloric stenosis (HPS) was suspected. Upon admission to SMC, he was found to be profoundly malnourished and hypotonic, with ichthyotic scaling involving the extremities and lower abdomen. Laboratory investigations revealed pancytopenia, with a white blood cell count of 2.5 × 109/L, hemoglobin of 5.8 g/dL, and platelet count of 54 × 109/L, necessitating multiple transfusions (Reference ranges for infants ~1-3 months: WBC 5.0-19.5 × 109/L; hemoglobin 10.7-17.1 g/dL; platelets 190-660 × 109/L.).
Shortly after his PICU admission, the patient underwent an Auditory Brainstem Response (ABR) test, which revealed no responses to air or bone conduction and no cochlear microphonics, consistent with bilateral profound sensorineural hearing loss. Plans were made for cochlear implantation once his weight surpassed 5 kg. He was also noted to have bilateral otitis media, for which pressure equalization tubes were placed. Ophthalmologic evaluation revealed no signs of keratitis, retinitis, or other retinal pathology, and there were no Kayser–Fleischer rings observed.
The clinical course was further complicated by persistent vomiting, initially attributed to mild hypertrophic pyloric stenosis (HPS) confirmed by ultrasound. He underwent pyloromyotomy without intraoperative complications, yet vomiting persisted. Over the ensuing months, he required repeated admissions for ongoing emesis and poor weight gain. Nutritional strategies were repeatedly adjusted, progressing from standard formula to extensively hydrolyzed formula (Nutramigen LGG) and subsequently to elemental formula (Neocate LCP), with a carbohydrate supplement added to increase caloric density (target 120-180 kcal/kg/day) while maintaining adequate protein intake. Despite these interventions, he continued to vomit 3 to 5 times daily, with intermittent exacerbations characterized by several days of partially digested emesis. Upper endoscopy demonstrated mild erosive gastritis; omeprazole provided modest symptom improvement. However, ongoing caloric losses were difficult to quantify, and failure to thrive persisted. Gastrostomy tube placement has been considered, but a decision has been deferred because the expected benefit remains uncertain relative to procedural risk.
During longitudinal follow-up from the initial 8-week presentation through early childhood (current age 2.5 years), the patient demonstrated persistently elevated liver enzymes, with AST levels ranging from 150 to 500 IU/L and ALT from 100 to 270 IU/L. His coagulation profile, bilirubin, and abdominal ultrasound findings remained normal. Serum copper and ceruloplasmin levels were low-normal, but urine copper excretion was significantly elevated at 432 µg/g creatinine. These findings were interpreted as indicative of a defect in hepatic copper excretion. Given the rarity of this presentation, liver biopsy was deferred due to the expected minimal impact on management (Reference ranges: AST 20-60 U/L (age 1-3 years); ALT 5-55 U/L (age 1-19 years); serum copper 80-180 mcg/dL (13 months–10 years); ceruloplasmin 21.7 to 43.3 mg/dL (12 months-7 years); spot urine copper/creatinine ratio typically < 50 nmol/mmol creatinine (~<28 µg/g creatinine); ranges may vary by laboratory.).
During follow-up beyond infancy (ie, after the first year of life), his laboratory investigations also revealed a constellation of endocrine abnormalities. He exhibited hyperparathyroidism, with PTH levels rising from 90 to 294 pg/mL despite normal calcium and phosphorus levels. Early hypophosphatemia and hyperphosphaturia were treated with oral phosphate supplements, which were later discontinued once serum levels normalized. Vitamin D testing repeatedly revealed elevated 25-hydroxyvitamin D levels between 80 and 120 ng/mL and elevated 1,25-dihydroxyvitamin D at 100 pg/mL, despite the cessation of all vitamin D supplementation. These findings raised the possibility of partial tissue resistance to PTH or assay interference. Additionally, subclinical hypothyroidism was noted, with elevated TSH and normal T4. No pharmacologic intervention has been initiated for these endocrinopathies, and he is under regular endocrine follow-up (Reference ranges: intact PTH 11-61 pg/mL (age ≥ 1 year); 25-hydroxyvitamin D sufficiency ≥ 20 ng/mL and levels > 50 ng/mL may be associated with adverse effects; 1,25-dihydroxyvitamin D 24–86 pg/mL (<16 years); reference intervals are age-dependent and vary by assay and laboratory.).
Over time, the patient’s ichthyosis (Figure 1) became more extensive and was joined by annular erythroderma affecting the chest and face, likely exacerbated by sun exposure and friction from developmental movement. A skin biopsy confirmed ichthyosis. Treatment with topical urea-based emollients (Ulactin), petrolatum, and locally prepared salicylic acid ointments (3% for the trunk and 10% for the soles) resulted in moderate improvement. Notably, despite widespread involvement, the skin condition has not been associated with significant pruritus or secondary infection.

Shows the generalized erythematous ichthyosis with fine, adherent white scaling.
His hematologic abnormalities improved over time, and he has remained transfusion-free for over a year. Bone marrow aspirate was normal, and tests for hemophagocytic lymphohistiocytosis (HLH) were negative. Throughout his hospital course, he also experienced 2 episodes of line-associated bloodstream infection, including 1 caused by Klebsiella pneumoniae and another by Staphylococcus epidermidis, both managed successfully with appropriate antibiotic therapy.
A brain MRI performed at 9 months of age revealed mild supratentorial ventricular enlargement with myelination corresponding to a 5 to 6-month developmental stage. No hemorrhage or masses were seen. Developmentally, he has achieved the ability to roll over and maintain head control, with non-progressive hypotonia of the lower extremities. He is visually alert, smiles responsively, and appears to explore his environment. Although he babbles, he does not yet respond to auditory cues, likely due to his profound hearing loss. The acquisition of hearing aids has been delayed due to logistical constraints.
Given the multisystem presentation—ichthyosis with erythroderma, persistent vomiting and enteropathy with failure to thrive, profound sensorineural hearing loss, elevated liver enzymes with copper-handling abnormalities, endocrine disturbances, hematologic abnormalities, and developmental delay—molecular testing was pursued. The patient was found to be homozygous for a frameshift variant in AP1B1 (p.Leu166TrpfsTer38), confirming the diagnosis of KIDAR syndrome (keratitis–ichthyosis–deafness with annular erythroderma and reticulated hyperkeratosis), an ultra-rare autosomal recessive disorder of intracellular trafficking with secondary disturbances of copper metabolism. Fewer than 15 molecularly confirmed cases have been reported to date. In view of the complexity of his course, expert consultation has been initiated, and he remains under ongoing multidisciplinary follow-up to address nutritional, dermatologic, audiologic, endocrinologic, hepatic, and developmental needs.
Discussion
KIDAR syndrome (AP1B1-deficiency syndrome) is a recently recognized, ultra-rare autosomal recessive disorder characterized by ichthyosis and sensorineural hearing loss.2 -4,6,7 Systemic involvement is variable and may include enteropathy, neurodevelopmental delay, endocrine abnormalities, and abnormalities in copper (Cu) metabolism.1,3,6 The present case—a 2.5-year-old boy with a homozygous AP1B1 frameshift variant (p.Leu166TrpfsTer38)—closely matches the core phenotype and also illustrates additional multisystem complexity over time.
In prior reports, Vasconcelos et al. 6 reported a new patient and performed a systematic review of the molecularly confirmed literature, enabling comparison of clinical features across published cases. Our patient’s manifestations align with the core dermatologic phenotype: ichthyosis is a consistently reported feature, and erythroderma and palmoplantar keratoderma have been described in multiple reports.4,6,7 Similar to our patient, erythroderma often develops postnatally and may evolve over time. Key clinical and laboratory findings from published molecularly confirmed reports are summarized in Table 2.
Summary of Published Molecularly Confirmed AP1B1-Associated KIDAR/MEDNIK-Like Reports and Selected Clinical/Laboratory Findings.

Abbreviation: NR, not reported/not described in the publication.
Sensorineural hearing loss, another defining feature of KIDAR syndrome, was identified early in our patient by auditory brainstem response (ABR) testing. This manifestation has been consistently reported across genetically confirmed cases.2 -4,6,7 Cochlear implantation is commonly considered once weight and clinical stability permit—an approach planned for our patient as well.
Failure to thrive and persistent enteropathy are prominent in many reported individuals, including feeding difficulties and the need for specialized nutritional support.3,6 In our patient, vomiting persisted despite pyloromyotomy and multiple formula changes, mirroring the substantial gastrointestinal burden emphasized in published cases and reinforcing that enteropathy is a major contributor to morbidity in KIDAR syndrome.3,6
A notable systemic feature in our patient was the persistent elevation of hepatic transaminases. Hepatic involvement and abnormalities in copper metabolism have been reported in AP1B1-related disease, consistent with disrupted intracellular trafficking of copper-transporting ATPases (ATP7A and ATP7B).1,3,6 The combination of increased urinary copper excretion with relatively preserved serum copper and ceruloplasmin suggests disordered copper handling; prior reports similarly highlight abnormalities in copper metabolism in KIDAR/AP1B1 deficiency.1,3
Endocrine abnormalities were also prominent, including hyperparathyroidism with rising parathyroid hormone (PTH) levels despite normocalcemia and near-normal phosphate after correction of early hypophosphatemia, as well as subclinical hypothyroidism. While endocrine involvement is variably reported, disturbances in growth and mineral metabolism (including abnormalities in the growth hormone axis and other endocrine parameters) have been described in prior cases.3,6 The combination of persistently elevated PTH with elevated vitamin D metabolites in our patient raises possibilities such as partial PTH resistance, altered vitamin D metabolism, or assay-related interference; however, causal mechanisms remain speculative and warrant further study.
Mechanistically, AP1B1 encodes the β1 subunit of adaptor protein complex-1 (AP-1), a clathrin adaptor that directs sorting of transmembrane cargo between the trans-Golgi network and endosomes and is critical for polarized trafficking in epithelial and secretory cells. In AP1B1 deficiency, impaired intracellular trafficking of specific membrane proteins (including copper-transporting ATPases) has been demonstrated in patient-derived cells, supporting a broader model of receptor/transporter mislocalization.1,3
Within the calcium–phosphate–vitamin D axis, appropriate cell-surface expression and compartmentalization of the calcium-sensing receptor (CaSR) in parathyroid cells, as well as renal tubular receptors and transporters (eg, the PTH receptor PTH1R, endocytic receptors involved in vitamin D handling, and sodium-phosphate co-transporters), are essential for suppressing PTH secretion and maintaining phosphate homeostasis. We hypothesize that AP1B1-dependent trafficking defects—supported by prior evidence of disrupted intracellular sorting of membrane cargos in AP1B1 deficiency, including copper-transporting ATPases—could attenuate functional CaSR signaling and/or alter renal PTH responsiveness, resulting in an inappropriately elevated PTH despite normocalcemia, and contributing to early phosphate wasting.1,3 Severe enteropathy and nutritional disturbances may amplify this phenotype; however, the persistence of abnormalities beyond infancy despite correction of overt deficiencies suggests a potential intrinsic trafficking-related contribution. Future work—such as repeat PTH measurement on an alternative platform to exclude assay interference, assessment of PTH fragments/heterophile antibodies when indicated, and targeted evaluation of CaSR/PTH-pathway trafficking in patient-derived cells—may help clarify the mechanism.
From a clinical standpoint, evaluation of persistent PTH elevation in early childhood should include confirmation of age-appropriate calcium–phosphate homeostasis (total/ionized calcium, phosphate, magnesium, alkaline phosphatase), renal function, urinary calcium excretion, and vitamin D status, with consideration of renal ultrasound and skeletal assessment to screen for nephrocalcinosis and metabolic bone disease. Secondary causes (eg, vitamin D deficiency, malabsorption, and chronic kidney disease) should be excluded before labeling primary hyperparathyroidism or PTH resistance. In the absence of a clear secondary driver, ongoing pediatric endocrinology follow-up and longitudinal monitoring are appropriate. 9
More broadly, there is currently no disease-specific targeted therapy for AP1B1 deficiency, and management is largely supportive and multidisciplinary. Reported approaches include intensive nutritional support for enteropathy and failure to thrive, proactive dermatologic care (emollients, keratolytics, and, in selected cases, systemic retinoids), early audiologic rehabilitation with consideration of cochlear implantation, and surveillance for hepatic, endocrine, ophthalmologic, and developmental complications.3,6,8
Genetic counseling is essential once a molecular diagnosis is established. KIDAR syndrome follows autosomal recessive inheritance; therefore, parents are expected to be heterozygous carriers and each future pregnancy carries an estimated 25% recurrence risk. Carrier testing of parents and at-risk relatives, and discussion of reproductive options (prenatal diagnosis and/or preimplantation genetic testing, where available) should be offered. 3
Prognosis appears variable but is driven by the cumulative burden of multisystem morbidity—including recurrent infections, severe enteropathy, growth failure, and neurodevelopmental impairment—and by access to coordinated long-term care. As additional longitudinal data emerge, standardized follow-up frameworks may help optimize anticipatory monitoring and supportive interventions.3,6
Developmental delay is a consistent finding in the published literature. Our patient demonstrated global developmental delay and hypotonia, with mild ventriculomegaly and delayed myelination on MRI, as shown in Figure 2. These findings are broadly concordant with prior reports describing cerebral atrophy, thin corpus callosum, and other neuroimaging abnormalities in affected individuals. 6

Shows T2—brain MRI with supratentorial venticular enlargement with myelination corresponding to a 5 to 6 months.
Hematologic abnormalities, including early pancytopenia and transfusion dependence, were observed in our patient and are consistent with prior reports of anemia and/or thrombocytopenia in a subset of cases.2,6 Subsequent normalization of blood counts without evidence of leukemia or hemophagocytic lymphohistiocytosis supports a transient process, potentially related to early infection, inflammation, or systemic metabolic stress in the setting of AP1B1 deficiency.
In summary, this case both confirms and expands the evolving clinical spectrum of KIDAR syndrome. The combination of dermatologic, audiologic, gastrointestinal, hepatic, endocrine, hematologic, and neurologic involvement highlights the multisystem nature of AP1B1 deficiency. Persistent vomiting/enteropathy and endocrine dysregulation were particularly prominent in our patient and add to the growing recognition of phenotypic complexity. Continued aggregation of cases and systematic longitudinal follow-up are needed to refine genotype–phenotype correlations and to inform anticipatory monitoring and supportive management strategies.
Key contributions of this report include longitudinal tracking of endocrine and copper indices from early infancy (including persistent PTH elevation with high vitamin D despite discontinuation of supplementation), complementary copper data including urinary copper excretion alongside serial serum copper/ceruloplasmin measurements, and a detailed management course for severe vomiting/enteropathy in a premature infant, adding to the limited longitudinal data in prior reports.1,3,6
Conclusion
This case adds to the limited body of evidence on KIDAR syndrome by documenting complex, longitudinal multisystem involvement in a molecularly confirmed patient. It reinforces the need for heightened clinician awareness, early genetic diagnosis, and coordinated multidisciplinary care to optimize supportive management and to improve recognition of this ultra-rare disorder.
Footnotes
Acknowledgements
We express our deep gratitude to the medical ward staff for their invaluable support in completing this report.
Ethical Considerations
Our institution does not require ethical approval for reporting individual cases or case series.
Consent to Participate
Written informed consent was obtained from the parents for their child’s anonymized information to be published in this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.