
Abstract
The carbon catabolite repression 4-negative on TATA-less transcription complex subunit 3 gene (CONT3) plays a key role in regulating the mRNA transcription and protein translation of other genes. Mutations in CONT3 have also recently been implicated as a causative factor of intellectual developmental disorder with speech delay, autism, and dysmorphic facies (IDDSADF). However, to date, only a few CONT3 mutations have been reported to be associated with IDDSADF-related diseases. In the present case, we report a Chinese patient with developmental delay, verbal regression, and facial dysmorphism, in whom cerebral magnetic resonance imaging showed an expansion of the lateral ventricle. The patient was diagnosed with an IDDSADF-related disease caused by a de novo c.1616_1623del mutation in exon 14 of CONT3, which was confirmed by whole-exome sequencing and direct Sanger sequencing. This case report is the first known documentation of a pathogenic mutation at the c.1616_1623del locus of CONT3 in the worldwide population. It provides a critical theoretical basis for the specific gene-based diagnosis of IDDSADF-related diseases and expands the mutation profile of CONT3.
Keywords
Introduction
Intellectual developmental disability occurs in approximately 0.5% of the worldwide population, and is a common reason for undergoing a diagnostic assessment by pediatricians. 1 The etiology of intellectual impairment includes genetic, environmental, and management factors of the child and his/her family. Following the widespread clinical application of next-generation sequencing, there has been a notable increase in the genetic diagnosis of rare diseases, and genetic factors are considered causative of approximately 25% to 50% of identified cases of intellectual developmental disability. 2
In recent years, intellectual developmental disorder with speech delay, autism, and dysmorphic facies (IDDSADF, OMIM #618672) has been identified as a novel developmental defect caused by mutations in the carbon catabolite repression 4 (CCR4)-negative on TATA-less (NOT) transcription complex subunit 3 gene (CONT3; OMIM *604910). 3 This has an extremely rare occurrence, and few genetically confirmed IDDSADF cases have been documented in European and Asian populations. 4
Here, we report a Chinese patient with IDDSADF harboring a novel CONT3 mutation; we provide detailed information regarding the development and diagnosis of IDDSADF, and a literature review of genetically confirmed CONT3 mutation-related intellectual disability cases published to date.
Case presentation
Clinical history
The guardian of the patient provided informed consent for publication of the case, and the reporting of this study conforms to CARE guidelines. 5 The patient was the only male child of non-consanguineous parents with no relevant family medical history. No grave abnormalities were discovered during routine prenatal examinations, and the patient’s mother had no exposure to teratogens during her pregnancy. The patient was born at 40 weeks of pregnancy. His weight (2650 g) and height (50 cm) were within normal ranges, but mild asphyxia was noted at birth. The 1-, 5-, and 10-minute Apgar scores were 7, 9, and 9 after birth, respectively. He walked independently at 13 months of age. No other medical concerns were noted.
He presented with developmental delay and verbal regression shortly after the age of 2 years, and was referred to the outpatient department of our institution aged 4 years and 8 months. On physical examination, he presented with facial dysmorphism, including frontal bossing, a low bridge of the nose, a long and smooth philtrum, a thin upper lip, micrognathia, a prominent labiomental groove, and a short neck (Figure 1(a)). His short height (102 cm [10%–25% percentile]) and low weight (17 kg [25%–50% percentile]) were both in the mid-to-low range at the time of assessment. His head circumference (51 cm) was normal (50%–75% percentile). His speech was slurred, and he could not repeat sentences of more than 10 words. His limb tone and the strength of his extremities were normal. He could walk independently but could not jump over small obstacles. In addition, his scapulas were asymmetric, with the left being lower and more prominent than the right (Figure 1(a)).
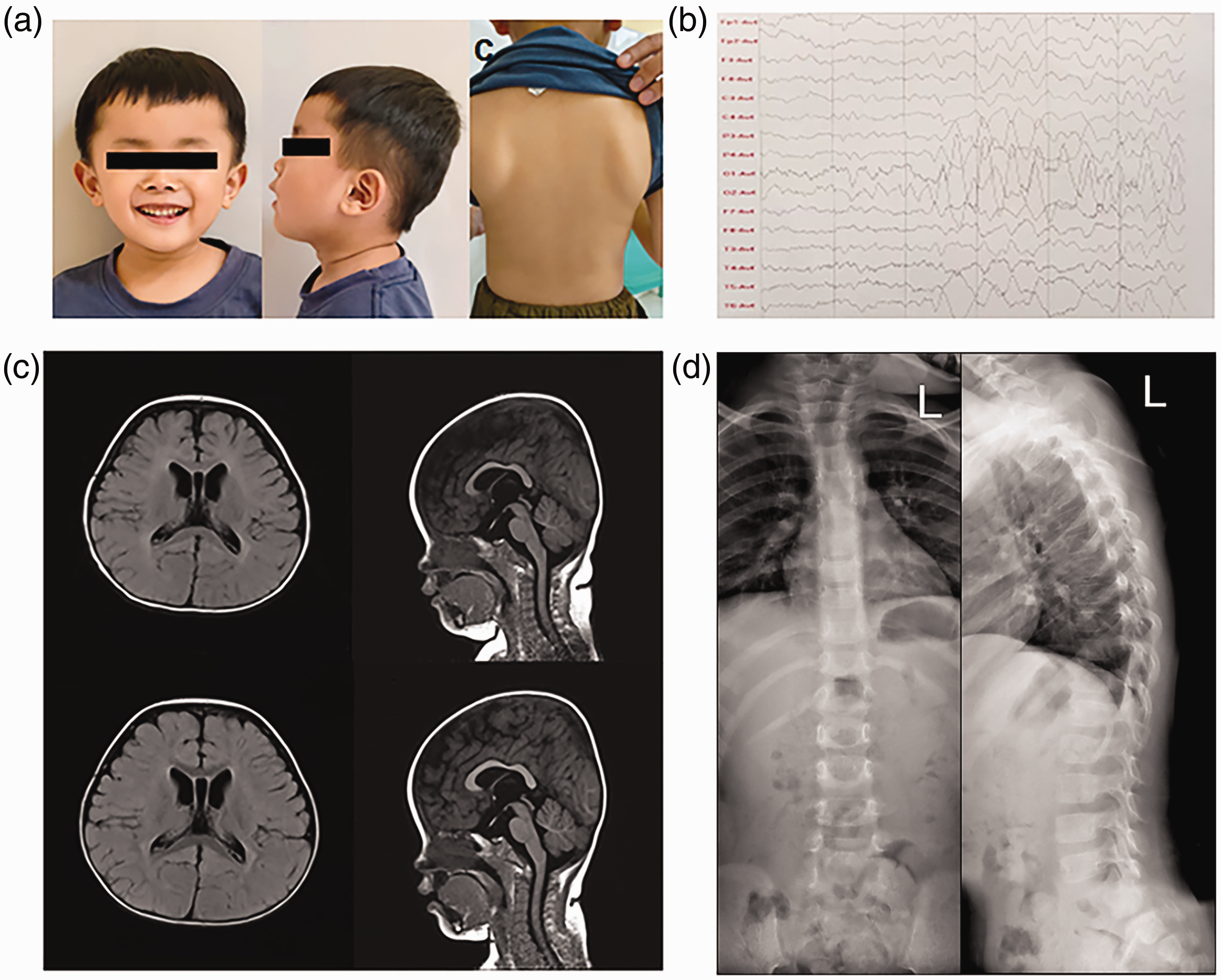
(a) Physical examination revealed abnormal characteristics, including facial dysmorphism seen in frontal (left) and lateral (middle) views. These included frontal bossing, a low nasal bridge, a long and smooth philtrum, a thin upper lip, micrognathia, and a prominent labiomental groove. Asymmetric scapulas were also observed (right). (b) Electroencephalography indicated slow background activity with an increased delta band activity. (c) Cerebral magnetic resonance imaging at the age of 10 months (top images) and 22 months (bottom images). Identical findings of ventriculomegaly were seen in transverse (top and bottom left) and coronal (top and bottom right) images and (d) no scoliosis was observed on the anteroposterior (left) and lateral (right) images of the spinal X-ray.
Electroencephalography of the patient’s brain indicated moderately slow background activity with an increased delta band activity (Figure 1(b)). Magnetic resonance imaging (MRI) showed an expanded lateral ventricle, but no partial agenesis of the corpus callosum or parenchymal atrophy. MRI results were comparable at 10 and 22 months of age (Figure 1(c)). An X-ray examination revealed no scoliosis (Figure 1(d)).
Routine biochemical and metabolic tests showed normal results, and the patient’s vision and hearing functions were also normal. He was HLA-B27-negative. The Wechsler Adult Intelligence Scale showed a verbal intelligence quotient of 52, an operational intelligence quotient of 56, and a total intelligence quotient of 49; these scores revealed a moderate intellectual deficiency and impairment in all fields of cognitive domains. The Peabody Picture Vocabulary Test revealed an intelligence quotient of 63, which was equivalent to that of a 25-month-old child. Findings from the Griffiths Development Scales-Chinese showed him to be equivalent to a child aged 26 to 38 months (Table 1), indicating a global developmental delay. Following parental consent, the patient was referred to locally available early intervention services for language skills and motor function training, but this has had little effect after 6 months.
Griffiths Development Scales-Chinese assessment of the patient.

Genetic analysis
Whole-exome sequencing was used to determine the genetic etiology of the patient’s developmental delay. A novel heterozygous variant (c.1616_1623del, p.Pro539Leufs*16) was detected in exon 14 of CONT3 (NM_014516.3), which was confirmed to be de novo by Sanger sequencing, as his parents do not have this mutation (Figure 2(a)). The variant is a frameshift mutation which is absent from ClinVar, 1000 genomes, DECIPHER, ExAC, and gnomAD databases. Based on predictions by the SWISS-MODEL protein structure homology-modelling tool, the variant is likely to result in a premature stop codon, thereby causing further loss of function of the protein (Figure 2(b)). The variant was shown to be pathogenic based on variant classification according to American College of Medical Genetics and Genomics criteria (PVS1 + PS2_Moderate + PM2_Supporting).

(a) Sanger sequencing of the patient and his parents. De novo truncating variant c.1616_1623del, p.Pro539Leufs*16 seen in CONT3 of the patient (top sequence) was verified by Sanger sequencing. Neither the mother (middle sequence) nor the father (bottom sequence) carried the variant. (b) Mutant (top) and wild-type (bottom) CONT3 proteins generated by the SWISS-MODEL tool show differences and (c) The cDNA structure of the human CONT3 transcript NM_014516.4 and the corresponding domain organization of human CONT3 protein (protein NP_055331.1). Positions are annotated from the first coding nucleotide. Numbers represent the first coding nucleotide of each exon and the last coding nucleotide (nucleotide 2262). Mutations reported by Martin et al., Meyer et al., Zhao et al., Lv et al., and the present case are marked in black, yellow, orange, blue, and red, respectively. Numbers below the figure represent the first and last coding amino acids for each of the protein domains and the last coding amino acid (aa 753).
Literature review
We reviewed relevant literature from PubMed by searching with the keywords “CONT3” or “IDDSADF,” and identified only 27 cases from 23 pedigrees caused by 21 different CONT3 mutations (Figure 2(c)). Table 2 shows a summary of the clinical phenotypes and genotypes for all 22 known CONT3 mutations including the present one. Of the 28 IDDSADF patients, 12 were men and 16 were women, and their ages ranged from 7 months to 55 years. Developmental delay (28/28), speech retardation (28/28), dysmorphic traits (28/28), behavioral anomalies (17/26), muscle hypotonia (13/28), and visual or hearing difficulties (15/26) were the most common clinical features of these patients. Other symptoms included coercive spondylitis, cardiac abnormality, and epilepsy. Imaging findings showed agenesis of the corpus callosum and enlargement of the ventricles. Regarding mutation types, 10 missense mutations, 17 truncating mutations, and one splice site mutation were identified, and distributed across the gene with no mutation hotspots identified (Figure 2(c)). Apart from being classified as de novo, the CONT3 mutations also revealed an autosomal dominant heredity among two unrelated families. 4
Clinical phenotypes and genotypes in individuals with IDDSADF.
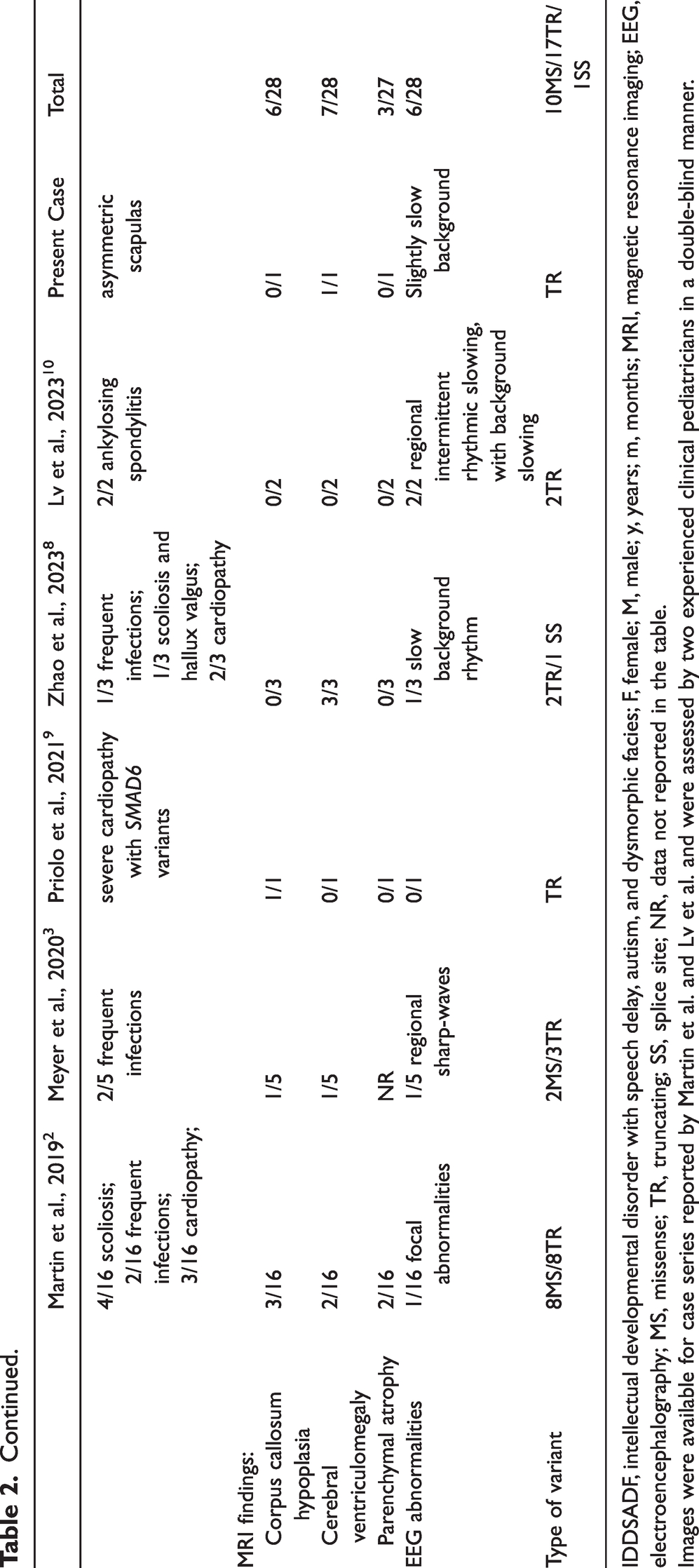
IDDSADF, intellectual developmental disorder with speech delay, autism, and dysmorphic facies; F, female; M, male; y, years; m, months; MRI, magnetic resonance imaging; EEG, electroencephalography; MS, missense; TR, truncating; SS, splice site; NR, data not reported in the table.
Images were available for case series reported by Martin et al. and Lv et al. and were assessed by two experienced clinical pediatricians in a double-blind manner.
Discussion
In the present report, we describe a patient with intellectual disability, speech delay, and dysmorphic features who was diagnosed with IDDSADF caused by a CONT3 mutation.
CCR4-NOT is a multi-subunit complex that mediates interactions between RNA polymerase II and other factors to regulate gene expression in different phases such as transcription initiation, extension, protein translation, and mRNA degradation. CONT3 encodes a subunit of the CCR4-NOT protein complex, which is involved in several biological activities. 6 In Drosophila, silencing components of the CCR4-NOT complex previously caused myofibrillar disarray and dilated cardiomyopathy, 7 while CONT3−/− knockout mice died prior to implantation because of the lack of internal cell growth, and CONT3+/− mice exhibited spontaneous and internal cardiac function deterioration. 8 In addition, an earlier study showed that CONT3 loss of function mutations were associated with T-cell acute lymphoblastic leukemias. 9
In 2019, Martin et al. 3 reported that de novo CONT3 variants caused a clinically variable developmental disorder with developmental delay, behavioral issues, intellectual impairment, and hypotonia, which was designated IDDSADF. CONT3 variants can significantly decrease mRNA levels, and splice site variants can lead to exon skipping. The mutations may also cause changes in the mRNA levels of other CCR4-NOT complex subunits. 10 Most CONT3 mutations are truncations leading to premature stop codons, indicating that haploinsufficiency may cause IDDSADF. The variant reported here would generate a truncated CONT3-M protein lacking downstream functional domains, including the C-terminal region containing the functionally relevant NOT1 anchor region, a connector sequence, and the NOT-box domain (Figure 2(c)). Truncated mutations may lead to degradation of the mutated mRNA, and an absence of protein synthesis following nonsense-mediated mRNA decay. Thus, they can greatly impair protein function leading to a severe phenotype. Missense mutations appear to cause less severe mental disorders because individuals harboring such mutations are able to attend mainstream education with supplementary learning support. 3 In the present case, our patient showed a moderate intellectual deficiency and a poor performance in mainstream education, but he is only young so his progress will be followed up.
Some highly distinctive facial phenotypes are recognized in patients with IDDSADF-related diseases, including a prominent forehead, a flat nasal bridge, flabby cheeks with a prominent nasolabial sulcus, a broad nasal tip with anteverted nares, a long and smooth philtrum, a thin upper lip, and a marked inferior lip with a prominent labiomental groove. 11 Our patient showed a high degree of overlap with these reported features. However, in some cases, these characteristics were not apparent, 3 so more evidence is needed to confirm whether these features are typical manifestations of CONT3 mutation-related IDDSADF.
In previous studies, two and five individuals with CONT3 mutations were diagnosed with ankylosing spondylitis with HLA-B27 positivity 12 and scoliosis, 3 respectively. These findings suggest an association between CONT3 variants and ankylosing spondylitis in HLA-B27-positive individuals. 13 Although our patient had an asymmetric scapula, his spine was normal and he was HLA-B27-negative, so a long-term follow-up is required. Some patients with CONT3 mutations report cardiac abnormalities. However, the underlying mechanism of this is unclear, and it remains unknown whether it results from a dualism of mutations. 11 In some rare cases, CONT3 variants were linked with unilateral/bilateral moyamoya angiopathy (MMA), 14 a vascular trait that confers an increased risk of stroke and starts in childhood, continuing through the fourth decade of life. Although the available data do not indicate that MMA is a major complication of IDDSADF, additional research is required to confirm the association.
In summary, we detected a de novo heterozygous mutation in a patient with CONT3-related neurodevelopmental diseases. His clinical presentation was identical to that reported previously, and our findings have expanded the CONT3 mutation spectrum. Screening for CONT3 variations in large cohorts of individuals with cognitive impairment, facial dysmorphism, and other symptoms of this rare condition may provide novel insights into underlying disease mechanisms. Future research should include an investigation of the complete clinical spectrum, genotype–phenotype correlation studies, and the functional analysis of pathogenic mechanisms.
Footnotes
Acknowledgements
We thank all members of the patient’s family for their support and cooperation.
Author contributions
MP was involved in data curation, formal analysis, investigation, and writing of the original draft. HL was involved in patient care, clinical diagnosis, and funding acquisition. LP participated in clinical diagnosis, scale assessment, formal analysis, and writing of the original draft. RS was involved in supervision, review, editing, and funding acquisition. All authors contributed to the article and approved the submitted version.
Data availability statement
The original contributions presented in the study are included in the article, further inquiries can be directed to the corresponding author.
Declaration of conflicting interests
The authors declare that there are no conflicts of interest.
Ethics statement and informed consent
This study was approved by the Ethics Committee of Huzhou Maternity & Child Care Hospital (approval number: 2022-R-002). Written informed consent was obtained from the mother of the patient, who is his legal guardian.
Funding
This study was supported by the Health Commission of Zhejiang Province (No. 2023KY323) and Huzhou Municipal Science and Technology Bureau (No. 2021GZB12).