
Abstract
Diabetes mellitus remains a growing global health burden, yet advances in mechanobiology highlight a pivotal role for mechanical cues in regulating pancreatic β-cell function. This review summarizes evidence that the extracellular matrix (ECM) surrounding pancreatic islets is not merely structural support but a dynamic mechanosignaling platform integrating biochemical and physical stimuli to govern β-cell fate. In healthy tissue, a compliant ECM enriched in collagen IV and laminin supports optimal insulin secretion and glucose responsiveness. In contrast, obesity- and diabetes-associated fibrosis induces excessive ECM deposition, aberrant crosslinking, and pathological stiffening, disrupting mechanosensing, glucose signaling, and β-cell maturation. Mechanical forces are transmitted through integrin-mediated focal adhesions and FAK activation, triggering MAPK, PI3K–AKT, and Rho GTPase pathways that regulate cytoskeletal tension, transcriptional programming, and survival. Mechanosensitive Piezo1 channels convert membrane stretch into Ca2+ influx, modulating insulin exocytosis, while the Hippo–YAP/TAZ axis interprets matrix stiffness to control β-cell proliferation and phenotypic stability. Chronic hyperglycemia further induces nuclear deformation and chromatin remodeling, impairing transcription factors such as PDX1 and accelerating β-cell dysfunction. These pathways intersect with inflammatory and oxidative stress signaling, driving apoptosis and progressive β-cell loss in type 2 diabetes. By reframing diabetes as a disorder of aberrant cell–matrix communication, this review highlights mechanobiology-guided strategies including stiffness-tunable matrices, viscoelastic hydrogels, and mechano-responsive scaffolds to restore a functional microenvironment. Targeting ECM remodeling, modulating Piezo1 activity, and fine-tuning YAP signaling, particularly with stem cell-derived β-cell replacement, offers a promising approach for restoring β-cell mass and achieving durable glycemic control.
Keywords
Introduction
Global burden of diabetes and β-cell failure
Diabetes mellitus (DM) is a highly prevalent and rapidly increasing chronic metabolic disease that currently lacks a curative therapy.1,2 DM is characterized by increased glucose levels in blood, known as hyperglycemia, which, if left untreated or uncontrolled, often leads to neuropathy, nephropathy, retinopathy, cardiovascular disease, stroke, peripheral vascular disease, and diabetic ulcers, significantly increasing mortality. 3 The global prevalence of DM was approximately 537 million in 2021 and is projected to rise to 643 million in 2030 and 800 million in 2045. 4 Worldwide, diabetes has a significant impact on morbidity and mortality. 5 Globally, 6.7 million deaths were recorded due to diabetes. The International Diabetes Federation estimates that global healthcare expenditures related to diabetes exceeded $966 billion in recent years, 4 highlighting the urgent need for disease-modifying and regenerative strategies that go beyond glycemic control to preserve or restore functional β-cell mass. 5
The pancreatic islet as a mechanosensitive microenvironment
Mechanobiological heterogeneity and measurement limitations.

Summary of reported stiffness ranges of healthy and diseased pancreatic tissues across species and measurement platforms. Values vary depending on anatomical region and technique, highlighting the need for careful interpretation when comparing mechanobiological data across studies.
β-cell physiology at the interface of metabolism and mechanics
Pancreatic β-cells are key regulators of glucose metabolism and energy homeostasis through their tightly controlled secretion of insulin that controls blood glucose levels. 20 Furthermore, β-cells maintain metabolic balance and promote cell growth. 21 Insulin enables glucose uptake by peripheral tissues, particularly skeletal muscle and adipose tissue, where it is stored as glycogen or lipid and utilized for energy metabolism. 22 When insulin action is impaired, persistent hyperglycemia develops, which, if left untreated, contributes to macrovascular complications such as cardiovascular disease and stroke, as well as progressive β-cell stress driven by glucolipotoxicity. Additionally, β-cells respond to incretin hormones, which enhance insulin release after a meal to improve the ability of the body to maintain a normal blood glucose level. 21 However, chronic exposure to elevated glucose and sustained secretory demand impose metabolic and mechanical stress on β-cells, ultimately driving β-cell dysfunction characterized by oxidative stress, inflammatory signaling, and impaired insulin secretion. 23 This progressive β-cell dysfunction is a defining feature of type 2 diabetes mellitus (T2DM). Glucotoxicity and lipotoxicity are major triggers of β-cell apoptosis and dedifferentiation, thereby reducing functional β-cell mass and promoting hyperglycemia.1,24 Uncontrolled hyperglycemia further exacerbates metabolic dysfunction and long-term macrovascular and microvascular complications. 1 Lifestyle interventions, including physical activity and weight loss, can partially restore β-cell function in individuals with T2DM, but do not directly address the underlying changes in the mechanical and structural properties of the islet niche. 25
Pancreatic stellate cells and fibrotic remodeling as mechanobiological drivers
Pancreatic stellate cells (PSCs) are specialized mesenchymal cells that constitute approximately 4–7% of the pancreatic parenchymal cell population.26,27 In their quiescent state, PSCs express nestin and glial fibrillary acidic protein and contain vitamin A–rich lipid droplets.6,28 They share phenotypic similarities with hepatic stellate cells but exhibit pancreas-specific functional characteristics. 6 PSCs have two distinct phenotypes, quiescent and activated. In the quiescent phase, PSCs maintain pancreatic homeostasis by regulating ECM remodeling and supporting the physiological architecture of islets.26,29 PSCs are activated under pathological conditions such as inflammatory cytokines, hyperglycemia, oxidative stress, or mechanical stress.7,30 This transformation involves several important changes, including induction of α-smooth muscle actin, loss of vitamin A droplets, increased proliferation and migration, secretion of proinflammatory and profibrotic factors, and enhanced ECM protein synthesis. 29 In diabetic conditions, several factors contribute to PSCs activation within islets. 30 Elevated glucose levels trigger PSC activation by inducing oxidative stress, increasing production of reactive oxygen species (ROS). 7 Hypoxia in diabetic islets enhances PSC activation through ROS-mediated mechanisms. 30 Administration of N-acetyl-L-cysteine can attenuate hypoxia-induced PSC activation, confirming the role of oxidative stress. 30 Transforming growth factor- β (TGF-β), mitogen-activated protein kinases (MAPK), platelet-derived growth factor (PDGF), and nuclear factor κB (NF-κB) are major pathways involved in PSC activation. 29 Gene expression profiling has identified Pdpn, Bad, and Fos as critical mediators of diabetes-induced PSC activation, 31 linking biochemical and mechanical cues to fibrotic ECM remodeling within the islet niche.
The β-cell microenvironment and emerging concepts in mechanobiology
β-cell function and survival are exquisitely sensitive to their surrounding microenvironment, which comprises the ECM, neighboring endocrine and stromal cells, vascular and immune cells, and soluble mediators. 10 The ECM of the islet niche is composed of collagen, fibronectin, heparan sulfate, chondroitin sulfate, and laminins, whose composition and organization determine both biochemical ligand presentation and mechanical properties. Mechanical features such as fibrosis, stiffness, and altered viscoelasticity influence β-cell activity by modulating integrin-mediated signaling and growth factor bioavailability.32,33 Disruption of ECM homeostasis activates mechanical and pro-fibrotic signaling pathways that drive apoptosis, insulin resistance, and progression of diabetes.
Mechanobiology, which investigates how physical and mechanical forces shape cell behavior, has revealed that changes in ECM stiffness, architecture, and tethering can profoundly alter β-cell morphology, polarity, and function. 34 In diabetes, progressive ECM remodeling and altered cell–matrix interactions lead to loss of β-cell identity and gene expression patterns. 1 Mechanotransduction, including integrin-mediated adhesion, cytoskeletal reorganization, and nucleus–cytoskeleton coupling, converts external forces into biochemical signals that regulate β-cell proliferation, survival, and insulin secretion.34,35 Central transcription factors such as pancreatic and duodenal homeobox 1 (PDX1) and MafA are influenced by mechanical forces, metabolic inputs, ECM stiffness, and mitochondrial dynamics.1,36,37 Thus, β-cell health is governed not only by metabolic and inflammatory cues but also by the physical properties and composition of the ECM.11,33 Pathological ECM remodeling in diabetes and obesity alters tissue stiffness and cell–matrix coupling, thereby impairing β-cell survival and function. 38
Limitations of current β-cell replacement strategies and the mechanobiology gap
Islet transplantation can restore endogenous insulin production but remains constrained by limited donor availability, immune rejection, and progressive loss of graft function.39,40 Transplanted islets often fail to achieve durable engraftment because they are introduced into an inhospitable microenvironment characterized by hypoxia, inflammation, and aberrant ECM remodeling.41,42 Stem cell-derived β cells (SC-β cells) generated from pluripotent stem cells and human-derived mesenchymal stem cells provide an alternative, potentially unlimited source of insulin-producing cells. 43 However, SC-β cells often exhibit immature phenotypes and are limited in vivo functionality when transplanted, due to a lack of a supportive microenvironment that replicates the mechanical and biochemical cues of the native islet microenvironment. 44 Recent clinical advances underscore both the promise and current limitations of SC-β cell therapy. ViaCyte’s PEC-Direct system demonstrated detectable C-peptide production in vivo, albeit with α-cell predominance, whereas Vertex’s VX-880 therapy significantly reduced HbA1c levels from 8.6% to 6.9% under immunosuppressive treatment (ClinicalTrials.gov: NCT04786262).45,46 These results validate the therapeutic potential of stem cell-based β-cell replacement but also highlight critical challenges, including incomplete cellular maturation, the need for robust immune protection, and the demanding requirement to manufacture therapeutically relevant cell numbers (on the order of ∼109 cells per patient). Notably, most transplantation strategies have yet to systematically address how fibrotic stiffening, altered viscoelasticity, and disrupted mechanotransduction in the diabetic pancreas limit engraftment and long-term function.
Accumulating evidence indicates that ECM mechanics and mechanotransduction pathways are central regulators of β-cell survival, proliferation, and insulin secretion.11,47 In diabetic and obese states, pathological ECM remodeling increases stiffness, fibrosis, and viscoelastic changes, which perturb β-cell mechanosensing via mechanosensitive ion channels such as Piezo1 and TRPV4, as well as nuclear effectors including Yes-associated protein (YAP)/transcriptional coactivator with a PDZ-binding domain (TAZ). 48 These mechanical perturbations contribute to β-cell dedifferentiation, dysfunction, and apoptosis, representing a previously underappreciated axis of β-cell failure. 49 Consequently, engineering biomimetic niches that recapitulate the mechanical properties of healthy islets or actively reverse fibrosis has emerged as a promising strategy to improve SC-β cell maturation, survival, and therapeutic efficacy.44,50
This review aims to integrate and examine the mechanobiological aspects of the pancreatic microenvironment and their implications for therapies using stem cell-derived β-cells. We compare β-cell function and ECM organization in healthy versus diabetes- and obesity-associated states and delineate how specific alterations in ECM composition, stiffness, and viscoelasticity interfere with β-cell mechanotransduction. We highlight major mechanobiological pathways—including YAP/TAZ, Piezo1, and TRPV4—as potential therapeutic targets. Furthermore, we discuss advanced bioengineering approaches, such as stiffness-gradient hydrogels, patient-derived stem cells, and mechano-pharmacological modulators (including agents such as semaglutide and retatrutide), that aim to mimic or restore physiological pancreatic mechanics. By integrating these concepts, we propose a translational framework for engineering personalized pancreatic microenvironments that support durable β-cell regeneration and function in diabetes.
Native islet microenvironment: Structural and mechanobiological foundations
Structural, cellular, and biomechanical organization of healthy pancreatic islets
The pancreatic islets of Langerhans represent highly organized micro-organs in which spatial architecture, cellular composition, and biomechanical properties are intricately coordinated to maintain glucose homeostasis.51,52 β-cells typically constitute 60–80% of the islet mass and are surrounded by glucagon-secreting α-cells, somatostatin-releasing δ-cells, pancreatic polypeptide-producing cells, endothelial cells, and stromal components, forming a cohesive endocrine unit that facilitates tightly regulated paracrine communication.52,53 The islets are richly vascularized, ensuring rapid nutrient and oxygen delivery, which is essential for β-cell function and survival. 54 The interaction between endocrine cells and intra-islet capillary network highlights the important role of the microenvironment in maintaining β-cell identity and function.52,55 The basement membrane-rich ECM, composed predominantly of collagen IV, laminin-411/511, fibronectin, and heparan sulfate proteoglycans, forms a compliant scaffold that actively supports mechanoresponsive signaling rather than serving as a passive structural support.56,57 Importantly, the healthy islet ECM exhibits a physiologically soft elastic modulus (0.2–1 kPa), generating a viscoelastic niche that buffers mechanical stress while enabling efficient force transmission essential for mechanosensing.11,15,58 This compliant environment preserves optimal focal adhesion turnover, cytoskeletal organization, and vesicle trafficking required for finely tuned insulin granule exocytosis. 11 The viscoelastic nature of the ECM permits stress relaxation and adaptive deformation, allowing β-cells to respond to fluctuating metabolic and mechanical demands without activating pathological stress responses.59,60 In this mechanically dynamic niche, β-cells maintain their identity, glucose responsiveness, and survival, underscoring the ECM as a functional regulator of endocrine homeostasis.
Mechanosensing and mechanotransduction in regulation of β-cells functions
β-cells continuously interpret biophysical signals from their surrounding ECM through mechanotransduction pathways that convert mechanical inputs into biochemical and transcriptional responses regulating proliferation, differentiation, and insulin secretion. 9 Integrin receptors form focal adhesion complexes linking the ECM to the actin cytoskeleton, enabling β-cells to detect alterations in matrix stiffness, topology, and viscoelasticity.61,62 This integrin–cytoskeletal network serves as a central mechanosensory interface, transmitting extracellular forces to intracellular signaling cascades including FAK, PI3K–AKT, MAPK, and RhoA/ROCK pathways, which collectively orchestrate cytoskeletal tension and β-cell functional maturation. Mechanosensitive ion channels, particularly Piezo1 and TRPV4, respond to membrane stretch and shear stress by modulating Ca2+ influx, thereby directly influencing insulin granule mobilization and secretion.63–65 Upon activation, these channels couple mechanical deformation to electrical excitability and insulin exocytosis, integrating physical force sensing with metabolic signaling. Ca2+ influx further activates downstream regulators, including YAP/TAZ transcriptional coactivators, which shuttle between the cytoplasm and nucleus in response to mechanical stimuli, controlling β-cell proliferation, survival, and phenotypic stability.9,66–68 Dysregulation of this mechanotransduction axis has been increasingly implicated in β-cell dysfunction and the pathogenesis of diabetes. 69
Calcium influx remains the principal trigger for insulin secretion following glucose-stimulated membrane depolarization. Mechanically activated Piezo1 and TRPV4 channels further fine-tune intracellular Ca2+ oscillatory dynamics, modulating the amplitude and frequency of Ca2+ spikes that govern pulsatile insulin release.63,69 Under physiological mechanical stress, β-cells exhibit rhythmic Ca2+ signaling that facilitates insulin granule docking and exocytosis through SNARE complex activation.70,71 Healthy ECM mechanics support cytoskeletal dynamics essential for vesicle transport and secretion, whereas pathological matrix stiffening disrupts Ca2+ homeostasis, impairs oscillatory signaling fidelity, and ultimately compromises insulin secretory function.11,72
Pathological remodeling in diabetes and obesity
Type 1 diabetes mellitus
Autoimmune destruction of β-cells
T1DM is an autoimmune disease that affects the insulin-producing β-cells, leading to hyperglycemia. 73 The pathophysiology of T1DM is quite complicated, involving genetic predisposition and environmental factors that initiate the autoimmune process. 74 The autoimmune process of β-cell destruction begins with the activation of autoreactive T cells. The interaction between β cell antigens and CD4+ T cells is crucial for the activation of helper T cells, which secrete pro-inflammatory cytokines such as Interleukin-12, that recruit other immune effector cells, including macrophages and dendritic cells, into the pancreatic microenvironment. 75 Activated CD4+ T cells stimulate and activate cytotoxic T cells by releasing interleukin-2 cytokines. These cytotoxic T cells migrate to the pancreatic islets and induce β cell apoptosis through the release of perforin, granzymes, ROS, and inflammatory cytokines.75,76 CD4+ and CD8+ T cells attack β cells by infiltrating into the pancreatic environment, releasing pro-inflammatory cytokines such as interferon-gamma (IFN-γ) and tumour necrosis factor-alpha (TNF-α), which further causes inflammation in the β cells and contributes to β cell apoptosis. 76 The released inflammatory cytokines activate the β cell apoptosis and disrupt insulin secretion. Cytokines like IFN-γ, IL-1β, and TNF-α are toxic to β cells and lead to β cell dysfunction. 76 Autoantibodies targeting β-cell components such as insulin, insulinoma-associated antigen 2, zinc transporter 8, and glutamic acid decarboxylase 65. These autoantibodies are the biomarkers used to detect T1DM in the early stages.74,76,77
Role of inflammation and immune response in T1DM progression
Inflammation plays a primary role in T1DM progression. The Inflammatory process begins with the infiltration of immune cells into the islet cells, a condition known as insulitis. 78 Inflammatory cytokines such as IL-1α, IL-1β, and IL-10 are significantly increased in the individuals with T1DM and promote disease progression. 79 These pro-inflammatory cytokines lead to β cell dysfunction and apoptosis through mechanisms including the infiltration of immune cells into pancreatic islets.79,80 This inflammatory environment is further aggravated by autoreactive T cells, which can lead to β-cell apoptosis. 81 The autoimmune attack is occurring due to the seroconversion of islet autoantibodies against insulin, zinc transporter 8, and/or glutamate decarboxylase, which are indicators of autoimmunity during the onset of T1DM. 80 Furthermore, this autoimmune response triggers Toll-like receptors (TLRs), which are components of the innate immune system. 81 Previous studies revealed that TLR2 and TLR4 are significantly elevated in individuals with T1DM, suggesting that innate immune signaling contributes to the development of the disease.81,82 Previous studies have revealed that the cytokine profile is dominated by proinflammatory cytokines. Elevated levels of IL-1β is associated with autoimmune damage to β cells, leading to β cell destruction, while IL-10, an anti-inflammatory cytokine, reduces inflammation but is insufficient to prevent the aggressive autoimmune response. 79 Pro-inflammatory and anti-inflammatory signals must be balanced; overproduction of inflammatory mediators can lead to sustained damage to β-cells, resulting in insulin deficiency. Therapeutic approaches are significantly influenced by understanding the inflammatory mechanisms underlying the progression of T1DM. New treatment strategies that involve modifying the immune response and targeting specific cytokines may have potential in preventing disease progression. 79 Treatments that enhance the production of anti-inflammatory cytokines and inhibit pro-inflammatory cytokines could potentially prevent the progression of T1DM. Furthermore, combining immunotherapy with β-cell therapy could effectively preserve β-cell function. 80
Mechanobiology of T1DM
The autoimmune inflammation characteristic of type 1 diabetes mellitus (T1DM) profoundly alters the mechanical homeostasis of the islet microenvironment, distinguishing it from the fibrotic stiffening observed in type 2 diabetes mellitus (T2DM). 12 Rather than matrix stiffening, T1DM is marked by pathological softening and structural destabilization of the islet niche, driven by immune-mediated extracellular matrix (ECM) degradation and inflammatory edema. This distinct mechanical phenotype has critical implications for β-cell survival and mechanotransduction.12,83 In mouse T1DM, proinflammatory cytokines such as IL-1β and TNF-α induce extensive remodeling of the islet ECM, particularly through increased hyaluronan (HA) synthesis and accumulation, resulting in osmotic swelling and mechanical softening of the tissue. 84 Atomic force microscopy (AFM) studies in the double transgenic DO11.10 × RIPmOVA (DORmO) mouse model of type 1 diabetes demonstrate that inflamed islets exhibit markedly reduced stiffness (∼284 Pa) compared with healthy islets (∼3039 Pa), in contrast to the increased stiffness observed in fibrotic T2DM islets. 12 This mechanically softened microenvironment promotes immune cell infiltration, disrupts mechanosensitive signaling, and compromises β-cell survival. Importantly, inhibition of HA synthesis attenuates immune infiltration and slows disease progression, highlighting a causal link between altered mechanics and autoimmune pathology.
In human T1DM, progressive degradation of the peri-islet basement membrane represents a critical mechanical failure point. Under physiological conditions, the basement membrane, which is rich in collagen IV and laminins, provides structural integrity and acts as a physical barrier against immune cell invasion. 85 However, in T1DM, this barrier is disrupted by increased activity of matrix metalloproteinases (MMPs), including MMP-3 and MMP-9, released by infiltrating macrophages and activated immune cells. 85 This ECM degradation is mediated by matrix metalloproteinases (MMPs), such as MMP-3 and MMP-9, which are secreted by infiltrating macrophages and induced by cytokine driven β-cell stress. 83 The resulting degradation of ECM components compromises integrin-mediated survival signaling and facilitates immune cell infiltration into the islet core. This loss of mechanical integrity renders β-cells highly susceptible to anoikis, inflammatory stress, and functional exhaustion.86–88 Taken together, these findings indicate that T1DM should be viewed not only as an autoimmune disorder but also as a disease involving mechanical disruption of the islet niche. Unlike T2DM, which is characterized by pathological stiffening, T1DM involves loss of peri-islet mechanical integrity and barrier function. Accordingly, therapeutic strategies should focus on restoring appropriate mechanical cues through immunoprotective and mechanically compliant biomaterials. Engineered matrices that mimic the viscoelastic properties of the native peri-islet environment may help preserve β-cell viability, support mechanotransduction, and improve long-term graft function in T1DM.
Type 2 diabetes mellitus
Metabolic dysregulation, insulin resistance, and organ complications
Type 2 diabetes mellitus (T2DM) is a chronic metabolic disorder characterized by sustained hyperglycemia. 89 Insulin resistance and loss of insulin secretion from β cells are the major causes of T2DM. 90 The progression of the disease is significantly influenced not only by metabolic stress but also by ECM remodeling and the development of a fibrotic microenvironment that alters tissue mechanics. If this condition is left untreated, it may lead to nephropathy, retinopathy, neuropathy, cardiovascular diseases, diabetic ketoacidosis, diabetic coma, and diabetic foot ulcers. Additionally, osteoporosis, myopathy, liver damage, and heart failure are associated with T2DM.89,91 One of the major characteristics of T2DM is insulin resistance, where the β-cells fail to respond to insulin, leading to β-cell dysfunction and death, 92 reducing the capacity of insulin to promote glucose uptake in muscle, liver, and adipose tissue, leading to hyperglycemia. 93 The development of insulin resistance leads to obesity, inflammation, and oxidative stress. Obesity is a major risk factor in developing T2DM; fat accumulation in the liver and muscle disrupts normal metabolic function, impairs insulin signaling, and leads to insulin resistance. 93 Inflammation is characterized by altered cytokine production, and the activation of inflammatory pathways associated with obesity leads to the release of pro-inflammatory cytokines such as IL-6 and TNF-α, that impairs insulin signaling pathways.93,94 Increased TNF-α expression in adipocytes leads to insulin resistance through JNK1 and IKK-related pathways that affect insulin signaling. 93 Oxidative stress, enhanced by excessive production of free radicals such as ROS, damages DNA, lipids, and proteins, resulting in loss of normal metabolic function. Long-term exposure to hyperglycemia is associated with increased synthesis of glycation end products, abnormal protein kinase C activation, and impaired oxidative phosphorylation.95,96 The accumulation of misfolded proteins in the endoplasmic reticulum (ER) can cause ER stress and disruption of insulin signaling. Mitochondrial dysfunction alters fatty acid oxidation and promotes lipotoxicity, which affects energy metabolism and leads to insulin resistance. 93
As insulin resistance progresses, β-cells compensate by increasing the secretion of insulin, which leads to β-cells dysfunction characterized by decreased insulin secretion, dedifferentiation of β-cells, and loss of β-cell mass due to β-cell apoptosis. The reduction in β-cell mass and failure is caused by metabolic stress, which decreases glucose uptake, contributing to disease progression. 97 The relationship between β-cell dysfunction and insulin resistance is complex. A high-fat diet causes obesity, which triggers insulin resistance and leads to β-cells secreting more insulin. When β-cells are unable to satisfy insulin demand, it results in hyperglycemia. Contrastingly, hyperglycemia can cause failure of β-cells through lipotoxicity, which damages these cells by increasing free fatty acids, and glucotoxicity, which impairs β-cell function due to elevated glucose levels. 92 Taken together, T2DM progression reflects a coupled metabolic–mechanobiological failure, in which chronic metabolic stress, inflammation, and oxidative injury progressively reshape the ECM and alter β-cell mechanosensing and survival.
Contributing roles of obesity and metabolic stress in T2DM pathogenesis
Metabolic disorders associated with obesity play a significant role in the development of insulin resistance and β-cell dysfunction. The relationship between T2DM and obesity has several mechanisms, including ectopic lipid accumulation, chronic inflammation, and ER and oxidative stresses. Excessive calorie intake leads to fat accumulation in adipose tissue, the liver, and muscle, disrupting normal metabolic functions, contributing to obesity and exacerbating insulin resistance.93,98 Elevated free fatty acid (FFA) levels in the blood can interfere with insulin signaling pathways, resulting in increased glucose synthesis in the liver and decreased glucose uptake in the muscles. 98 Furthermore, obesity is characterized by chronic inflammation. A high-fat diet induces obesity and triggers macrophage infiltration and the release of pro-inflammatory cytokines such as IL-1β, IL-6, and TNF-α, impairing insulin signaling and further contributing to insulin resistance.94,99 The activation of inflammatory pathways such as JAK/STAT, JNK, and NF-κB leads to metabolic dysregulation, causing IL-1-mediated β-cell dysfunction and apoptosis, ultimately resulting in hyperglycemia. 99 Importantly, these inflammatory and metabolic insults also activate stromal cells and fibroblasts, driving increased ECM deposition and crosslinking. As a result, obesity not only perturbs endocrine signaling but also progressively converts the β-cell niche into a mechanically stiff, fibrotic microenvironment that further aggravates insulin resistance and β-cell failure.
Obesity-driven inflammation, collagen I/fibronectin deposition, and ECM mechanics
Obesity contributes to the onset of T2DM by creating a pro-inflammatory environment that promotes systemic inflammation and alters the microenvironment of β-cells. 100 The expansion of adipose tissue increases levels of free fatty acids and cytokines, which impair β-cell function and promote apoptosis. 101 This creates a vicious cycle where obesity increases insulin resistance and reduces the ability of β-cells to produce insulin. 100 Adipose tissue releases various cytokines that induce insulin resistance and activate fibrogenic pathways, leading to fibrosis in organs such as the liver and kidneys. 102 Chronic inflammation in β-cell environment is characterized by immune cell infiltration and increased cytokine secretion, which activates fibroblasts and excessive deposition of ECM proteins like collagen I and fibronectin. Enhanced ECM cross-linking through lysyl oxidase further increases matrix stiffness and disrupts normal cellular structure, leading to fibrosis. 42 These fibrotic and mechanical alterations impair integrin-mediated signaling, distort mechanotransduction, and ultimately compromise β-cell survival and glucose-stimulated insulin secretion. Moreover, these inflammatory and fibrotic changes complicate the management of diabetes by exacerbating tissue remodeling and metabolic dysregulation, but also highlight the necessity to develop treatment strategies that simultaneously target inflammation-driven fibrosis and β-cell dysfunction in patients with diabetes. 103
PSC-β cell crosstalk and fibrotic islet microenvironment
The interaction between PSC and β-cell is mediated by multiple mechanisms involving direct cell contact and paracrine signaling.104,105 Activated PSCs release various substances that directly influence the β-cell function and survival. Exposure of β-cells to conditioned media from hypoxia-induced PSCs significantly reduces β-cell survival and increases apoptosis, highlighting the detrimental effects of PSC activation on β-cell health. 30 PSCs enhance β-cell function under certain conditions. Co-culture studies reveal that PSCs from healthy mice promote glucose-stimulated insulin secretion (GSIS) from Min6 cells by secreting IL-6. 104 A previous study highlighted that Wnt5a plays a significant role in the interaction between PSCs and β-cells. PSCs regulate insulin secretion through Wnt5a-induced calcium signaling and FoxO1-PDX1-GLUT2 pathways, resulting in increased intracellular calcium levels and enhanced pFoxO1, PDX-1, GLUT2, and pCamKII expression in β-cells. 105
PSC activation leads to excessive accumulation of ECM, resulting in a fibrotic microenvironment that progressively impairs islet function.7,106 This fibrotic process involves excessive production of collagen I and III, and fibronectin, disrupting normal islet architecture and β-cell interactions. 107 The fibrotic response creates a self-perpetuating mechanobiological cycle in which increased matrix stiffness further activates PSCs, thereby amplifying tissue remodeling and fibrosis.108,109 The role of fibrosis in β-cell function is complex, with increased matrix stiffness directly disrupting GSIS through mechanosensitive pathways. 30 Furthermore, fibrotic tissue acts as a diffusion barrier that limits oxygen and nutrient supply to β-cells, increasing cellular stress. 30 Fibrotic remodeling disrupts the vascularization of normal islets and weakens the function and survival of β-cells. 30 Clinical data highlights the important role of pancreatic fibrosis in diabetes progression, where increased fibrosis is associated with a greater risk of developing pancreoprivic diabetes after pancreatectomy, 106 highlighting the importance of addressing fibrotic mechanisms in the management of diabetes. Therefore, PSCs emerge as central stromal regulators that couple inflammation, hypoxia, and mechanical stress to ECM remodeling, positioning them as key mechanobiological targets in strategies aiming to preserve or restore β-cell function.
β-cell microenvironment and mechanobiological states
The microenvironment of the pancreatic β-cell plays an important role in maintaining its function, proliferation, and survival. This environment is influenced by the composition of ECM and inflammatory cytokines and their interactions with adipocytes.10,52 Understanding the characteristics of the β-cell dysfunction is essential for comprehending the its microenvironment in different metabolic states, such as obesity and fibrosis, compared to lean and healthy conditions. Previous studies highlighted that the interactions in the β-cell microenvironment affect its physiology and pathophysiology in diabetes. Mechanobiological characteristics of healthy versus obese/fibrotic conditions, including key signaling pathways and ECM changes, are depicted in Figure 1. Mechanobiological characteristics of healthy versus obese/fibrotic β-cell microenvironments. 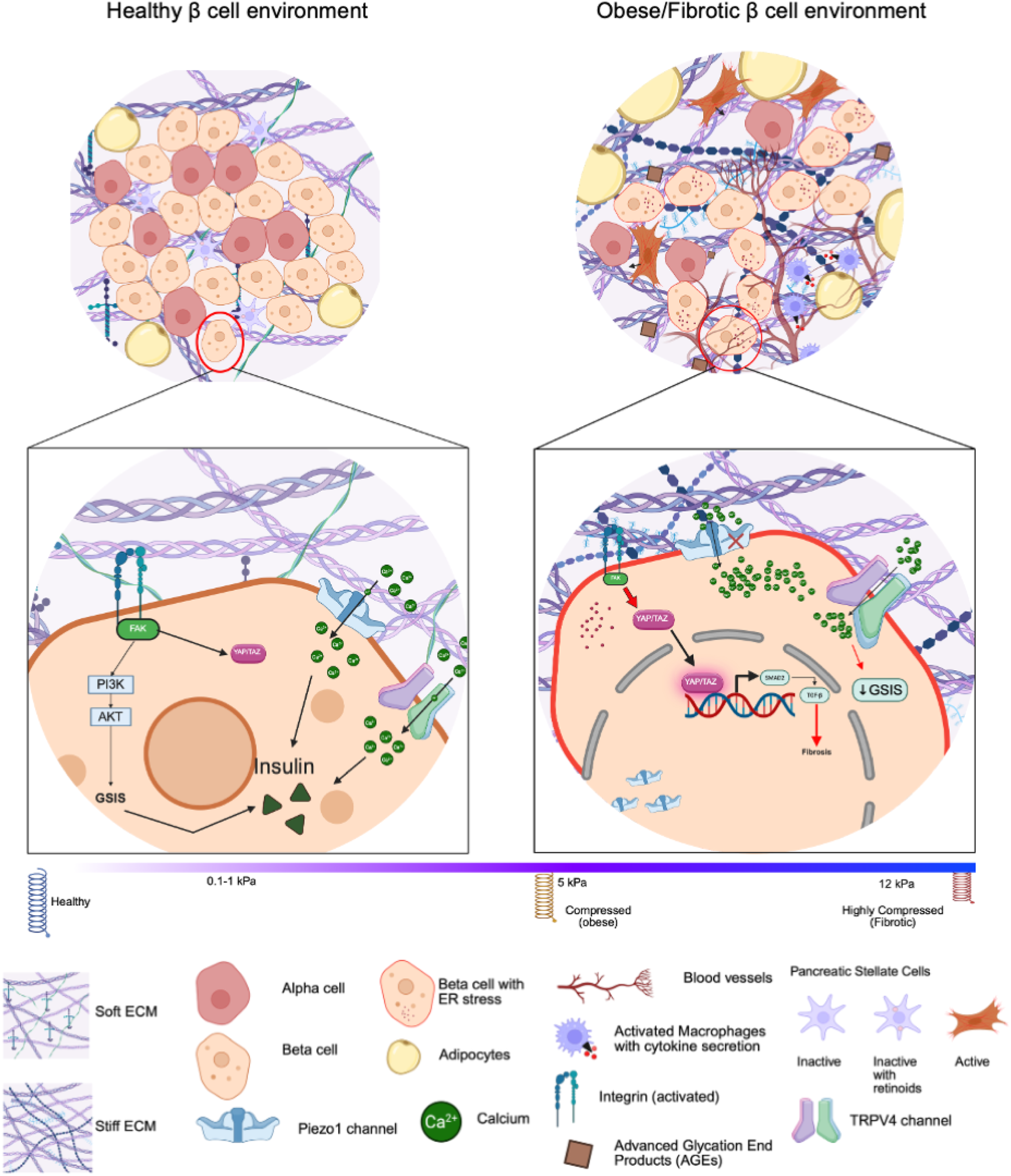
As illustrated in Figure 1, the healthy β-cell microenvironment is characterized by a compliant, loosely organized ECM composed of a thin basement membrane and sparse collagen fibers, generating a soft mechanical niche (≈0.1–1 kPa). Functional β-cells, surrounded by α-cells, quiescent pancreatic stellate cells (PSCs) containing retinoid droplets, and metabolically normal adipocytes, exhibit robust insulin secretion supported by balanced mechanotransduction. In this state, YAP/TAZ remain predominantly cytoplasmic, Piezo1 and TRPV4 channels finely regulate physiological Ca2+ influx, and integrin–FAK complexes maintain stable cell–ECM adhesion and force transmission, collectively preserving cytoskeletal organization, calcium homeostasis, and efficient glucose-stimulated insulin secretion (GSIS).
In contrast, the obese/fibrotic β-cell microenvironment undergoes pathological remodeling marked by ECM stiffening (5–12 kPa), dense collagen bundles, accumulation of advanced glycation end-product (AGE)-mediated crosslinks, increased hyaluronan deposition, and elevated collagen VI expression. β-cells within this environment exhibit pronounced dysfunction, including ER stress, inflammation, and reduced insulin granule density, accompanied by activated PSCs and hypertrophic adipocytes that further amplify fibrosis and inflammatory signaling. Mechanosensors become dysregulated, with aberrant nuclear translocation of YAP/TAZ, pathological Piezo1-mediated Ca2+ overload and mislocalization, and hyperactivation of integrin–FAK signaling, collectively driving maladaptive mechanotransduction and transcriptional dysregulation. This mechanically constrained niche establishes a fibrotic feedback loop that exacerbates cellular stress, impairs GSIS, and accelerates diabetes progression.
Beyond mechanical alterations, the ECM also provides essential biochemical cues that influence β-cell behavior. Collagen IV and laminin, produced by pericytes and endothelial cells, are critical for maintaining islet architecture and promoting insulin secretion.52,110 The dynamic nature of ECM properties allows adaptation to physiological fluctuations; however, during metabolic stress, pro-inflammatory cytokines disrupt these interactions, leading to β-cell dysfunction and apoptosis. 10 In obesity, chronic inflammation and immune cell infiltration further distort the microenvironment, creating a hostile niche that promotes β-cell death and impaired regenerative capacity. 111 Notably, islet macrophages have been reported to exert both inflammatory and regenerative roles under specific contexts, underscoring the complexity and plasticity of the β-cell microenvironment. Collectively, these findings highlight that the β-cell niche exists along a mechanobiological spectrum, where shifts in ECM composition, stiffness, and inflammatory signaling drive the transition from a supportive regenerative environment to a fibrosis-dominated, mechanically hostile state that impairs β-cell survival and function. Understanding this mechanobiological reprogramming is essential for designing therapeutic strategies aimed at restoring functional β-cell mass in diabetes.
Characteristics of the β-cell microenvironment in obese versus lean states
The β-cell microenvironment differs between obese and lean individuals. Obesity is associated with a fibrotic microenvironment characterized by increased ECM deposition, chronic inflammation, and altered mechanical properties. Contrastingly, lean microenvironment exhibits reduced ECM stiffness and inflammatory signaling.112–114 In obese individuals, ECM deposition increases and becomes stiffer due to the accumulation of collagen and other fibrous proteins,
112
impairing the β-cell function and decreasing insulin secretion by disrupting intracellular signaling pathways, which correlates with reduced β-cell responsiveness to glucose, resulting in impaired insulin release.
115
Additionally, chronic inflammation causes the release of inflammatory cytokines that promote fibrosis and lead to the β-cell apoptosis.
112
Β-cell mass increases in obese individuals compared to that in lean individuals.
116
However, this hyperplasia is often insufficient to overcome insulin resistance over time, leading to a subsequent decline in β-cell mass and function. Additionally, newly formed β-cells may not maintain normal insulin secretion capabilities.
92
Contrastingly, the lean individuals exhibit a sufficient ECM composition characterized by optimal levels of fibronectin, collagen, and laminin that support insulin secretion and β-cell adhesion, proliferation, and survival. Additionally, lean individuals have reduced inflammation, which preserves β-cell function and lowers the risk of apoptosis.
114
These observations underscore that ECM composition and mechanics, together with inflammatory status, define distinct mechanobiological states in obese versus lean pancreata, which in turn strongly influence β-cell fate and regenerative capacity. Cellular and vascular changes in the pancreatic niche under healthy and diseased conditions are summarized schematically in Figure 2. Progressive remodeling of the β-cell niche from a healthy to a fibrotic and inflammatory microenvironment. This schematic illustrates the dynamic transition of the pancreatic islet microenvironment from a physiologically compliant state to a mechanically dysregulated and fibrotic niche during diabetes progression. In the healthy condition (left), β-cells are organized within a soft, loosely structured ECM, surrounded by α- and δ-cells, quiescent pancreatic stellate cells (PSCs), and minimal immune cell presence. The ECM network is thin and spatially permissive, supporting balanced cell–cell and cell–matrix interactions, efficient paracrine signaling, and maintenance of β-cell identity and insulin secretory function. In contrast, the pathological condition (right) is characterized by ECM densification, increased fiber crosslinking, and elevated matrix stiffness, accompanied by activation of PSCs, recruitment of inflammatory immune cells, and adipocyte accumulation. These changes promote the formation of a pro-fibrotic and pro-inflammatory microenvironment, disrupting structural integrity and increasing mechanical confinement of β-cells. The altered niche impairs β-cell morphology and function, promotes cellular stress, and weakens intercellular communication, thereby contributing to progressive β-cell dysfunction and reduced insulin secretion. This figure highlights how mechanical remodeling and altered stromal–immune interactions synergistically drive the conversion of a regenerative β-cell microenvironment into a hostile, fibrosis-dominated niche, reinforcing the need for therapeutic strategies targeting ECM mechanics and cellular crosstalk in diabetes. This figure was created using BioRender.com.
Influence of ECM components on β-cell function
ECM is composed of proteins and glycoproteins play a major role in structural support and cellular signaling. Laminin, collagen, fibronectin, and heparan sulfate proteoglycans are the key components of the ECM and influence β-cell function. Each component has major and unique roles in modulating β-cell function. 33 Collagen provides structural support to the pancreatic islets and influences cellular signaling pathways and promotes β-cell proliferation and survival by binding to integrin receptors on β-cells.33,117 Laminin, particularly laminin 411 and laminin 511, plays a critical role in β-cell survival and proliferation. It binds with integrin and non-integrin receptors to enhance insulin secretion, promoting β-cell survival and proliferation.33,52,117 Fibronectin has multiple roles, including adhesion, differentiation, and migration. It binds to integrin receptors on the surface of β-cells, promoting proliferation and enhancing survival.33,117 The characteristics of the β-cell microenvironment are important for understanding how metabolic dysfunction impacts the health of the pancreas. The difference between obese/fibrotic and lean/soft environments emphasizes how crucial the mechanical properties and composition of the ECM are in regulating β-cell function.
Impact of inflammation, adipokines, and multicellular interactions on β-cells
Inflammatory cytokines such as IL-1β, IL-6, TNF-α, and IFN-γ play a significant role in β-cell stress, dysfunction, and apoptosis.118,119 Exposure to pro-inflammatory cytokines such as IL-1β and IFN-γ induces ER stress, leading to the accumulation of misfolded proteins and altering UPR signaling, triggering apoptosis in β-cells and resulting in β-cell dysfunction, impairing their insulin secretion and production.118,120 These inflammatory cytokines can alter gene expression in β-cells. Exposure to these pro-inflammatory cytokines can change chromatin structure and activate cis-regulatory elements, leading to β-cell dysfunction. Alteration in the β-cell regulatory landscape is associated with increased to autoimmune attacks in genetically predisposed individuals. 118 Chronic exposure to inflammatory mediators increases β-cell immunogenicity and decreases insulin production, making β-cells more recognizable targets for autoreactive T cells, which leads to their destruction and death during an autoimmune response. 121
The interaction between adipocytes and islets is complex and plays an important role in metabolic regulation. Adipose tissue secretes adipokines, including adiponectin, leptin, and resistin, which affect β-cell function. Under normal conditions, adiponectin enhances insulin secretion and β-cell survival, while leptin inhibits insulin secretion. 122 Adipocytes in obese individuals secrete pro-inflammatory cytokines, which create an inflammatory microenvironment that can cause β-cell stress and lead to β-cell dysfunction. 123 This inflammatory condition is exacerbated by immune cell infiltration into adipose tissue, which leads to impaired insulin resistance and glucose metabolism. 124 The interaction between adipose tissue and pancreatic β-cells involves hormonal signaling and other factors, such as the release of FFA from adipocytes. Increased FFA levels can induce lipotoxicity, leading to β-cell apoptosis and decreased insulin secretion. 122 Peripancreatic adipose tissue (PAT) is a small fat mass that surrounds the pancreas. PAT plays a role in hepatic steatosis and insulin resistance in obese individuals. 125 The communication between pancreatic β-cells and PAT modulates the function of β-cells during metabolic disorders. The interplay between adipokines and cytokines affects the β-cell microenvironment. Chronic inflammation can impair insulin secretion and promote apoptosis due to cellular stress. Additionally, dysregulation of adipokine signaling can lead to β-cell dysfunction by affecting β-cell physiology and through secondary effects due to inflammation. Addressing adipokine dysregulation and inflammation may be crucial for maintaining β-cell function and preventing disease progression. Addressing both adipokine dysregulation and inflammation may therefore be crucial not only for metabolic control but also for preserving a mechanically permissive microenvironment that supports β-cell resilience.
Multicellular interactions in disease progression
The interaction between macrophages and PSCs creates a profibrotic and proinflammatory environment that contributes to the development of diabetes. 28 PSCs secrete high amounts of Th2 cytokines such as IL-4, IL-5, and IL-13, which promote alternative M2 macrophage activation, increasing TGF-β and PDGF expression from M2 macrophages, creating a positive feedback loop that activates more PSCs. 28 Alternatively activated macrophages in the pancreas exhibit increased expression of IL-10, CD206, and TIMP2, and reduced expression of activation markers such as TNF-α. These macrophages increase the expression of matrix metalloproteinase 9, which contributes to ECM remodeling and exhibits immunosuppressive properties that may support diabetes progression. 28 The interaction between macrophages and PSCs is regulated by IL-4 receptor α signaling, as demonstrated by the inability of macrophages lacking IL-4Rα to undergo PSC-mediated alternative activation, suggesting that targeting IL-4/IL-13 signaling pathways may disrupt the pathological interaction between macrophages and PSCs.28,126
The interaction between α, β-, and δ-cells will be disrupted in diabetes, with PSCs contributing to the modulation of these interactions. Β-cells usually secrete inhibitory factors, including ZN2+, γ-aminobutyric acid, and ATP, along with insulin, to regulate α cell function. 8 Conversely, glucagon, glucagon-like peptide-1 (GLP-1), and acetylcholine are secreted from α cells to enhance insulin secretion from β-cells through paracrine signaling. 8 Previous studies revealed that glucagon binding to its receptors, such as G protein-coupled receptors and GLP-1R, on the surface of β-cells enhances GSIS. 127 δ-cells secrete somatostatin, which suppresses insulin and glucagon secretion, serving as a negative feedback mechanism to control hormone release. 8
Under diabetic conditions, high glucose and lipotoxicity impair β-cells, reducing insulin secretion and paracrine signals that inhibit α-cells, leading to increased glucagon secretion even under hyperglycemic conditions, aggravating the disease condition. 8 The ability of α-cells to enhance β-cells may be diminished due to decreased glucagon secretion or impaired paracrine signaling, further reducing the insulin secretion. Impaired somatostatin secretion or δ-cell dysfunction can worsen hyperglycemia by increasing the glucagon secretion, or cause hypoglycemia by excessively inhibiting α-cell activity, disrupting glucose homeostasis. 128 PSC activation disrupts this balance by altering the islet microenvironment and blocking cell-to-cell interactions. The fibrotic ECM secreted by activated PSCs can create a physical barrier between endocrine cells, impairing efficient paracrine signaling. Additionally, PSC-derived inflammatory mediators may influence α- and δ-cell function, disrupting glucose homeostasis.
PSCs interact with pancreatic endothelial cells to regulate the vascularization of islets. 126 PSC activation during fibrosis progression results in vascular remodeling, which is characterized by a reduction in capillary density and disrupted vessel architecture. This vascular dysfunction impairs the oxygen and nutrition supply to islets, leading to hypoxia that activates PSCs. 30 The interaction between endothelial cells and PSCs is bidirectional. In diabetes, endothelial dysfunction triggers PSC activation by releasing proinflammatory and profibrotic signaling molecules. Conversely, activated PSCs secrete factors that promote dysregulated angiogenesis and endothelial dysfunction, maintaining the pathological cycle.
These multicellular and matrix-mediated interactions emphasize that diabetes progression is not solely a β-cell–centric process but emerges from coordinated crosstalk among immune cells, stromal cells, vasculature, adipocytes, and endocrine cells within a progressively stiffening and fibrotic ECM. This systems-level mechanobiological view is essential for designing regenerative and stem cell-based interventions that can successfully operate within, or actively remodel, the diseased microenvironment.
Mechanotransduction in β-Cells: From ECM stiffening to nuclear reprogramming
Mechanical remodeling of the islet microenvironment in diabetes
Fibrotic remodeling in diabetic β-cells significantly increases tissue stiffness, with elastic moduli ranging from 5 to 20 kPa, and decreases viscosity compared to normal β-cells.12,129 This shift from a soft, stress-relaxing matrix to a dense, viscoelastically impaired ECM is caused by increased deposition of ECM proteins, including collagen, resulting from obesity-induced inflammation, hyperglycemia, and pro-fibrotic cytokines such as TGF-β.130–132 Under diabetic conditions, ECM stiffness is increased and viscoelasticity is decreased, reducing the ability of the matrix to dissipate mechanical stress and increasing force transmission to β-cells, which leads to restricted cell deformation, disrupted ECM–β-cell interactions, and impaired mechanotransduction pathways that are important for insulin secretion and β-cell survival.11,133 These changes promote β-cell dysfunction and apoptosis, increasing T2DM progression and contributing to diabetic retinopathy and nephropathy. 1 Activated fibroblasts deposit ECM and mediate inflammation, linking classical fibrotic signaling to mechanical dysfunction of the islet niche. 134 Understanding these mechanisms is essential for developing therapies that target fibrosis to improve diabetes outcomes.
Pathological stiffening of the islet ECM disrupts the normal function and regulation of mechanosensitive ion channels in β-cells, particularly Piezo1 and TRPV4. Aberrant activation or dysfunction of Piezo1 impairs Ca2+ homeostasis and disrupts cellular volume regulation.11,48 Under hyperglycemic conditions and a stiffer matrix in T2DM, Piezo 1 levels increase abnormally and translocate from the plasma membrane to the nucleus. This mislocalization decouples membrane tension from ion channel activity, impairing membrane excitability and Ca2+ influx, which affects insulin secretion. 52 Another mechanosensitive channel, TRPV4, which responds to osmotic and mechanical changes, regulates intracellular Ca2+ levels and inflammatory pathways.135–137 Abnormal stiffening of the matrix enhances the interaction between Piezo1 and TRPV4, leading to impaired Ca2+ signaling and increased cellular stress, increasing the risk of β-cell dysfunction and apoptosis.138,139 Simultaneously, increased matrix stiffness activates the mechanosenstive transcriptional co-regulator YAP, resulting in its translocation into the nucleus. 140 When YAP enters the nucleus, it disrupts normal gene expression, causing β-cells to lose their identity and functional maturity, and to dedifferentiate, which aggravates β-cell failure. 141 Thus, aberrant Piezo1 and TRPV4 signaling together with YAP activation establish a self-reinforcing mechanobiological loop of β-cell dysfunction, dedifferentiation, and apoptosis that drives diabetes progression.
The combined effects of mechanobiological stress and chronic inflammation degrade β-cell health and survival throughout the progression of diabetes. 142 Mechanical stress caused by ECM stiffening impairs β-cell homeostasis by disrupting Piezo1-mediated ion influx, resulting in abnormal cell swelling or shrinking and reduced cell viability. Abnormal ECM stiffness impairs Ca2+ oscillations, which are essential for proper insulin secretion, compromising secretory efficiency at the cellular and tissue levels. 143 β-cells initially increase insulin secretion to compensate for insulin resistance; however, sustained secretory demand in a stiff, inflammatory microenvironment leads to β-cell dysfunction, characterized by impaired insulin secretion, dedifferentiation, and loss of β-cell mass.92,144 Chronic mechanical stress and inflammation further increase β-cell stress by promoting apoptosis and elevating inflammatory cytokine release, which results in fibrosis and additional β-cell death.145,146 Chronic changes in the mechanical microenvironment caused by ECM remodeling, sustained inflammation, and oxidative stress trigger apoptosis and lead to β-cell dedifferentiation. 147 The loss of β-cell structure and mass reduces the functional β-cell reservoir, which is essential for glucose regulation, emphasizing the influence of mechanobiology in the development and progression of diabetes. The mechanical characteristics of the β-cell environment can be quantitatively mapped using high-resolution biophysical techniques, including atomic force microscopy (AFM) for nanoscale stiffness measurements,148,149 Brillouin microscopy for non-invasive profiling of viscoelastic properties, 150 and traction force microscopy (TFM) for evaluating cell-generated forces and ECM remodeling. 151 While these approaches have substantially advanced our understanding of β-cell mechanobiology, each method presents important limitations that must be considered when interpreting results in a physiological or translational context. Despite the advanced precision of current high-resolution techniques, significant limitations remain when translating findings from in vitro models to in vivo human islet physiology. Although atomic force microscopy (AFM) provides nanoscale resolution, its measurements are restricted to surface mechanics, assessing only the outer layer of islets rather than the internal multicellular architecture. In addition, the contact-based nature of AFM may introduce mechanical artifacts or non-physiological deformation.149,152 AFM also lacks sufficient temporal resolution to capture rapid mechanotransduction events, such as dynamic insulin secretion.
Similarly, TFM is largely limited to two-dimensional culture systems and therefore fails to recapitulate the complex three-dimensional organization of the islet niche, where cells experience multidirectional forces within a viscoelastic extracellular matrix. 153 In vivo, pancreatic β-cells are additionally subjected to mechanical influences such as capillary perfusion, hydrostatic pressure, and neural inputs, which are not adequately modeled in conventional in vitro platforms. 154
Although AFM and TFM remain valuable tools for mechanistic investigation, their translational relevance is limited. In contrast, Brillouin microscopy has emerged as a promising non-contact approach capable of mapping viscoelastic properties in intact human tissues. Meanwhile, magnetic resonance elastography (MRE) currently represents the only non-invasive technique suitable for in vivo assessment of pancreatic stiffness in clinical settings.15,155,156 Taken together, these limitations emphasize the need for integrative, multiscale approaches that combine complementary mechanical measurement techniques. Such strategies will be essential for accurately capturing the complex biomechanical landscape of the pancreatic niche and for translating mechanobiological insights into clinically relevant therapeutic strategies.
Mechanical stimuli and β-cell function
Mechanosensitive ion channels in the β-cells, such as PIEZO1, are activated by mechanical forces, leading to Ca2+ influx that triggers insulin release. When the PIEZO1 channel is activated, it facilitates glucose-induced insulin secretion by allowing Ca2+ influx.69,157 Mechanical stress and forces can induce deformation of the nucleus, which affects gene transcription related to insulin secretion and production.158,159 When cells undergo mechanical stress, the force is transmitted to the nucleus through the cytoskeleton, resulting in alterations in its shape and structure. This alteration can activate mechanosensitive transcription factors such as YAP and myocardin-related transcription factor A, allowing them to enter the nucleus and trigger changes in gene expression associated with insulin production.
160
Mechanical forces trigger chromatin rearrangement, facilitating the entry of transcriptional regulators to DNA and influencing gene expression essential for insulin secretion. Additionally, conditions such as ER stress, worsened by mechanical stressors, can disrupt β-cell activity and insulin secretion by activating signaling pathways such as inositol-requiring enzyme 1α (IRE), activating transcriptional factor, and IRE-JNK signaling pathway, leading to cellular dysfunction.
142
This mechanism emphasizes the impact of mechanical stimuli on the β-cell structure and function, including insulin release, highlighting the crucial role of mechanotransduction in regulating cellular activities. These mechanical stimuli can also influence metabolic pathways in β-cells. Alterations in ECM stiffness have been linked to dysfunction in glucose metabolism and reduced insulin secretion. By modulating metabolic enzymes such as phosphofructokinase (PFK), mechanical forces can affect insulin secretion dynamics.
47
ECM stiffness regulates insulin secretion through PFK activity; however, increased stiffness can lead to insulin dysfunction.
47
Thus, mechanical forces profoundly regulate insulin secretion by coordinating nuclear deformation, chromatin accessibility, metabolic signaling, and mechanotransductive pathways. Figure 3 summarizes the integration of soluble and insoluble cues that generate mechanical stimuli, activating integrins, Piezo1, TRPV4, and Hippo signaling pathways to regulate β-cell survival and proliferation. Mechanoresponsive signaling pathways. Schematic overview of key mechanotransduction cascades in β-cells, including integrin–FAK–RhoA/ROCK, MAPK (ERK), PI3K–AKT, and YAP/TAZ pathways. JNK, c-Jun N-terminal kinase; ERK, extracellular signal-regulated kinase; YAP, Yes-associated protein; MKK, mitogen-activated protein kinase kinase; MAPK, mitogen-activated protein kinase. This figure was created using BioRender.com.
Mechanosensors and signaling pathways in β-cells
β-cells possess diverse mechanosensors, including primary cilia, ion channels, glycocalyx, and integrins, that detect mechanical signals and regulate cellular activity. 9 Primary cilia are crucial for detecting mechanical signals and affect the regulation of β-cell activity. 161 Integrins play an important role in mechanosensing by responding to the stiffness of the ECM, which influences the activation and functionality of β-cells. 9 Additionally, Piezo1 channels are important ion channels that detect mechanical forces and facilitate the entry of Ca2+, which is essential for insulin secretion and the functioning of β-cells. 69 When mechanical forces are exerted, these sensors activate various intracellular signaling pathways that contribute to β-cell function. Together, these mechano-receptors form a distributed “sensing network” that couples ECM properties to intracellular biochemical programs and transcriptional outputs. The activation of piezo1 initiates a cascade of intracellular reactions that enhance calcium signaling and electrical activity, both of which are essential for insulin secretion. 69 Furthermore, primary cilia and integrins play an important role in mechanotransduction pathways that alter cellular response to mechanical stimuli, affecting the differentiation and proliferation of β-cells.9,161
Role of Piezo1 channels in regulating β-cell functions
Piezo1 is a mechanosensitive, non-selective channel that is activated mechanically and plays an important role in mechanotransduction. It responds to mechanical forces such as membrane stretching or pressure, allowing the entry of specific ions such as Ca2+, K+, and Na+ into the cell.162–164 When the cell membrane interacts with mechanical stress, Piezo1 gets activated, resulting in conformational changes that open the channel and facilitate ion influx, which is important for insulin secretion.165,166 In β-cells, Piezo1 plays a crucial role in glucose-induced insulin secretion. When glucose levels rise, intracellular glucose metabolism increases osmotic pressure, causing β-cells to stretch and activate the Piezo1 channel, resulting in Ca2+ influx that triggers insulin granule exocytosis. 69 Specific Piezo1 channel agonists, such as Yoda1, can stimulate Ca2+ signaling and enhance insulin secretion from β-cells lines. 167 Piezo1 knockout impairs glucose tolerance and reduces insulin secretion. 69
In T2DM, Piezo1 expression increases but its functional capacity decreases due to translocation from the plasma membrane to the nucleus under elevated glucose conditions, uncoupling mechanical strain from channel opening and impairing electrical activity. Overall, mechanotransduction mediated by Piezo1 is important for effective insulin secretion and may contribute to the dysfunction observed in T2DM. 69 When Piezo1 channels play a role in Ca2+ influx in response to mechanical stimuli, other mechanisms contribute to Ca2+ signaling in β-cells. Ca2+ influx pathways include voltage-gated calcium channels (VGCC) and ATP-regulated potassium (K-ATP) channels. VGCCs are essential for mediating Ca2+ influx during glucose-induced insulin secretion, allowing Ca2+ ions to enter the cell and leading to exocytosis of insulin-containing secretory granules. 168 Activation of VGCCs occurs when the cell membrane depolarizes following K-ATP channel closure due to increased ATP levels from glucose metabolism. 169 Ca2+ influx through VGCC is crucial for insulin secretion.169–171 Unlike piezo1, which responds to mechanical stimuli, VGCCs are activated by changes in membrane potential. 170 K-ATP channels are essential for connecting cellular metabolism to β-cell electrical activity. When glucose and ATP levels are elevated, the closure of the K-ATP channel occurs, causing membrane depolarization and activating VGCCs, which leads to insulin release. 171 Thus, Piezo1 provides a rapid mechanosensitive Ca2+ entry route that complements the metabolically driven K-ATP–VGCC axis, together shaping the amplitude and timing of insulin secretion.
Role of YAP signaling pathway in β-cell function
Yes-associated protein (YAP) is a central transcriptional coactivator of the Hippo signaling pathway that regulates tissue homeostasis, cell proliferation, and survival.172–174 In β-cells, YAP activity is tightly controlled by upstream kinases such as large tumor suppressor kinases (LATS1/2), which phosphorylate YAP and retain it in the cytoplasm. When Hippo signaling is suppressed, YAP translocates into the nucleus and interacts with TEAD family transcription factors to activate genes associated with cell growth, survival, and metabolic adaptation.172,173 Physiologically, transient YAP activation promotes β-cell proliferation and enhances resistance to metabolic stress, supporting its potential role in regenerative responses. Indeed, YAP overexpression has been shown to increase β-cell mass and preserve insulin-producing capacity. 66 However, sustained or excessive YAP activation disrupts normal differentiation programs, underscoring the importance of tightly regulated signaling. In particular, prolonged YAP activity impairs the maturation of pancreatic progenitor cells into fully functional β-cells, highlighting the delicate balance between proliferation and differentiation required for maintaining pancreatic homeostasis. 67
YAP also regulates metabolic functions critical for glucose homeostasis and β-cell survival. By modulating gluconeogenic pathways, YAP influences glucose tolerance and represents a potential therapeutic target for diabetes.66,173 In β-cells, YAP exhibits pro-proliferative and antiapoptotic effects. YAP overexpression enhances β-cell proliferation without compromising insulin secretory capacity, thereby supporting β-cell mass expansion. 66 However, precise control of YAP activity is essential, as excessive or prolonged activation poses safety concerns, including tumorigenesis and fibrosis. 175 YAP exhibits context-dependent regenerative behavior: transient nuclear localization promotes β-cell proliferation and survival, whereas sustained activation leads to dysfunction. 66 Persistent YAP signaling is a known driver of pancreatic tumorigenesis, and in pancreatic ductal adenocarcinoma (PDAC), YAP acts as a critical oncogenic factor that sustains tumor growth even in the absence of KRAS signaling.176,177 In fibrotic microenvironments, increased matrix stiffness promotes YAP nuclear localization in pancreatic stellate cells, inducing myofibroblastic differentiation, α-SMA expression, and collagen deposition. 178 In contrast, YAP activity is markedly reduced in mature β-cells, where its downregulation is associated with maintenance of a differentiated and functional state. 67 Importantly, accumulating evidence indicates that sustained YAP activation suppresses key endocrine transcription factors, thereby maintaining cells in a progenitor-like state and preventing terminal differentiation. 179
Sustained activation of YAP signaling can induce β-cell dedifferentiation, shifting their cellular behaviour from insulin secretion toward proliferation. To achieve controlled activation, inducible systems such as doxycycline-responsive (Tet-ON/OFF) platforms enable reversible and temporally precise YAP regulation. Similarly, optogenetic tools such as OptoYAP allow light-triggered, reversible nuclear translocation of YAP with millisecond precision, enabling transient activation that may promote β-cell proliferation followed by redifferentiation and functional maturation. 180 In parallel, biomaterial-based approaches offer additional control over YAP activity. Hydrogels with tunable stress-relaxation properties can modulate YAP signaling by permitting cell spreading while preventing sustained mechanical tension that drives fibrotic YAP hyperactivation. 181 Furthermore, YAP-modulating agents, including targeted siRNAs or small molecules such as verteporfin, can be delivered using engineered nanoparticles functionalized with β-cell–specific ligands such as GLP-1 receptor ligands to minimize off-target effects on pancreatic stellate cells or exocrine tissue. 182 Collectively, these strategies highlight the importance of temporally controlled YAP activation to promote β-cell proliferation while preserving functional identity.
The interaction between YAP and the mechanical environment is central to its regulatory function in β-cells. YAP acts as a mechanosensor that integrates signals from the ECM to influence cellular fate decisions.9,183 Variations in ECM stiffness modulate YAP activity, with increased mechanical tension promoting nuclear translocation and transcriptional activation, while softer substrates restrict YAP activity and favor cellular quiescence. 140 By sensing mechanical signals, β-cells can adapt their growth and functions in response to their microenvironment, which is essential for maintaining tissue homeostasis. YAP plays a crucial role in mechanotransduction by regulating genes that are important for focal adhesion formation and cytoskeletal dynamics. 184 By modulating the Hippo–YAP axis, mechanical cues directly influence β-cell morphology, differentiation, and function, underscoring the importance of biomechanical regulation in maintaining β-cell homeostasis and highlighting YAP as a key therapeutic target in diabetes. 185
The YAP/TAZ pathway is therefore a central regulator of mechanotransduction, processing mechanical stimuli such as environmental stiffness and cellular morphology to influence β-cell behavior.186,187 In stiff ECM environments, YAP/TAZ proteins translocate into the nucleus, where they activate genetic pathways governing cell proliferation and ECM remodeling. This process is mediated by biochemical signaling cascades that link cytoplasmic rigidity to nuclear dynamics. 186 Dysregulated YAP/TAZ signaling in β cells can result in fibrosis and impaired insulin secretion. 173 Increased ECM stiffness activates YAP/TAZ through integrin/FAK signaling, driving pathological microenvironment stiffening via enhanced ECM production. 188 YAP/TAZ activation increases the fibrotic gene expression, leading to ECM deposition.
Role of integrin-mediated signaling in β-cell function
Integrins play an important role in mechanotransduction by linking the ECM to the cytoskeleton. Mechanical stimulation activates integrins, which activate FAK and initiate a signaling cascade involving Rho family GTPases. This pathway is crucial for cytoskeletal rearrangement and for cellular responses to mechanical stimuli. 189 Integrin-mediated mechanotransduction begins at adhesion sites where integrins connect to the ECM and F-actin cytoskeleton, facilitating the conversion of mechanical forces produced by actin and myosin-II. 190 Mechanotransduction involves several mechanisms. Initially, mechanical stimuli induce conformational changes in integrins, increasing their binding affinity for ECM ligands. This interaction activates FAK and Src kinases, leading to downstream signaling pathways that regulate RhoA activity, which is important for cytoskeletal dynamics.191,192 Furthermore, the mechanical forces at these adhesion sites can modulate the activity of mechanosensitive proteins, triggering immediate cellular responses and long-term changes in gene expression. 190 Disruption of integrin-mediated signaling is associated with various diseases, emphasizing its significance in maintaining tissue homeostasis and cellular function. 190 In β-cells, inappropriate activation of integrin–FAK–RhoA signaling on stiff or glycated matrices contributes to altered cell shape, impaired granule trafficking, and defective insulin release.
Role MAPK signaling pathway in β-cell function
Mechanical forces such as stress and stretch can activate MAPK, particularly ERK1/2, through integrin-mediated signaling, promoting cell survival and proliferation in β-cells, which are important for insulin production.189,193 The MAPK pathway interacts with other signaling pathways, resulting in a complex interplay of signals from mechanical stimuli and growth factors. This interaction is crucial for various cellular activities, including the regulation of apoptosis and differentiation. The intricate mechanisms involve multiple upstream signals that converge on downstream MAPKs, regulating transcription factors and other proteins to produce cellular responses. 194 Thus, MAPK signaling represents a key biochemical hub through which β-cells integrate mechanical and hormonal cues.
Role PI3K-AKT pathway in β-cell function
The PI3K-AKT pathway regulates both the β-cell mass and function. It is activated by various signals, including insulin, which triggers the phosphorylation of phosphatidylinositol-4,5-bisphosphate to produce phosphatidylinositol-3,4,5-triphosphate (PIP3).2,195 PIP3 recruits and activates AKT, a serine/threonine kinase that promotes cell survival and inhibits apoptosis. 195 Mechanical signals can increase PI3K activity, leading to AKT activation and promotion of β-cell survival. 2 Furthermore, the PI3K-AKT pathway regulates insulin secretion through several targets that influence glucose metabolism and cellular growth. 195 Its diverse functions include the regulation of apoptosis, metabolism, and cell cycle progression. 195 Targeting the PI3K-AKT pathway is a therapeutic strategy that improves β-cell function and inhibits apoptosis in T1DM. 196 By identifying specific modulators of this pathway, strategies that improve β-cell health without inducing excessive proliferation can be developed, which could lead to oncogenic risks. 196 Overall, the PI3K-AKT pathway may be a promising target for therapeutic approaches that maintain and preserve β-cell function.
Nuclear mechanotransduction, PDX1, chromatin remodeling, and therapeutic targeting
PDX1 is a crucial transcription factor essential for the development, maintenance, and function of pancreatic β-cells responsible for insulin production. 197 It is indispensable for preserving β-cell identity and transcriptional programs governing insulin biosynthesis and secretion. PDX1 regulates insulin gene expression and participates in early pancreatic morphogenesis, influencing both endocrine and exocrine lineage specification. 198 Its expression is tightly controlled through multiple signaling networks and cis-regulatory regions; areas I–III coordinate embryonic developmental processes, whereas area IV is key for mature β-cell function and glucose responsiveness. 199 Dynamic regulation of PDX1 throughout the cell cycle reflects its central role in modulating β-cell proliferation, differentiation, and functional adaptation to environmental stimuli. 200
Mechanical forces propagating from the extracellular matrix through the cytoskeleton induce deformation of the nucleus and reorganization of chromatin architecture, thereby influencing transcriptional accessibility and gene expression.158,159 These biomechanically induced chromatin alterations regulate the recruitment and activity of transcription factors such as PDX1 at insulin regulatory loci, linking nuclear mechanics to β-cell fate determination. Mechanotransduction-associated mediators, including vascular endothelial growth factor (VEGF) and endothelial nitric oxide synthase (eNOS), further modulate PDX1 activity under mechanical stress. 198 VEGF supports β-cell survival and vascular integrity in mechanically strained environments, while eNOS-derived nitric oxide confers cytoprotective effects during biomechanical challenge.182,185 Additionally, transient receptor potential (TRP) channels contribute to Ca2+-dependent transcriptional modulation by responding to mechanical deformation and integrating mechanosensitive signaling with insulin secretion dynamics.198,201,202 Collectively, these findings position PDX1 as a central integrator of mechanical, metabolic, and transcriptional governance of β-cell identity.
Importantly, this mechanoregulation of nuclear architecture and transcriptional identity provides a direct foundation for therapeutic intervention. Mechanotransduction pathways are critical regulators of β-cell function and insulin signaling, and their dysregulation contributes directly to the pathogenesis of diabetes. Mechanical stimuli derived from the ECM strongly influence β-cell behavior, and alterations in matrix stiffness significantly impair β-cell proliferation and functional competence. 115 Modulating ECM properties and targeting mechanotransductive signaling cascades therefore offers a strategic avenue to enhance β-cell survival and restore insulin secretory capacity in diabetic conditions. Furthermore, mechanical forces such as shear stress modulate insulin signaling pathways and improve peripheral insulin sensitivity, highlighting the systemic relevance of biomechanical regulation. 159
Several promising mechanotransduction-based therapeutic targets emerge from this framework. Modulation of YAP/TAZ signaling has been shown to enhance β-cell proliferation and augment insulin secretion, 67 while targeting Rho GTPase signaling improves cytoskeletal organization and insulin responsiveness. Stem cell-based strategies further harness these principles to replace dysfunctional β-cells with newly generated insulin-producing cells. Induced pluripotent stem cells (iPSCs) can be differentiated into β-like cells,203,204 and incorporation of optimized mechanical cues during differentiation significantly improves maturation and functional competence. The integration of mechanotransduction modulation with stem cell therapy thus represents a powerful approach to improve engraftment, survival, and physiological insulin output.
In parallel, several pharmacological agents demonstrate mechanobiology-informed protective effects. Small molecules such as ALK5 inhibitors, harmine, and 5-ludotubercidin promote β-cell proliferation and protect against dedifferentiation by suppressing apoptotic and stress-responsive signaling.205,206 Antioxidants such as N-acetyl cysteine reduce oxidative damage induced by glucotoxicity and lipotoxicity, preserving β-cell viability and limiting apoptosi. 206 For type 1 diabetes mellitus (T1DM), immunomodulatory strategies aimed at selectively targeting autoreactive T cells, including monoclonal antibody-based interventions, offer protection against autoimmune β-cell destruction while preserving systemic immune competence.207,208
Finally, enhancement of β-cell function through established pharmacological interventions such as metformin and GLP-1 receptor agonists, combined with lifestyle modifications, remains a cornerstone strategy for diabetes management. Nevertheless, emerging mechanotransduction-centered approaches, particularly when integrated with stem cell-based regenerative therapies, represent a next-generation therapeutic paradigm capable of restoring β-cell identity, functionality, and long-term viability. Advancing this mechanobiology-driven strategy may yield transformative therapeutic platforms for improving glycemic control and clinical outcomes in diabetes.
Calcium signaling, stem cell-derived β-cells, and mechanobiology-guided therapeutic platforms
Role of calcium influx mechanisms in regulating β-cell functions
Calcium signaling pathways and insulin secretion
Ca2+ plays an important role in various cellular activities, particularly in β-cells.
209
The regulation of intracellular calcium concentration is essential for insulin secretion. The closure of K-ATP channels due to increased ATP production from glucose metabolism causes Ca2+ influx, triggering insulin release from β-cells (Figure 4).
210
Ca2+ is a secondary messenger in β-cells, activating several kinases and phosphatases that regulate cellular functions such as cell proliferation, apoptosis, and gene expression.209–211 Dysregulation of Ca2+ homeostasis can impair insulin secretion, leading to disease progression. Chronic elevation of cytosolic calcium levels can induce β-cell apoptosis and dysfunction, underscoring the need to maintain optimal calcium levels within β-cells.211,212 Calcium influx mechanism. Overview of major Ca2+ entry and release pathways in β-cells, including K-ATP channels, voltage-gated calcium channels (VGCCs), mechanosensitive Piezo1 channels, and transient receptor potential (TRP) channels. VGCC, voltage-gated calcium channels; TRP, transient receptor potential channels. This figure was created using BioRender.com.
Calcium release from the ER through ryanodine receptors and inositol triphosphate receptors further increases insulin production during high glucose levels. This phenomenon, termed calcium-induced calcium release, occurs when a Ca2+ influx triggers further release of calcium, enhancing the overall calcium signal necessary for effective insulin release.209–212
The Piezo1 channel is an important component of mechanotransduction, responding to mechanical stimuli through membrane tension rather than changes in membrane potential. 213 Piezo1 differs from other calcium channels, such as VGCCs and TRP channels, because of its unique mechanosensitivity. Its distinct activation mechanism enables it to respond to mechanical forces, including shear stress and membrane stretch. During osmotic stress, Piezo1 mediates calcium influx, helping cells regulate their volume through mechanisms such as regulatory volume decrease (RVD). 143 VGCCs play an important role in insulin release in response to elevated glucose levels. Once glucose enters β cells, it is metabolised, leading to an increase in ATP levels, closure of K-ATP channels, membrane depolarization, and opening of VGCCs. 169
Role of TRP channels in calcium influx and insulin secretion
TRP channels play an important role in calcium signaling and insulin secretion in β-cells. They act as molecular sensors, responding to mechanical stress, temperature, and osmotic changes. TRP channels participate in Ca2+ signaling through various pathways that respond to hormonal cues or changes in osmotic pressure. TRPC1, TRPC3, and TRPC4 are involved in insulin secretion. TRPC1 and ORai1 mediate Ca2+ influx and are regulated by stromal interaction molecule 1. 214 Additionally, PKC-α activation can promote insulin secretion through TRPC1 phosphorylation. 215 TRPC3 activation can enhance insulin secretion and lead to Ca2+ influx and membrane depolarization, whereas TRPC4 is activated by leptin and phosphorylated by PI3K, promoting Ca2+ influx and increasing insulin secretion. 214 TRPC6 specifically mediates Ca2+ entry, maintaining cell volume through RVD.209,216
TRPV family channels also contribute to β-cell function. TRPV1 activation is triggered by insulin secretion, leading to neuronal release of calcitonin gene-related peptide and substance P, which modulate insulin levels to maintain a balance between neuropeptides and glucose concentration. TRPV2 translocates to the plasma membrane in response to glucose stimulation and mediates Ca2+ influx, enhancing β-cell proliferation and insulin secretion. 214 TRPV2 translocation can be inhibited by somatostatin and diazoxide, blocking glucose-stimulated insulin secretion, whereas suppression of TRPV2 activation by tranilast impairs glucose-stimulated insulin secretion and cell proliferation. 217 TRPV4 activation increases Ca2+ influx and insulin secretion but can impair cell viability and induce ER stress. 217
Among TRPM channels, TRPM2 activation enhances Ca2+ influx and membrane depolarization, which in turn activates VGCCs. ROS-activated TRPM2 promotes mitochondrial dysfunction and leads to β-cell apoptosis, 214 facilitating a sustained flow of Ca2+ entry that interacts with intracellular calcium to maintain optimal insulin release. TRPM5 is an intracellular calcium-dependent channel activated by increases in intracellular calcium; it depolarizes the membrane through sodium influx, opening VGCCs to promote glucose-stimulated insulin release. TRPM5 activated by PKC can further stimulate these channels to enhance their depolarizing effect and insulin secretion. 214 TRPM7 enhances Ca2+ influx under certain conditions and indirectly influences β-cell function. The ability of TRP channels to modulate Ca2+ homeostasis makes them promising targets for diabetes treatment. Selective modulation of these channels could potentially enhance insulin secretion and preserve β-cell function.
Stem cell-derived β-cells and the impact of mechanical cues
Engineering the biomechanical microenvironment for functional maturation of stem cell-derived β-cells
Human pluripotent stem cells, such as embryonic stem cells (ESCs) and iPSCs, are promising sources of β-cells through differentiation methods that mimic the natural development of the pancreas.43,218,219 Additionally, human tonsil-derived mesenchymal stem cells are novel autologous β -cell sources with immunomodulatory properties and differentiation potential toward endocrine lineages.220–222 However, the immaturity of SC-β cells, which results in reduced glucose sensitivity and insulin secretion compared to adult β-cells, is a limitation.223,224 The persistent immaturity is correlated with the absence of key microenvironmental factors, particularly with biochemical signals essential for endocrine cell specification, development, and maturation.225,226
Substrate stiffness plays an important role in β-cell development and function. Softer matrices, with elastic moduli similar to those of the natural pancreatic ECM around 0.5 kPa, enhance endocrine differentiation, whereas stiffer substrates over 5 kPa promote fibrotic or dedifferentiated phenotypes.227,228 Viscoelastic materials that allow stress relaxation over time more accurately mimic physiological environments, promoting the cytoskeletal remodeling and focal adhesion dynamics conducive to β-cell development.60,229–231 Dynamic mechanical stimuli, such as cyclic stretching or fluid shear stress, further modulate gene expression and insulin secretory responses by activating mechanosensitive pathways, including Piezo1 and YAP/TAZ signaling.189,232
Hydrogels engineered with precise stiffness, gradient, and viscoelastic nature can provide an environment similar to that of the physiological β-cell microenvironment.233–236 These platforms facilitate the exploration of mechanical memory, where stem cells preserve lineage-specific markers following exposure to a specific stiffness environment. 237 The ability of hydrogel biomaterials to alter their stiffness demonstrates mechanical memory in skeletal muscle stem cells. 237 Advanced biofabrication methods, such as three-dimensional bioprinting and the fabrication of stiffness gradient gels, allow for the precise organization of cells and ECM components, mimicking β-cell architecture and vascularization.234,238,239 These constructs, combined with microfluidic organ-on-chip devices, simulate physiological flow and nutrient transport, essential for long-term β-cell function and survival. 240 These platforms supports real-time observation of mechanotransduction and functional markers in precisely controlled mechanical environments, increasing the optimization of SC-β cell therapies.9,241,242
Macroencapsulation and next-generation islet replacement systems
One of the most recent advancements in islet transplantation involves efficient macroencapsulation systems that eliminate the need for prevascularization. These novel biomaterials can be transplanted subcutaneously without requiring prevascularization at the transplant site. The engineered environment mimics essential biological and mechanical signals, promotes effective immunoprotection, and supports rapid graft engraftment. These materials demonstrated a clear outcome; they improve insulin output and glycemic control while reducing fibrotic encapsulation and inflammation compared to conventional transplantation techniques. This significant development represents a minimally invasive and clinically effective islet replacement therapy for individuals with diabetes. 243 This development represents a minimally invasive and clinically promising islet replacement therapy for individuals with diabetes.
Strategic engineering of biological microenvironments
Bioengineered ECM-mimetic and dynamic matrices for β-cell support
Bioengineered pancreatic microenvironments aim to mimic biochemical and mechanical cues of the physiological ECM.244,245 Hydrogels functionalized with basement membrane components such as collagen IV and laminin, combined with RGD peptide motifs, enhance β-cell adhesion and survival through integrin receptors42,246 and have tunable storage (G’) and loss (G”) moduli, allowing precise control of stiffness and viscoelasticity to replicate the properties of healthy β-cells of approximately 0.5–1 kPa.247,248 Such ECM-mimetic matrices enhance mechanotransduction signaling and promote insulin gene expression in stem cell-derived β-cells.9,11,58
Integrating viscoelastic and enzymatically degradable crosslinkers enables dynamic matrix remodeling by encapsulated cells, which is important for proliferation, migration, and functional maturation.231,249,250 Hydrogels fabricated using hyaluronic acid and polyethylene glycol (PEG) with MMP-sensitive crosslinkers mimic the natural remodeling of pancreatic ECM, allowing β-cells to modulate their mechanical and biochemical microenvironment. 251 This adaptability promotes long-term insulin secretion and enhances engraftment potential post-transplantation.54,252
Advanced engineering strategies for optimizing β-cell microenvironments
Pharmacologically targeting mechanotransduction pathways within engineered microenvironments can further optimize β-cell function. YODA1, a small-molecule Piezo1 agonist, enhances Ca2+ influx and insulin secretion in vitro by mimicking physiological mechanical stimuli.69,167 TRPV4 modulators or agonists, such as GSK1016790A and 4α-Phorbol 12-13-dicaprinate (4α-PDD), modulate Ca2+ signaling and inflammatory pathways, and is another method for mechanical regulation.135,253 Additionally, lysyl oxidase inhibitors reduce pathological collagen crosslinking, thereby decreasing ECM stiffness and fibrosis in diabetic models and potentially restoring a favorable microenvironment for β-cell proliferation and survival. 254
To overcome immune rejection and hypoxia-related graft loss, bioengineered microenvironments integrate immunoprotective coatings, such as PEGylation, zwitterionic polymers, or alginate encapsulation with reduced antigenicity. 255 Incorporating oxygen-generating particles or perfluorocarbons enhances β-cell survival in extrahepatic sites with poor vascularization.256,257 Integrating these approaches with mechanical tuning results in a multifaceted niche that facilitates long-term engraftment and the functional integration. 34
Targeting pancreatic stellate cells (PSCs) and fibrosis in mechanobiology-guided therapy
PSCs play an important role in the progression of pancreatic fibrosis and β-cell dysfunction. Targeting their activation and function has therapeutic potential. 6 Preclinical studies have demonstrated the efficacy of various approaches, including mechanotransduction regulators, anti-fibrotic agents, and antioxidant therapies. Considering how matrix stiffness contributes to PSC activation and β-cell dysfunction, therapies targeting mechanosensitive pathways may offer novel therapeutic strategies. The use of Piezo1 channel modulators considerably rescues GSIS in stiffer matrix environments. 11
Targeting major fibrotic pathways, such as Hedgehog, TGF-β, and PDGF signaling, can reduce the activation of PSC and ECM deposition.29,258 In preclinical studies, inhibiting the hedgehog signaling pathway using cyclopamine considerably reduced collagen synthesis, fibroblast proliferation, and myofibroblast differentiation. 258 PSC activation can be suppressed by glutathione through inhibition of the ROS/TGF-β/SMAD signaling cascade. 7 Clinical studies have revealed a reduction of glutathione levels in erythrocytes from patients with T2DM, supporting antioxidant therapy. 7 Additionally, N-acetyl-cysteine helps protect β-cells from oxidative stress-induced damage and inhibits the activation of PSCs. 30
Integrating PSC intervention in diabetes stem cell therapy
Stem cell-based therapies for diabetes must consider PSCs in disease pathogenesis and therapeutic outcomes. Several methods involve targeting PSCs with regenerative techniques such as reprogramming PSCs, microenvironment optimization, ECM engineering, and therapeutic combinations. Previous research suggests that PSCs exhibit stem cell characteristics, express various stem cell markers, and have the potential to differentiate into multiple cell types. 6 Under certain conditions, PSCs can differentiate into insulin-producing β-cells, providing potential insights into β-cell replacement therapy. Common signaling pathways between pancreatic stem cells and PSCs, including PI3K, TGF-β, MAPK, and Wnt/β-catenin, are potential mechanisms influencing PSC differentiation. 6
Optimizing the tissue microenvironment is crucial for successful stem cell therapy to support the function and engraftment of transplanted cells. This approach includes reducing the activation of PSC and fibrosis before transplantation, modulating inflammation through M2 macrophage polarization, and promoting vascularization to enhance engraftment and cell survival.126,259 Developing biomaterial scaffolds that mimic the characteristics of healthy pancreatic ECM can promote the regeneration of native β-cells and the function of transplanted cells.117,260 Human pancreatic ECM-based hydrogels may improve the survival and function of islets. 260
The most promising strategy is to combine stem cell-based transplantation with PSC-targeted interventions. For example, combining pluripotent stem cell-derived β-cells with mesenchymal stromal cells has significantly enhanced vascularization and cardiac function. 261 Mesenchymal stromal cells exert anti-inflammatory and antifibrotic effects by promoting vascularization and may also promote β-cell maturation.
Technical challenges in differentiating stem cells into functional β-cells
The differentiation of patient-derived stem cells into functional β-cells is complex and not yet fully understood. 119 Despite substantial progress, several challenges hinder the production of mature β-cells, including inefficient differentiation protocols, immature β-cell phenotypes, suboptimal microenvironments, and tumorigenicity risks. Current protocols for differentiating iPSCs and human embryonic stem cells into pancreatic progenitor cells are often inefficient, resulting in a low yield of mature insulin-producing β-cells. 119 Additionally, patient-derived stem cells are ineffective at differentiating into functional β-cells, which may lack proper glucose responsiveness.262,263
A further challenge lies in maturation: stem cell-derived β-cells may fail to express key markers associated with mature β-cell activity, such as NKX6.1 and MAFA, which are required for appropriate insulin secretion. 263 The β-cell microenvironment strongly affects the differentiation process, with factors such as growth factors, cell–cell interactions, and ECM composition influencing the efficiency and outcomes of differentiation protocols. Co-culturing pancreatic stromal cells with stem cell-derived progenitors can enhance their maturation into functional β-cells. 262
Tumorigenicity remains a major concern, as undifferentiated stem cells can form teratomas and tumors if not rigorously removed before transplantation, representing a serious safety risk. 262 Addressing these technical issues related to maturation, differentiation efficiency, and safety is essential for developing effective and clinically viable β-cell replacement therapies.
Integrate mechanobiological platforms for β-cell functional profiling
Recent advances in live-cell imaging and biosensors enable real-time imaging of β-cell volume and membrane tension, which is essential for understanding mechanotransduction. 264 Fluorescent tools such as Flipper-TR and genetically encoded sensors allow visualization of real-time changes in plasma membrane mechanics under physiological and pathological conditions.265,266 These tools help reveal how mechanical stimuli influence ion channels such as Piezo1, Ca2+ influx, and insulin secretion in live β-cells.9,190
The integration of single-cell transcriptomics, epigenomics, and proteomics with mechanical phenotyping is revolutionizing our understanding of β-cell heterogeneity and mechanobiology.267,268 Mechano-transcriptomic methods combine cell stiffness measurements using AFM or micropipette aspiration with RNA sequencing to correlate mechanical properties with gene expression profiles.269,270 This comprehensive strategy has revealed that mechanosensitive pathways, including Piezo1 and YAP/TAZ, play critical roles in β-cell differentiation and dysfunction within diabetic ECM microenvironments.9,271
High-throughput platforms incorporating stiffness-gradient hydrogels or microfabricated arrays enable systematic screening of pharmacological agents and biomaterials for their effects on β-cell mechanotransduction and function. 9 These microarrays expose cells to precisely controlled variations in matrix stiffness and viscoelasticity, allowing analysis of mechanical responses such as Ca2+ signaling, YAP nuclear localization, and insulin secretion.272,273 Such screening systems facilitate rapid identification of optimal mechanopharmacological strategies to enhance the maturation and engraftment of SC-β cells and to design next-generation, mechanobiology-informed therapies for diabetes.
Conclusion
β-cell function, survival, and fate are increasingly recognized to be governed not only by biochemical signaling but also by the mechanical and biological properties of the pancreatic extracellular matrix (ECM). Under physiological conditions, β-cells reside within a compliant, viscoelastic niche that supports coordinated mechanotransduction through key sensors and pathways such as YAP/TAZ, Piezo1, and TRPV4. These coordinated mechanical cues promote efficient insulin secretion, support β-cell maturation, and help maintain systemic metabolic homeostasis. In contrast, diabetic and obese states are characterized by pathological ECM remodeling, including fibrosis and increased stiffness, which disrupt mechanosensitive signaling, impair Ca2+ homeostasis, and drive β-cell dedifferentiation and dysfunction. This altered biomechanical environment is further amplified by chronic inflammation, dysregulated adipokine signaling, and immune cell infiltration, establishing a deleterious feedback loop that accelerates metabolic dysfunction and progressive β-cell loss.
Emerging therapeutic strategies increasingly integrate biomaterial engineering with targeted pharmacological interventions to restore a favorable β-cell microenvironment. Regenerative approaches, including stem cell-derived β-cell (SC-β) transplantation, require precisely engineered niches that recapitulate the biochemical and mechanical features of healthy pancreatic ECM. Tunable hydrogels, viscoelastic matrices, and mechanomodulatory compounds such as YAP/TAZ regulators or Piezo1 modulators represent promising tools to overcome maturation barriers and enhance graft survival. In parallel, pharmacological agents including GLP-1 receptor agonists and antifibrotic therapies may synergistically normalize ECM stiffness, suppress inflammatory signaling, and support long-term β-cell function.
Despite these advances, critical knowledge gaps remain regarding how specific mechanical parameters govern β-cell fate decisions in vivo. In particular, defining quantitative thresholds of ECM stiffness and viscoelasticity that support glucose-stimulated insulin secretion (GSIS) represents an essential unmet need. Addressing this gap will require systematic evaluation using stiffness-gradient hydrogels, tunable viscoelastic platforms, and pre-vascularized 3D culture systems that recapitulate the dynamic transition from fibrotic to compliant microenvironments. Integration of engineered, patient-derived β-cells into these platforms will enable direct assessment of interindividual variability in mechanosensitivity and therapeutic responsiveness.
Looking forward, the convergence of mechanobiology, biomaterials science, and precision medicine offers a powerful framework for developing next-generation diabetes therapies. By combining quantitative mechanobiological measurements with targeted genetic and pharmacological modulation of pathways such as YAP/TAZ, Piezo1, and TRPV4, it will be possible to establish rational design principles for restoring β-cell function. Such integrative approaches hold promise for translating mechanistic insights into clinically actionable strategies, ultimately enabling personalized and durable regenerative therapies for diabetes.
Footnotes
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Soonchunhyang University Research Fund and the National Research Foundation of Korea funded by the Ministry of Science and ICT (MSIT) [grant numbers: RS-2019-NR040068].
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.