
Abstract
Late-onset Alzheimer’s disease (LOAD) is the most common age-related dementia, and its etiology remains unclear. Recent studies have linked abnormal neuronal aging to LOAD. Neurons are non-proliferative, and thus, majority of aged neurons must be rejuvenated through repairing or eliminating damaged molecules to regain their healthy status and functionalities. We discovered a surge of oxidative stress in neurons at middle age in mice. A rapid upregulation of neuronal rejuvenation is vital, while astrocyte-expressed interleukin33 (IL33), an IL1-like cytokine, is critical for this process. Thus, IL33-deficiency cripples the neuronal rejuvenation mechanisms, such as repairing DNA double strand breaks, eliminating damaged molecules by autophagy or by glymphatic drainage. IL33-deficient mice develop tau deposition and age-related dementia following a path similar to LOAD. We hypothesize that any interferences on IL33-initiated rejuvenation process for aged neurons after middle life is a potential risk for LOAD development.
Abnormal Neuronal Aging in Late-Onset Alzheimer’s Disease (LOAD)
Etiology of sporadic late-onset Alzheimer’s disease (LOAD), which is the most common age-related dementia, remains a medical mystery. However, decades’ dedicated studies have revealed several promising hypothetic mechanisms for causing LOAD. Like many human diseases, LOAD may be caused by genetic predisposition and environmental influence on gene expression pattern. Currently, ApoE*ε4 allele of polymorphic apolipoprotein E (ApoE) gene is considered a genetic risk for development of LOAD, probably due to interrupted lipid homeostasis in astrocyte. 1 This alone, however, is not sufficient to cause LOAD. In fact, transgenic mice for over-expression of ApoE4 do not show any signs of aging related dementia. 2
As compared to familial AD, characteristics of LOAD is age-related, slow development over a long asymptomatic period followed by a rapid progress toward dementia. Although they are hallmarks for AD, amyloid β plaques (senile plaques) and neurofibrillary tangles (accumulation of abnormal tau) are not unique to AD, as they also appear in aged brains. Mounting evidence supports a critical role of abnormal or accelerated neuronal aging in LOAD development. 3 Neuronal aging is a process of accumulation of damaged molecules largely caused by oxidative stress or excessive metabolic wastes. Several ways may accelerate cellular aging. One is the defects in cells’ or tissues’ abilities to eliminate metabolic wastes or to repair damaged molecules. Neurons are non-proliferative and, neurogenesis in adults is limited. Thus, majority of aged neurons must undergo “rejuvenation” of themselves by repairing or eliminating damaged molecules to regain healthy status and functionalities even at old age. 4 Multiple homeostatic mechanisms for tissues or cells, for example, autophagic digestion and glymphatic disposal of damaged molecules, have been identified as essential parts for neuronal “rejuvenation.”5-7 Defects in neuronal rejuvenation will accelerate neuronal aging, leading to aging-related neurodegeneration or dementias including LOAD. Numerous studies have indeed linked deficiencies in those rejuvenation mechanisms to amyloid plaques and tau deposition in animal models at cellular or molecular levels.5-7 Again, neuronal rejuvenation mechanisms, in turn, are controlled by both genetics and environmental influences. How are those mechanisms for neuronal rejuvenation regulated? Among many potential answers, one is cytokines’ regulation. Brain cytokines may regulate neuroinflammation, which has been postulated as a potential cause of AD. A recent study has shown that deficiency of IFNβ pathway in brains causes neurodegeneration with Lewy-body, which mimics human Parkinson’s disease. Our serial investigations also demonstrated the involvement of another cytokine interleukin33 (IL33) in neuronal rejuvenation and development of aging related dementia. Our discoveries may constitute a foundation for us to postulate a novel hypothesis for abnormal neuronal aging in LOAD development.
Interleukin33 (IL33) in Tissue Homeostasis
IL33, a relatively new member of interlukin1 (IL1) gene family, is first expressed as a nuclear protein, and released as a mature cytokine after cleavage. Similar to other IL1 cytokines, the receptor for mature IL33 is a heterodimer composed of special ST2 and coreceptor IL-1RacP. IL33/ST2 triggers activation of NFκB transcription pathway (Figure 1A). Due to structural similarity of cytokine domain and their receptors between IL33 and IL1, early studies have solely focused on whether IL33 possesses functions similar to IL1 or IL1-cytokines in inflammations and immune response. Studies have identified surprisingly diverse functions for IL33, ranging from viral infection alarming, activation of innate immunity, pro- or anti-inflammation to Th2 polarization. On the other hand, recent studies have found IL33’s unusual constitutive expression in a wide range of tissues including the brains, suggesting its potential roles beyond “traditional” functions of IL1 family in immune defense or inflammation. IL33 has been implicated in the injury healing in central nervous system (CNS) and in other non-inflammatory diseases, suggesting that IL33 may serve an important role in tissue preservation and repair in response to injury. Mounting evidence supports a critical role of IL33/ST2 axis in vascular health with diagnostic and/or prognostic value in cardiovascular diseases, for example coronary artery disease, myocardial infarction, atherosclerosis, ischemic stroke, and diabetes. 8 Our early studies have discovered IL33’s unique regulatory role in removal of degenerative ovarian tissue, and its deficiency greatly shortens ovarian functional life. 9 In summary, unlike other IL1 cytokines, IL33 participates in tissue homeostasis during tissue aging, injury or modifications. 10
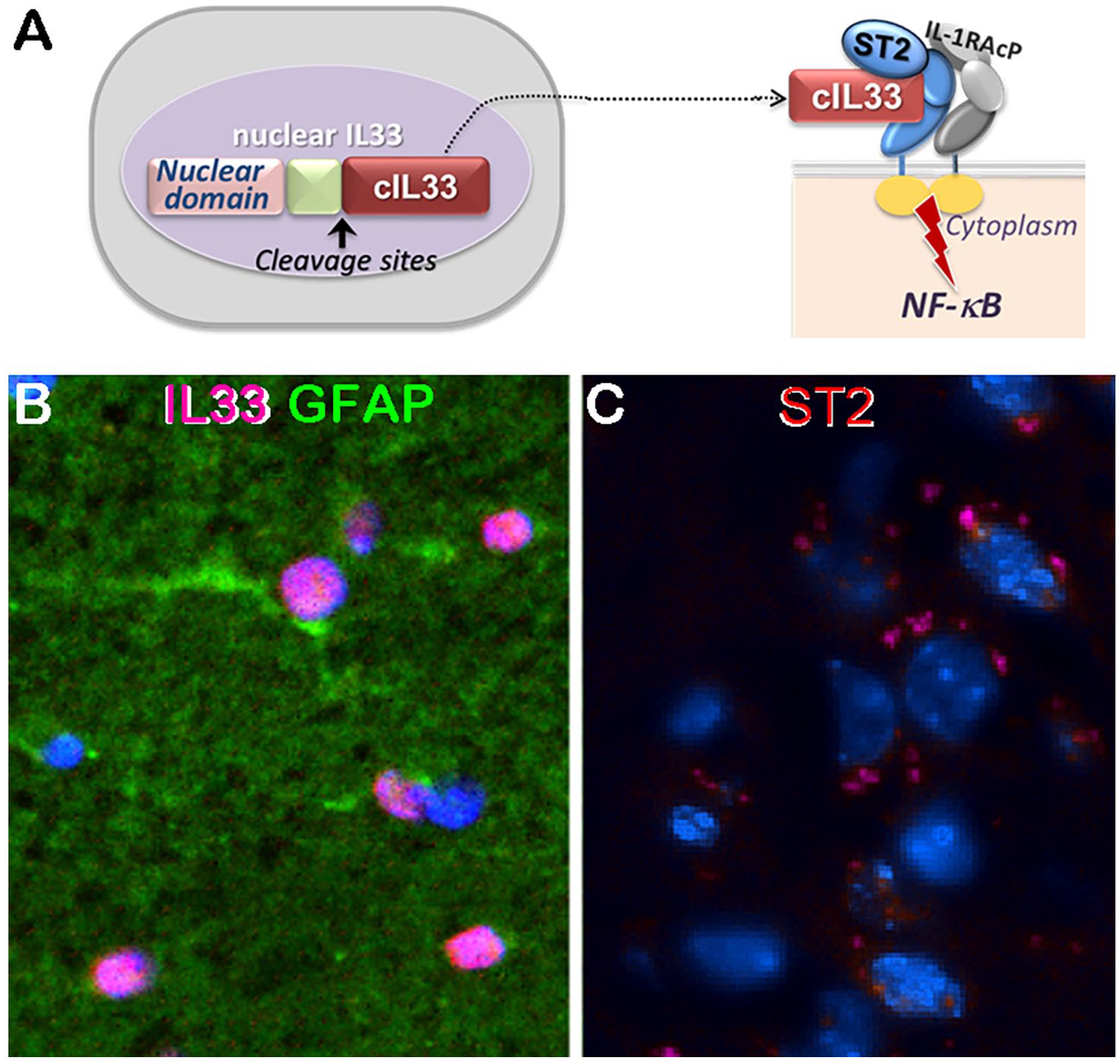
Interlukin33 (IL33) and its receptor ST2 in brains: (A) diagram depicts IL33/ST2/NFκB axis. nIL33, nuclear IL33; cIL33, cytokine IL33, (B) nuclear IL33 (red) in many astrocytes (GFAP, green), and (C) ST2 (red) expression in a group of neurons identified by their nuclear morphology.
Interleukin33 Is a Master Regulator for Rejuvenation of Aged Neurons
IL33 has been genetically linked to human AD, and shows a beneficial effect in reduction of amyloid β loads in a mouse AD model,11,12 suggesting its potential involvement in AD. We further found a high density of astrocytes in the brains with nuclear IL33, and their number increases with age far beyond reproductive maturity (Figure 1B). In aged mouse brains (older than 65 weeks), up to 75% of astrocytes in certain regions express nuclear IL33. 13 Importantly, we also demonstrated the release of mature IL33 in brains especially at old age. The high density of IL33-expressing astrocytes and constant release of mature IL33 suggested its involvement in physiological, rather than pathological (e.g. inflammatory), events. It is well known that CNS is an immunoprivileged site, where any types of inflammation will not be tolerized in order to avoid grave pathological consequence. Which physiological events in the brains may IL33 participate or play a role in? One possible event is neuronal aging and/or their rejuvenation. To test our hypothesis, we compared neuronal aging between IL33-sufficient and -deficient mouse (Il33-/-), or by intervention with recombinant IL33. Neuronal aging can be partially measured by quantity of oxidative damages. We first uncovered a sudden surge of accumulation of oxidatively damaged molecules in neurons at middle age (35-40 weeks in mice) in IL33-deficient mice. The most notable is rapid accumulation of DNA double-strand-breaks (DSBs) in neurons in the cortex and hippocampus (Figure 2A). On the other hand, wild type littermates showed no DSBs at all until old age (70 weeks) but still at a much lower level. A similar time course was also observed for proteins or lipids with oxidative damages or modifications in the cortex and hippocampus of IL33 deficient mice. 13 Those results suggested a sudden acceleration of neuronal aging. Interestingly, human brains age accelerates after 30. 14 As of non-proliferative nature, rejuvenation of stressed or aged neurons by repair or removal of oxidative damages is vital to maintains normal brain function throughout whole lifespan. During the oxidative surge, neurons or brains first unleash an antioxidative response to minimize oxidative sources, for example, reactive oxygen species. Brains will then eliminate or repair the damaged molecules to rejuvenate the stressed neurons. Although Il33-/- brains can initiate anti-oxidative response (unpublished data, Lou et al), they lose the ability to eliminate or repair the damaged molecules. 13 We identified defects in 3 mechanisms post the oxidative surge at middle age in Il33-/- brains, that is, (1) repair of DNA DSBs in neurons, (2) autophagic digestion of damaged or old molecules in neurons, 13 and (3) glymphatic drainage of damaged molecules, for example, p-Tau or PHF tau, from brains to the circulation. 15 Importantly, numerous studies have linked these 3 defects to AD or related dementias in both humans and AD animal models. People have vividly described glymphatic system as brains’ “garbage truck.” 7 Its defect leads to accumulation of unwanted “garbage” such as amyloid-β peptides and abnormal tau. Our most recent study showed that aquaporin4 (molecular water pump) expressed on astrocytes generates 2 flows to drain neuronal wastes from brains, while IL33 regulates their expression. 15 IL33 deficiency causes a significant loss of aquaporin4 in astrocytes after middle age (Figure 2C), and thus, impairs much needed glymphatic drainage. The defects in those mechanisms in Il33-/- mice lead to a rapid accumulation of DSBs and abnormal tau in neurons (Figure 2A and D). 13 Buildup of unrepaired molecules after middle age, in turn, initiates a chronic path to neurodegeneration, which is evidenced by loss of synapse connections and neurites and neurodegeneration after 40 weeks, tau deposition after 65 weeks (Figure 2), followed by impaired cognition and memory loss after 70 to 80 weeks in Il33-/- mice.13,15 Similar to AD, those pathological changes largely limit to the cortex and hippocampus, and their development timeline resembles the course of sporadic LOAD. Our recent studies revealed expression of ST2 on stressed neurons (Figure 1C). However, NFκB pathway in neurons may turn on only neuronal rejuvenation mechanisms, since significant neuroinflammation was absent in aged Il33-/- brains. It is possible that that neurodegeneration may be the cause of, but not the result of neuroinflammation. In summary, IL33 deficiency cripples mechanisms for rejuvenation of stressed neurons, leading to tau abnormality, and chronic neurodegeneration and AD-like symptoms in Il33-/- mice.

IL33-deficient (IL33-/-) mice develop pathological changes in the cortex and hippocampus. IL33-/- mice show accumulation of DNA DSBs (by TUNEL, green, A) in neuronal nuclei, loss of synapses (by synaptophysin, red, B) and neurites (tubulinβ3, in both A and B), and loss of astrocyte aquaporin4 (red, C) at 45 weeks, and neuronal deposition of abnormal tau (PHF, D) at 65 weeks, followed by cognition impairment and memory loss after 70 to 75 weeks. 13
Hypothesis: Deficiency in Rejuvenation of Aged Neurons May Cause Age-Related Dementias
Aging-related neurodegeneration in Il33-/- mice bears several hallmarks for human LOAD, that is, (1) late-onset, (2) loss of synapses and neurites in the cortex and hippocampus at early stage, (3) deposition of abnormal tau in neurons and neuron death, and (4) impaired cognition and memory at old age. We believe our IL33-deficiency mouse model could be a reliable tool for studying LOAD. Current animal models for AD are largely based on mutant genes such as amyloid precursor protein (APP), PSEN1, or microtubule-associated proteins (MTAP). However, LOAD is unrelated to mutations in those genes. From this point of view, the advantage of Il33-/- model is that abnormal tau deposition and age-related neurodegeneration better mimics human LOAD. Although this model does not develop amyloid β plaques (that is one important hallmark for AD), it is expectable since structure of mouse APP lacks the domain for aggregation. It would be interesting to test if unmutated human APP could cause senile plaques in IL33 deficient mice. In summary, our findings lead to the following hypothetic mechanism for LODA development (Figure 3). Neurons at middle age undergo a rapid surge of oxidative stress. Astrocytes may sense oxidative stress in nearby neurons and release mature IL33, which, in turn, turns on multiple mechanisms in the stressed neurons to remove or repair oxidative damaged molecules, including abnormal tau and amyloid β. The neurons return to healthy status. Therefore, interruptions in IL33 expression or other steps in the cascade for rejuvenation of stressed neurons may lead to LOAD or other age-related dementias.

Hypothetic cascade for IL33-initiated neuronal rejuvenation; Step1. Stressed or aging neurons stimulate astrocytes; Step2. Astrocytes cleave nuclear IL33 to release cytokine IL33; Step3. Through ST2, IL33 triggers activation of NFκB pathway, which in turn, upregulates neuronal rejuvenation mechanisms that is repair of DSBs, autophagic digestion of damaged molecules and aquaporin4 mediated glymphatic drainage of neuronal wastes. Defects in any steps in the rejuvenation cascade, therefore, lead to chronic neurodegeneration, tauopathy, and age-related dementias.
Footnotes
Funding:
The author disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by NIH R21AG067311 (to YL), NIH R01DK077857 (to YL), and NIH R01HD049613 (to YL).
Declaration of Conflicting Interests:
The author declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Significance Statement
Neurons at middle age face a surge of oxidative stress. IL33 initiate multiple mechanisms to rejuvenate stressed neurons, and thus their defects may cause age-related chronic neurodegeneration and dementia.