
Abstract
The effects of cerebral ischemia/reperfusion on phosphorylation of microtubule-associated tau proteins were assessed in a canine model of cardiac arrest. As tau proteins are phosphorylated by kinases involved in different transduction signal pathways, their phosphorylation state is an excellent marker of neuronal homeostasis and microtubule dynamics. Canine brain tau proteins were characterized by immunoblotting using phosphorylation-dependent antibodies and antisera raised against different amino- and carboxy-terminal tau sequences. The present study reports a complete dephosphorylation of tau proteins during ischemia, which is shown by a higher electrophoretic mobility and the almost (if not total) disappearance of phosphorylation-dependent monoclonal antibody labeling. After 2-hour restoration of spontaneous circulation, a decrease in the electrophoretic mobility was observed, and after 24 hours of reperfusion, a full restoration of the phosphorylation was visualized using phosphorylation-dependent monoclonal antibodies directed against Ser/Thr-Pro sites. However, one particular phosphorylation site involved in tau binding to microtubules, located on Ser262/356, was never fully significantly rephosphorylated, suggesting that microtubule metabolism was still affected after 24 hours of reperfusion. Thus, the sequential and differential recovery of tau phosphorylation after ischemia followed by reperfusion is a useful marker with which to monitor neuronal integrity after brain ischemia.
Keywords
Tau proteins belong to the microtubule-associated protein family. They are found primarily in neurons. In the human adult brain, alternative splicing of three exons (exons 2, 3, and 10) of a single gene generates six tau isoforms (Goedert et al., 1989a, b ), which range from 352 to 441 amino acids with an apparent molecular mass from 45 to 74 kDa, when separated by polyacrylamide gel electrophoresis in the presence of sodium dodecyl sulfate (SDS-PAGE). From a structural point of view, tau amino-terminal domain corresponds to the projection domain and may interact with cytoskeletal elements and mitochondria (Hirokawa et al., 1988; Rendeon et al., 1990). Tau proteins bind to microtubules through repetitive regions in their carboxy-terminal part (Lee et al., 1989). This binding is regulated by the degree of Ser/Thr phosphorylation of tau proteins. One of the critical sites of phosphorylation in tau binding to microtubules is Ser262, a non-Ser/Thr-Pro site (numbering according to the longest human tau isoform in brain, 441 amino acids) (Biernat et al., 1993; Schneider et al., 1999). A number of phosphorylation-dependent monoclonal antibodies have been developed against many different tau phosphorylation sites (for review, see Buée and Delacourte, 1999). The different states of tau phosphorylation result from kinase and phosphatase activities. Phosphorylation sites are also found on native tau from biopsy-derived samples, but they are rapidly dephosphorylated after death, due to postmortem phosphatase activity (Matsuo et al., 1994). By SDS-PAGE, this tau dephosphorylation is characterized by a decrease in apparent molecular mass (Baudier and Cole, 1987; Matsuo et al., 1994).
Prolonged cerebral ischemia, as occurs in focal ischemia models, results in disruption of the neuronal cytoskeleton. This can occur by proteolysis of microtubules and other components; however, tau protein immunoreactivity is relatively unaffected (Pettigrew et al., 1996). Cytoskeletal disruption can also occur through the effects of ischemia on protein phosphorylation that control cytoskeletal assembly and stability (Dewar et al., 1994; Pettigrew et al., 1996). Recent studies have indicated that tau dephosphorylation is an early event that occurs during ischemia but does not persist during reperfusion regardless of the neurological outcome (Burkhart et al., 1998; Shackelford and Yeh, 1998). Furthermore, tau proteins are normally highly phosphorylated, and all phosphorylation sites may not be similarly affected by ischemia as they involve activity of different kinases/phosphatases. In fact, phosphorylation of particular sites on tau proteins may reflect microtubule dynamics and activation of different transduction signals. In the present study, we have investigated more especially the rephosphorylation period of tau protein after the ischemic insult followed by a short time of reperfusion in a clinically relevant canine model of cardiac arrest and resuscitation (Rosenthal et al., 1992). We observed a rapid dephosphorylation of tau during ischemia that was more or less recovered after 24-hour restoration of spontaneous circulation (ROSC) according to the different tau phosphorylation sites.
MATERIALS AND METHODS
Animal experiments
All animal experiments were conducted in accordance with guidelines established by the Institutional Animal Care and Use Committee of the George Washington University Medical Center. The canine cardiac arrest and resuscitation model of global brain ischemia and reperfusion was performed essentially as described by Rosenthal et al. (1992) and utilized chloraloseanesthetized female adult beagles. Four animal groups were used in this study: sham-operated, nonischemic control animals (n = 4); animals that underwent 10 minutes of electrically induced ventricular fibrillation cardiac arrest without ROSC (n = 4); and animals subjected to 10 minutes of cardiac arrest, resuscitation, and either 2 hours of ROSC (n = 3) or 24 hours of ROSC (n = 4). Previous studies have demonstrated a moderate to severe degree of neurologic impairment in these animals subjected to 10-minute global cerebral ischemia and 24-hour reperfusion (Rosenthal et al., 1992; Liu et al., 1998), based on a standardized neurologic deficit scoring system similar to that used by Bircher and Safar (1985) and others (Zwemer et al., 1994). These animals typically exhibit normal respiration but show marked alterations of their levels of consciousness and are incapable of standing or drinking. We and others have demonstrated substantial delayed cell death in the hippocampus and cortex (for example, 30% in cortical layer V pyramidal cells) following 24 to 96 hours of reperfusion without histologic evidence of acute death after only 2 hours of reperfusion (Bogaert et al., 1996; Radovsky et al., 1995).
At the end of the appropriate experimental period, a craniotomy was performed on anesthetized animals, and samples from the frontal cerebral cortex were immersed in liquid nitrogen within 5 seconds after excision and then stored at −80°C. Homogenates were generated using 100 mg of frozen tissue/mL of Laemmli sample buffer and boiled at 100°C for 10 minutes.
Cell culture and transfection
COS-7 cells were grown in 25-cm2 flasks in Dulbecco's modified Eagle's medium (Life Technologies, Cergy Pontoise, France) with 10% fetal calf serum (Boehringer Mannheim) in a 5% CO2 incubator at 37°C. The cDNA of the six human tau isoforms was cloned in pSG5 vector (Stratagene; a kind gift of Dr. M. Goedert, Cambridge, U.K.). Tau cDNAs were transiently transfected in COS cells using the diethylaminoethyldextran method. Forty-two hours after transfection, cells were treated or not by okadaic acid (OA; Sigma, St. Louis, MO, U.S.A.), an inhibitor of protein phosphatases 1 and 2A, for 6 hours, as previously described (Mailliot et al., 1998; Sautière et al., 1994). Cells were harvested in ethylenediaminetetraacetate solution at 4°C and centrifuged. Cell pellets were homogenized in Laemmli sample buffer (Laemmli, 1970) and boiled for 10 minutes.
Antibodies
Antibodies used in the present study are summarized in Fig. 1, and numbering of the epitopes is given according to the longest human tau isoform. Phosphorylation-dependent monoclonal antibodies included 12E8 recognizing phosphorylated Ser262 and to a lesser extent phosphorylated Ser356 (Seubert et al., 1995), AD2 directed against phosphorylated Ser396 to Ser404 (Buée-Scherrer et al., 1996), AT180 labeling phosphorylated Thr231, AT270 recognizing phosphorylated Thr181, AT8 directed against phosphorylated Ser202/Thr205 (Goedert et al., 1994, 1995), and Tau-1, which binds amino acids 189 to 207 only when they are dephosphorylated (Szendrei et al., 1993). With the exception of 12E8, all other phosphorylation-dependent monoclonal antibodies (AD2, AT8, AT180, and AT270) recognize Ser/Thr-Pro sites. Well characterized polyclonal antibodies against different regions of tau proteins were also used and consisted of E2, E3, and E10 directed against sequences encoded by exons 2, 3, and 10, respectively (Mailliot et al., 1998; Sergeant et al., 1997). Finally, M19G is a well characterized antiserum, directed against the first 19 amino acids of the tau sequence encoded by exon 1 (Buée-Scherrer et al., 1996; Sautière et al., 1994). All polyclonal antibodies recognize their epitope independently of the tau phosphorylation state.
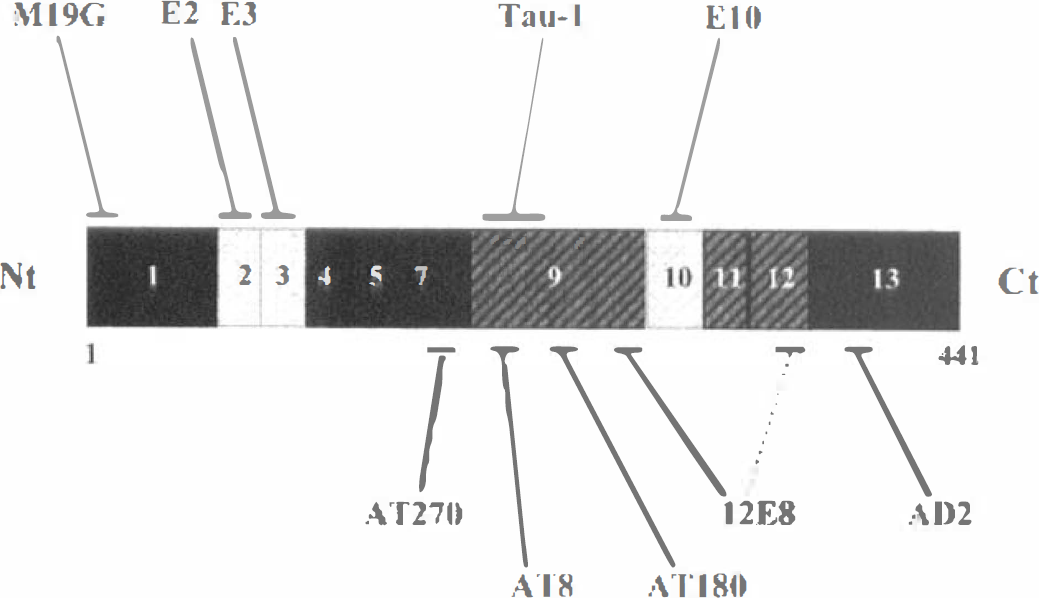
Epitopes of antitau antibodies used in the present study. Schematic representation of the longest human tau isoform [2+3+10+ (hT40), 441 amino acids, numbered according to the longest human brain tau isoform]. Alternatively spliced exons 2, 3, and 10 are shown in light type. The microtubule binding domain includes the gray hatched regions of exons 9 to 12. M19G, E2, E3, and E10 recognize tau proteins independently of their phosphorylation state. Tau-1 binds to its tau epitope when it is not phosphorylated. All other phosphorylation-dependent monoclonal antibodies recognize their epitopes when they are phosphorylated. With the exception of 12E8, all other phosphorylation-dependent monoclonal antibodies (AD2, AT8, AT180, AT270) recognize Ser/Thr-Pro sites.
Immunoblotting
Electrophoresis and immunoblotting were performed as previously described (Buée-Scherrer et al., 1996; Mailliot et al., 1998). In brief, samples were loaded onto SDS-PAGE gels. Two vertical electrophoresis systems were used: a minigel system from Pharmacia Biotech (using 10% SDS-PAGE) and Bio-Rad Protein II (10 to 20% slab gel). After transfer to nitrocellulose (Amersham), membranes were blocked with 5% skim milk and incubated with the primary antibody overnight at 4°C. Horseradish peroxidase-conjugated antibody was used as secondary antibody, and reaction product was detected using the Amersham ECL Western blotting system.
Quantitative analysis
All quantitative analyses were performed as described by Sergeant et al. (1997). A correction factor was introduced to adjust tau immunoreactivity among samples and obtain approximately equally strong immunoreactivity (if binding occurred at all) of the various samples, based on binding of phosphorylation-independent antibody E10 and/or M19G to the various samples. Each immunoblotting experiment was repeated at least three times. Optical densities were expressed in arbitrary units. Statistical analyses were done using the nonparametric Mann—Whitney U test.
RESULTS
Effect of ischemia/reperfusion on canine brain tau proteins
By immunoblotting using the polyclonal antibodies M19G, E2, E3, and E10, we investigated the nature of tau isoforms in our canine model of global cerebral ischemia/reperfusion. No high molecular mass tau variants were detected in normal canine brain. Two major tau variants were labeled at 72 to 74 kDa with antibodies E2, E3, and E10 (data not shown). The same bands as well as other lower molecular mass variants between 68 and 74 kDa were visualized with M19G (Fig. 2, lane 2) and co-migrated with the longest human tau isoform (2+3+10+) transfected in COS cells treated with OA (Fig. 2, lane 1). Moreover, these 72- to 74-kDa canine variants co-migrated with the 72- to 74-kDa variant found in human brain (Sergeant et al., 1997). These data suggested that canine cerebral tau variants were phosphorylated in normal conditions to the same extent as human brain tau proteins.
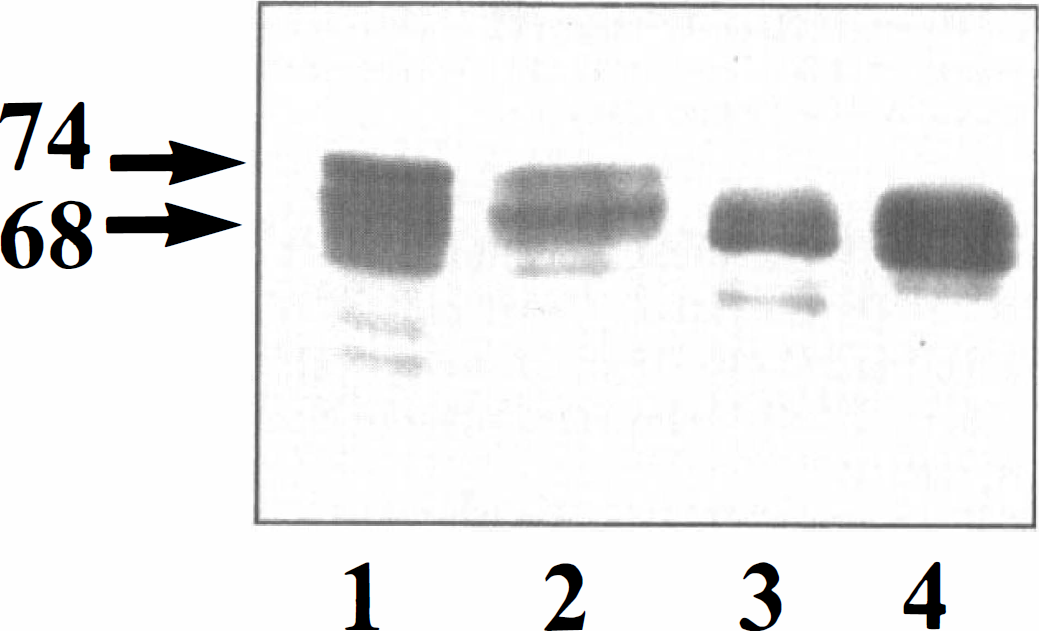
Tau isoform in the canine model of brain ischemia/reperfusion. Immunoblotting was performed using polyclonal antibody M19G. This phosphorylation-independent antibody labeled different tau variants between 68 and 74 kDa (arrowheads on left). High molecular mass tau variants at 72 to 74 kDa were detected in COS cells transfected with 2+3+10+ cDNA (hτ40) and okadaic acid-treated cells (lane 1) as well as in control dog cells (lane 2). Lower molecular mass tau variants around 68 kDa were found in dog brains after ischemia (lane 3) and in transfected non-okadaic acid-treated COS cells (lane 4). A similar electrophoretic pattern was obtained with all other polyclonal antibodies. A major isoform at 72 to 74 kDa is labeled in the control canine cerebral cortex, and its electrophoretic mobility increased after 10 minutes of ischemia.
After 10-minute global cerebral ischemia, a shift in the electrophoretic mobility of these variants occurred and the labeling was visualized around 68 kDa (Fig. 2, lane 3), as observed in transfected non-OA-treated COS cells (Fig. 2, lane 4), indicating that dephosphorylation had occurred. Whatever the conditions, tau variants were detected by all tau polyclonal antibodies (M19G, E2, E3, and E10) and gave the same electrophoretic profiles.
Effect of ischemia/reperfusion on cerebral cortex tau phosphorylation
To investigate tau protein phosphorylation state during the ischemia/reperfusion protocol, different phosphorylation-dependent monoclonal antibodies (12E8, AD2, AT8, AT180, AT270, and Tau-1) were used. In control samples, all phosphorylation-dependent monoclonal antibodies, with the exception of Tau-1 antibody, labeled two broad bands around 74 kDa (as shown for AD2 antibody in Fig. 3A, lane 2). Transfected human tau 2+3+10+ from OA-treated COS cells showed the same tau immunoreactivity (Fig. 3A, lane 1). After ischemia alone, a loss of immunoreactivity was observed. AD2 and 12E8 did not label ischemic samples anymore (Fig. 3A, lane 3), whereas AT270 showed a much weaker staining at 68 kDa. The human tau 2+3+10+-transfected non-OA-treated cells were not labeled anymore by AD2 antibody (Fig. 3, lane 4). In the case of ischemia followed by 2-hour ROSC, phosphorylation-dependent tau immunoreactivity was much more intense than that observed with samples obtained after ischemia in the absence of reperfusion. In particular, a 72-kDa band was very evident (Fig. 3A, lane 5). After 24 hours of ROSC, this band was still apparent, but the 74 kDa was much more intense [especially for AD2 (Fig. 3A, lane 6), AT8, and AT270]. Conversely, Tau-1 antibody strongly labeled ischemic samples around 68 kDa, whereas in normal and 24-hour ROSC conditions, Tau-1 poorly labeled the 72-kDa tau variant. Tau-1 did not label transfected human tau 2+3+10+ from OA-treated cells (Fig. 3B, lane 1), but it labeled transfected non-OA-treated cells (Fig. 3B, lane 4).
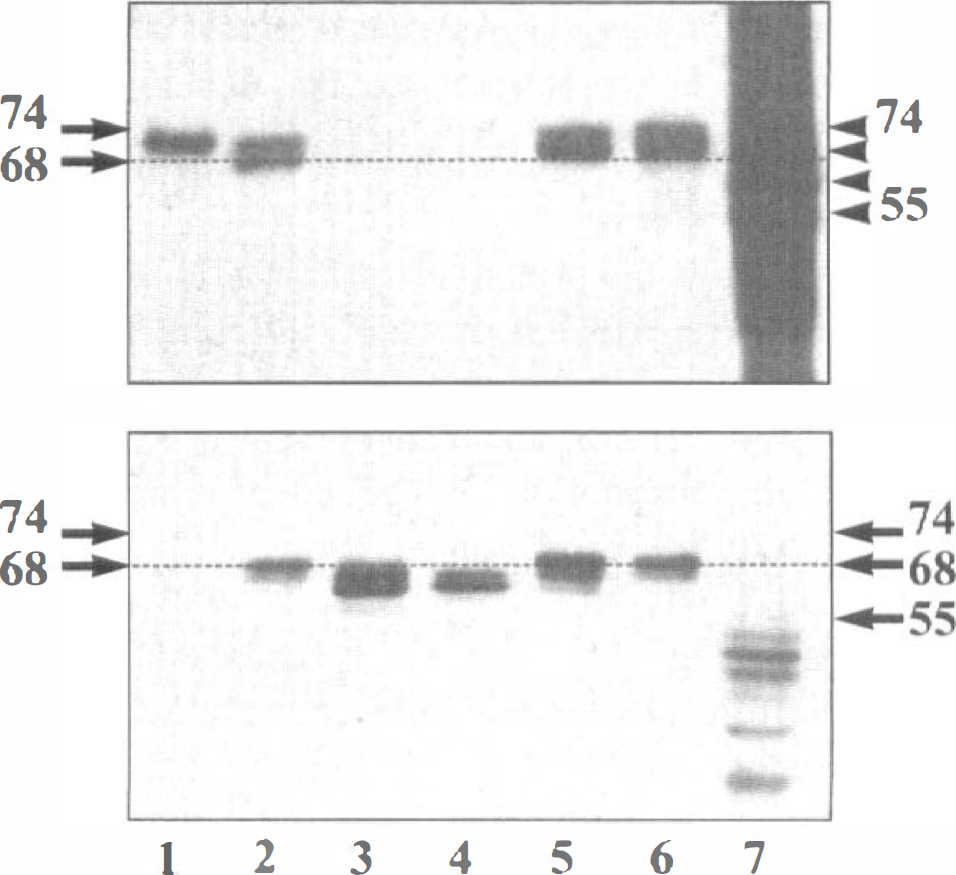
Tau phosphorylation in the canine model of brain ischemia/reperfusion. Immunoblotting was performed using monoclonal antibodies AD2 (
Quantitative analysis of tau immunoreactivity
Quantitative analysis of tau phosphorylation was performed by immunoblotting using monoclonal antibodies AD2, AT8, AT180, AT270, 12E8, and Tau-1 (Fig. 4). All values were expressed after correction of the tau amount estimated by E10 immunoreactivity. This correction allowed for elimination of possible variations among samples due to differences in tau content. Furthermore, it should be noted that no increase in the amount of low molecular mass tau products consistent with proteolysis was observed in any conditions.
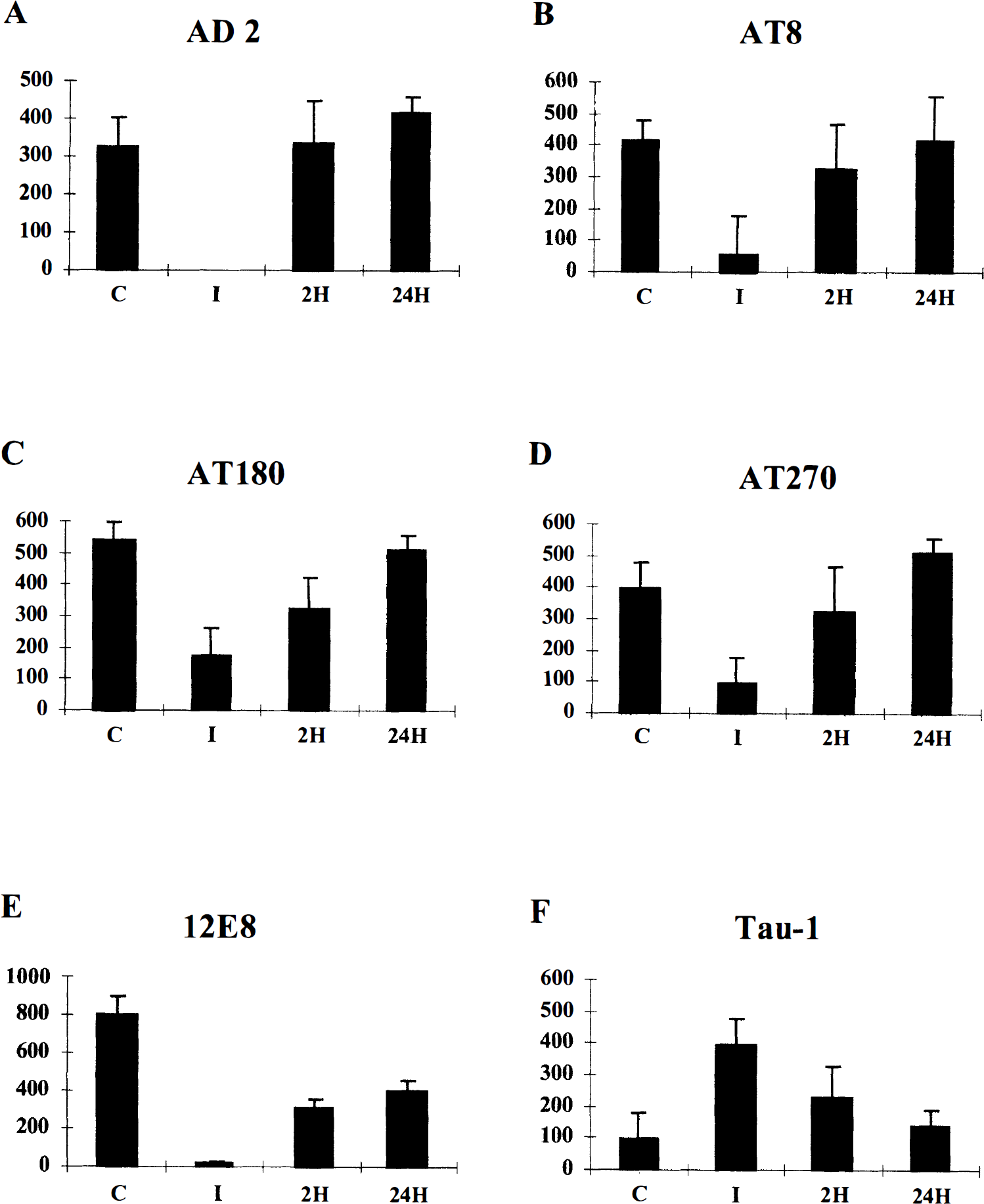
Quantification of phosphorylation-dependent monoclonal antibody immunoreactivity. Tau phosphorylation was quantified using AD2 (
In control dogs, high levels of AD2 immunoreactivity were observed. In ischemic conditions, this immunoreactivity was significantly reduced (Table 1; Mann-Whitney U test, P < 0.05) and actually appeared to be completely eliminated. After 2 hours of ROSC, phosphorylation at the AD2 epitope was restored and there was no significant difference in immunoreactivity compared with samples from control dogs. After 24 hours of ROSC, levels of AD2 immunoreactivity were comparable with those in control conditions. Similar results were obtained using the AT8, AT180, and AT270 antibodies; however, −25% of control immunoreactivity was still apparent after 10 minutes of cerebral ischemia, and the restoration of the initial level of phosphorylation was more progressive for these antibodies. The pattern of 12E8 immunoreactivity was similar to that of AD2 in the sense that there was a complete loss of signal after ischemia alone. However, unlike the immunoreactivity with either AD2 or AT270, the 12E8 immunoreactivity continued to be significantly lower than that of control tissue at both 2 and 24 hours of reperfusion (Table 1; P < 0.05). Conversely, Tau-1 immunoreactivity was lower in control samples, whereas it increased during ischemia and dwindled again after 2 and 24 hours of ROSC (Table 1).
Statistical analysis of tau phosphorylation
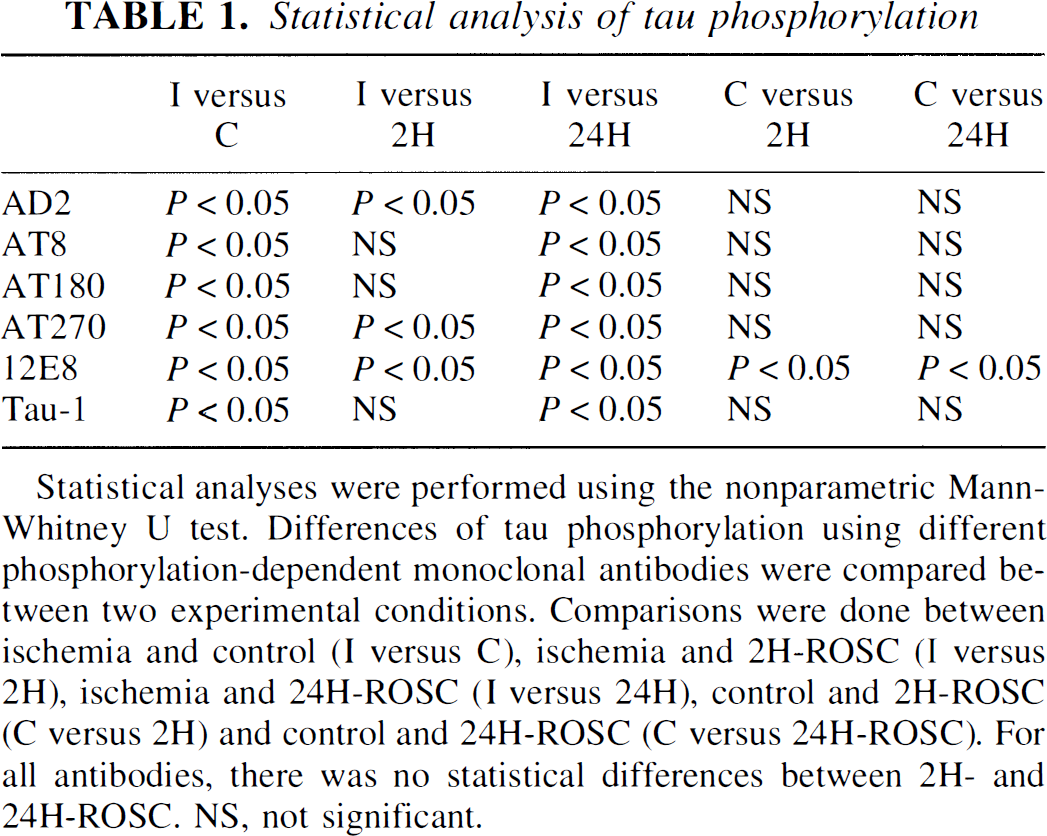
Statistical analyses were performed using the nonparametric Mann-Whitney U test. Differences of tau phosphorylation using different phosphorylation-dependent monoclonal antibodies were compared between two experimental conditions. Comparisons were done between ischemia and control (I versus C), ischemia and 2H-ROSC (I versus 2H), ischemia and 24H-ROSC (I versus 24H), control and 2H-ROSC (C versus 2H) and control and 24H-ROSC (C versus 24H-ROSC). For all antibodies, there was no statistical differences between 2H- and 24H-ROSC. NS, not significant.
DISCUSSION
In the present study, we demonstrated that rapid and extensive tau dephosphorylation occurs in the frontal cortex in a clinically relevant cardiac arrest model of global cerebral ischemia. These results corroborate those of Shackelford and Yeh (1998) on rapid tau dephosphorylation during transient rat cerebral ischemia. The use of a large variety of phosphorylation-dependent monoclonal antibodies allowed us to easily study tau phosphorylation at different well defined sites, demonstrating that it was site specific. Furthermore, tau variants in canine brain are in fewer number than in rodent brain where numerous bands are labeled with AD2 and Tau-1 antibodies (Shackelford and Nelson, 1996; Shackelford and Yeh, 1998). The most sensitive epitope to dephosphorylation was that recognized by AD2, which includes residues Ser396 and Ser404. After resuscitation and cerebral reperfusion, most phosphorylation sites, including those of AT8/Tau-1, AT180, and AT270, were completely rephosphorylated. However, the sites recognized by 12E8, located primarily on Ser262 and to a lesser extent on Ser356, were not fully rephosphorylated, even after 24 hours of reperfusion. Our study therefore demonstrates that all the phosphorylation sites on tau proteins do not react to the same extent to the ischemic injury: There is a sequential and differential recovery of tau phosphorylation after ischemia and reperfusion according to the different tau phosphorylation sites we consider. This moderates the conclusions of all previous studies on tau phosphorylation during and after ischemia, which explained that tau proteins were rapidly dephosphorylated during ischemia and then recovered their initial phosphorylation state after reperfusion, regardless of the neurological outcome. But all of these studies focused only on a few tau phosphorylated residues, mainly those recognized by AD2 and Tau-1 antibodies.
Tau dephosphorylation during ischemia
If we consider tau dephosphorylation during ischemia, it may result from specific activation of phosphatases after different ischemia-related phenomena including apoptosis and cytoskeletal alterations. Recently, apoptosis has drawn attention in ischemic neuronal death. Bcl-2 family proteins, DNA fragmentation, and release of caspase-9 from mitochondria have been involved in ischemia (Krajewski et al., 1999; MacManus et al., 1999; Matsushita et al., 1998). It should be noted that activation of protein phosphatase 2A and dephosphorylation of tau protein were reported as early phenomena of the execution phase of apoptosis (Mills et al., 1998). Finally, one of the most striking alterations that occurs during ischemia and reperfusion is an increase in intracellular levels and redistribution of calcium. This leads to an overactivation of glutamate receptors and has deleterious effects on cytoskeleton integrity by modulating calcium-dependent kinases/phosphatases and proteases; this generates proteolysis of microtubules and dysregulation of the tau kinase/phosphatase balance. Moreover, elevated intracellular calcium may compromise mitochondrial function, leading to tau dephosphorylation at Tau-1 site through calcineurin activation (Norman and Johnson, 1994). As mitochondria may bind to microtubules through tau interactions (Rendeon et al., 1990), tau phosphorylation is likely to be modified after changes in either microtubule dynamics or mitochondria metabolism. Conversely, altered microtubule structure-impaired fast axonal transport of mitochondria is believed to play a role in the delayed mitochondrial degeneration observed after cerebral ischemia (Abe et al., 1995). Thus, a number of ischemia-related phenomena could explain tau dephosphorylation, which is a characteristic feature reported in a number of studies on ischemia.
Sequential and differential restoration of tau phosphorylation
Among the Ser-Pro and Thr-Pro sites analyzed in the present study, the AD2 site was completely dephosphorylated during ischemia. After 2-hour ROSC, a full restoration of phosphorylation was already observed. Among tau phosphorylation sites, AD2 is a Ser/Thr-Pro one. It is thus interesting to note that phosphorylation of tau proteins on Ser/Pro or Thr/Pro residues does not significantly modify tau-microtubule interactions (Schneider et al., 1999). This could explain why an almost complete recovery of these sites is obtained after 24-hour ROSC, because they are less implicated in the restoration of the microtubule network integrity and thus of a normal intracellular trafficking than the non Ser-Thr/Pro sites Ser262/356.
Finally, one of the most striking findings of our present study was the incomplete restoration of tau phosphorylation on Ser262/356 residues even after 24-hour ROSC. Interestingly, Ser262/356 are located within the microtubule binding domains of tau. Moreover, Ser262 phosphorylation alone has been shown to dramatically reduce tau affinity for microtubules (Biernat et al., 1993; Schneider et al., 1999). Nevertheless, this particular site is insufficient to eliminate tau binding to microtubules (Seubert et al., 1995). After ischemia, microtubule metabolism and dynamics are severely affected (Minger et al., 1998; Pettigrew et al., 1996). Thus, during reperfusion, tubulin polymerization into microtubules is likely to be a major process necessary for restoration of normal axonal transport. The persistent abnormally low level of Ser262 phosphorylation we have observed may result in a prolonged shift in the tubulin-microtubule equilibrium toward the polymerized state. On the other hand, the mechanisms responsible for changes in tau phosphorylation and differential rephosphorylation during cerebral ischemia and reperfusion likely involve changes in the activities of specific phosphatases and kinases. Some of the kinases responsible for Ser262/356 phosphorylation have already been identified and include cyclic AMP-dependent protein kinase, calcium/calmodulin-dependent protein kinase II, phosphorylase K, and mitogen-activated protein kinase (Drewes et al., 1997; Illenberger et al., 1998; Litersky et al., 1996; Paudel, 1997). In a reversible model of spinal cord ischemia in rabbits, tau proteins have been found to be partially dephosphorylated in response to ischemia with a time course that closely correlates with the period necessary for installation of permanent paraplegia. Moreover, loss of calcium/calmodulin-dependent protein kinase II activity was shown to be an early marker of ischemia in the same model (Shackelford and Nelson, 1996). Thus, during the reperfusion period in our model, the inhibition of calcium/calmodulin-dependent protein kinase II may explain the low degree of Ser262 phosphorylation that facilitates tau binding to microtubules.
In conclusion, the differential phosphorylation of tau proteins is a physiological neuronal marker that may contribute to monitor transduction signals (restoration of full phosphorylation on Ser/Thr-Pro sites). Moreover, tau phosphorylation is an indicator of neuronal metabolism as some non-Ser/Thr-Pro phosphorylation sites on tau proteins such as Ser262/Ser356 reflect microtubule dynamics. During reperfusion, tau proteins are not fully rephosphorylated at Ser262/Ser356, which may indicate a chronic alteration of microtubule dynamics and therefore abnormal axonal transport. A defect in axonal transport of mitochondria and other cell constituents might contribute to delayed neuronal damage observed after reperfusion. Finally, as tau isoforms are differentially expressed in subpopulations of neurons in human brain, tau proteins are of particular interest to define subpopulations of neurons selectively vulnerable to ischemia.
Footnotes
Acknowledgments
The authors are indebted to Dr. Michel Goedert (Cambridge, U.K.) for tau isoforms cDNAs, Dr. Dale Schenk (Athena Neuroscience) for 12E8 antibody, and Dr. Eugeen Vanmechelen (Innogenetics) for AT antibodies. AD2 was developed through a collaboration between UMR 9921 (CNRS Sanofi/Diagnostic Pasteur) and INSERIVI. The authors thank Pr. Roméo Cecchelli and Drs. Thierry Bussière, Valérie Buée-Scherrer, Marie-Laure Caillet, and Patrick R. Hof for helpful discussions.