
Abstract
Background:
Traumatic brain injury is a significant public health issue that results in serious disability in survivors. Traumatic brain injury patients are often intoxicated with alcohol when admitted to the hospital; however, it is not clear how acute intoxication affects recovery from a traumatic brain injury. Our group has previously shown that binge alcohol prior to traumatic brain injury resulted in long-term impairment in a fine sensorimotor task that was correlated with a decreased proliferative and neuroblast response from the subventricular zone. However, whether binge alcohol prior to traumatic brain injury affects the proliferative response in the hippocampal dentate gyrus is not yet known.
Methods:
Male rats underwent binge alcohol (3 g/kg/day) by gastric gavage for 3 days prior to traumatic brain injury. Cell proliferation was labeled by BrdU injections following traumatic brain injury. Stereological quantification and immunofluorescence confocal analysis of BrdU+ cells in the hippocampal dorsal dentate gyrus was performed at 24 hours, 1 week and 6 weeks post traumatic brain injury.
Results:
We found that either traumatic brain injury alone or binge alcohol alone significantly increased dentate gyrus proliferation at 24 hours and 1 week. However, a combined binge alcohol and traumatic brain injury regimen resulted in decreased dentate gyrus proliferation at 24 hours post-traumatic brain injury. At the 6 week time point, binge alcohol overall reduced the number of BrdU+ cells. Furthermore, more BrdU+ cells were found in the dentate hilar region of alcohol traumatic brain injury compared to vehicle traumatic brain injury groups. The location and double-labeling of these mismigrated BrdU+ cells was consistent with hilar ectopic granule cells.
Conclusion:
The results from this study showed that pre-traumatic brain injury binge alcohol impacts the injury-induced proliferative response in the dentate gyrus in the short-term and may affect the distribution of newly generated cells in the dentate gyrus in the long-term.
Keywords
Introduction
Traumatic brain injury (TBI) is a major health problem with as many as 2.8 million cases reported each year in the United States.1,2 Patients face a long road to recovery and many will live with permanent deficits in sensory and motor functions and with an increased risk for other psychiatric and neurological disorders.3 -5 Furthermore, several clinical studies showed up to nearly half of TBI patients present to the emergency department with elevated blood alcohol levels above the legal limit.6 -9 It is noted however, that these studies only examined a limited TBI population, and the level of alcohol intoxication at time of injury may change if different criteria were examined, including age, sex or cause of TBI. In the United States today, binge drinking is the most common mode of alcohol misuse. 3 According to the National Institute on Alcohol Abuse and Alcoholism (NIAAA), the definition of binge drinking is typically 5 drinks for men and 4 drinks for women during a 2-hour session, resulting in a blood alcohol level greater than 80 mg/dl. 10 In experimental studies, alcohol given acutely or in a binge-like paradigm resulted in worse cognitive performance, impairment in spatial processing and decreased recognition memory in rats.11 -13 Furthermore, in a rat model of TBI and binge alcohol, our group has shown that skilled sensorimotor recovery was worse in alcohol-treated animals. 14 This correlated well with clinical findings in a population of TBI patients who were intoxicated at the time of injury.15 -18 Subsequently, we showed that binge alcohol decreased TBI-induced subventricular zone (SVZ) cell proliferation, attenuated SVZ neuronal differentiation, and decreased the number of newborn migratory neurons to the olfactory bulbs. 19
Neurogenesis is thought to be a compensatory mechanism observed to be up-regulated in the hippocampal dentate gyrus (DG) 20 and the SVZ 21 after a TBI. These newly produced neurons have been associated with recovery of function following brain injuries.22,23 On the contrary, post-traumatic neurogenesis could be contributing to significant pathology following a CNS injury, such as producing an epileptic focus. 24 The goal of this study was to determine how a focal TBI in the forelimb motor cortex affects proliferation in the DG and if a prior alcohol binge would change this result. We report that binge alcohol before a TBI led to a reduced cell proliferative response from the DG and increased ectopic proliferation in the hilar region of the DG at 6 weeks. Therefore, the aberrant cellular responses following binge alcohol could result in negative ramifications on long-term hippocampal functions.
Materials and Methods
Animal subjects
Animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of Edward Hines Jr. Veterans Affairs Hospital (permit #H13-001). Brain tissue sections encompassing the dorsal dentate gyrus from rats used in our previous study 19 were utilized in the current study. Therefore, all animal procedures, including alcohol administration, controlled cortical impact, measurement of blood alcohol level, bromodeoxyuridine (BrdU) administration, perfusion, tissue processing, and histology were performed identically as our previous study. 19 Briefly, we used 2 month-old adult male Sprague Dawley rats (Envigo, Indianapolis, IN; see Table 1). Rats were housed in a fully accredited animal care facility with a 12-hour light/dark cycle and food and water was available ad libitum. Rats were number coded and randomized before all procedures and codes were revealed only after data analysis.
Experimental groups. .

Animal number (n) and groups for the 3 different time points examined (24 hours, 1 week, and 6 weeks, respectively). Animals were randomly assigned to each of the 4 experimental groups (1-4), either with alcohol or vehicle and TBI or sham surgery.
Alcohol administration
We administered alcohol to rats by gastric gavage for 3 consecutive days as described in our previous study, 19 (3 g/kg/dose/day at 9 AM, i.g. using Everclear (Luxco, St. Louis, MO) diluted to 40% alcohol by volume (ABV) with distilled water). Control animals received an equal dose of distilled water by gastric gavage. We observed no adverse effects following the above procedure.
TBI by controlled cortical impact (CCI)
One hour following the last alcohol administration, CCI procedure was performed as described in our previous studies.14,19 Rats were anesthetized (5% isoflurane (75 mg/ml) in 100% oxygen at induction, then 2%-3% to maintain anesthesia), and placed in a stereotaxic frame, with a skin incision and trephination (5 mm diameter) performed directly above the right forelimb area of the sensorimotor cortex (1 mm anterior, 1.5 mm lateral from bregma) 25 (see Figure 1). CCI parameters were as follow: 3 mm diameter, 2.5 m/s velocity, 2 mm depth, 250 ms dwell time (Impact One, MyNeurolab, St. Louis, MO). Sham animals underwent the same duration of anesthesia, and received identical skin incision and trephination procedure.

(A) Experimental design, (B) representative image of a brain at 6 weeks post TBI, with the lesion outlined by a dotted line, and (C) corresponding Nissl stained serial coronal sections of a brain 6 weeks post TBI.
Blood alcohol level (BAL) quantification
BAL measurement was performed identically as reported in our past studies.14,19 Blood samples by tail venipuncture were separated by centrifugation to obtain serum then de-proteinized per the manufacturer’s instructions (Pointe Scientific, Canton MI, USA). The following assay parameters were used: sample/reagent ratio (1:201), 5 minutes incubation (30°C), reading (30°C, absorbance 340 nm). All measurements were done in duplicate and then averaged; unknown samples were run at the same time as known ethanol samples.
Bromodeoxyuridine (BrdU) administration
Preparation of BrdU solution (20 mg/ml in sterile saline) and injection schedule were performed identically to our previous study. 19 All rats received BrdU at 100 mg/kg (i.p.) dose immediately following surgery (sham or CCI). Rats in the 1 and 6 week groups were given 1 BrdU injection (100 mg/kg, i.p.) daily and this continued until each animal received a total of 7 injections.
Perfusion, tissue processing, and histology
The following procedures were performed identically to our previous studies.14,19 Rats were overdosed (phenytoin/pentobarbital, 390 mg/kg, i.p.) and transcardially perfused (at 4°C, first with heparinized saline, then 4% paraformaldehyde (PFA)). Brains were processed as follow: post fixed (4°C in 4% PFA), cryoprotected (30% sucrose in PBS, pH 7.4), cryosectioned (coronal, 40 μm thickness), storage (−20°C in ethylene glycol), antigen retrieval (99°C-100°C, 10 mM sodium citrate, pH 6.0, 15 minutes). See Table 2 for primary and secondary antibodies used. All incubation times and washes were performed identically as described in our previous publication. 19 Phosphate-buffered saline (PBS) (pH 7.4, 0.2% Tween-20, 5% normal goat serum) was used to dilute antibodies to the correct working concentrations (see Table 2). Primary antibodies were incubated overnight (4°C) and secondary antibodies were incubated at room temperature for 2 hours. Nickel-enhanced diaminobenzidine (DAB) staining was performed exactly as described by manufacturer’s instruction (Vector Laboratories). All sections were mounted on gelatin-subbed slides and coverslipped with either Fluoromount G or Permount mounting medias.
Summary of antibodies used for immunofluorescence and immunohistochemistry.

Stereology
All stereological counting procedures were performed similarly to previous published protocols from our lab.19,26 The following parameters were used: 6 sections/animal (dorsal dentate gyrus, 480 μm apart, between −2.0 mm posterior and −4.8 mm posterior to bregma). 27 The granule cell layer (GCL) was outlined by tracing (2.5X objective), and the sampling grid size (64 × 150 μm) and the dissector window size (25 × 25 μm) were used. Counting was performed under high magnification (40X/0.75 NA objective, MBF Bioscience Leica DM400B microscope) but with upper and lower focal planes excluded to avoid oversampling. Cell counting and analysis were performed using StereoInvestigator software version 9.0.
Confocal imaging
To identify cellular fates of newborn cells, we examined tissue sections on a Leica SPE confocal microscope (Leica Microsystems Inc., Buffalo Grove, IL). Using a 10x objective, we identified representative equivalent areas of the DG from a series of stained sections. We subsequently used a 63x/1.3 NA oil immersion objective to confirm double labeling of BrdU+ cells. All image acquisition settings were kept constant and image stacks were imported into Leica Application Suite X and compressed to maximum intensity Z projections.
Analysis of BrdU+ dentate gyrus cell distribution
In order to determine the distribution of newborn cells within the hippocampal dentate gyrus (1 week and 6 week survival groups), we used two 40 μm sections per animal encompassing the middle of the dorsal dentate gyrus (−3.14 mm to −3.30 mm posterior to bregma). Outline tracings of the GCL were performed in StereoInvestigator software version 9.0 in a similar manner as the stereology protocol above (Figure 6). The thickness of the contour encompassing the GCL was kept at approximately 60 μm and was further sub-divided into 2 zones: GCL1 was adjacent to the subgranular zone (SGZ) and GCL2 was adjacent to the molecular layer (ML). Furthermore, two 30 μm contours encompassing the hilar zone adjacent to the SGZ were constructed: Hilus1 was most adjacent to the SGZ and Hilus 2 was furthest away. This method was adapted from previous reports.28,29 The number of BrdU+ cells was determined in each of these zones by manual counting on a MBF Bioscience Leica DM400B microscope under high magnification of 40X/0.75 NA objective.
Statistical analysis
All data analysis was performed using either Minitab version 17 (Minitab, Inc. State College, Pennsylvania, USA) or Graphpad Prism version 5.0 (GraphPad Software, San Diego California, USA). Statistical methods including data transformations, ANOVA and WITH-IN group analysis were performed similarly to a previous study. 19 All data transformations were performed using the Box-Cox procedure, and were necessary to satisfy the Normality-and-Constant-Variance requirements.30,31 The 24 hour data was square-root transformed, the 1 week data was natural log transformed, and the 6 week data was square-root transformed. All data was analyzed by 2-way ANOVA (F tests, general linear model). 32 We looked for main effects and interactions between treatment and injury groups. Furthermore, we performed WITH-IN group analysis (regression analysis) to determine if certain group means were significantly different. 33
Results
Binge alcohol given before TBI results in high blood alcohol level (BAL), however, did not affect lesion size
As reported in our previous study, 19 the alcohol gavage protocol resulted in 156.1 mg/dl ± 8.3 mg/dl mean BAL. Previous reports using the same rat strain resulted in similar BAL.14,34 Furthermore, binge alcohol did not significantly alter the lesion size as measured at any time point post-TBI as reported in our previous study. 19
TBI stimulated bilateral proliferation in the GCL of the dentate gyrus at 24 hours post TBI and binge alcohol decreased this response
After a TBI, cell proliferation in the dentate gyrus has been shown to markedly increase (see Ngwenya and Danzer 20 for review). In our model of TBI, we also saw the dentate gyrus respond robustly through up-regulation of the number of proliferating cells in the GCL starting as early as 24 hours after injury (Figure 2A, A’-A’’’’). There was a statistically significant Injury (TBI) × Treatment (alcohol) interaction for ipsilesional F(1,21) = 14.55; P < .005 and contralesional F(1,21) = 26.89; P < .005 BrdU+ cell counts. Pre-planned multiple comparisons (WITH-IN group regression analysis) showed that the mean number of proliferating cells in the GCL of the Vehicle/TBI group was higher than that of the Vehicle/Sham group for both ipsilesional (F(1,21) = 17.41; P < .005) and contralesional (F(1,21) = 24.88; P < .005) hemispheres (Figure 2A), indicating that TBI alone induced an approximately 10 fold increase in cell proliferation in the GCL. However, when alcohol was administered prior to the TBI, proliferation was reduced bilaterally in the GCL by about 3 fold (Figure 2A, A’’’’). The mean number of proliferating cells in the Alcohol/TBI group was lower than that of the Vehicle/TBI group in ipsilesional (F(1,21) = 7.89; P = .012) and contralesional (F(1,21) = 18.77; P < .005) hemispheres. Interestingly, the number of proliferating cells in the Alcohol/Sham group was 3 fold higher than that of the Vehicle/Sham group in ipsilesional (F(1,21) = 6.76; P = .018) and contralesional (F(1,21) = 9.40; P = .007) hemispheres, (Figure 2A, A’’), indicating that alcohol alone (no TBI) stimulated proliferation in the GCL bilaterally at 24 hours after the last binge.

Binge alcohol and TBI alone and in combination altered BrdU+ cell number in the granule cell layer at 24 hours and 1 week post TBI. (A and B) plots of un-transformed BrdU+ cells count in the ipsi and contra-lesional GCL at 24 hours and 1 week post-TBI respectively: (A) at 24 hours after injury, binge alcohol significantly increased DG proliferation in sham groups and decreased proliferation in TBI groups bilaterally, (B) at 1 week after injury, binge alcohol and TBI alone significantly increased GCL proliferating cells. (A’-A’’’’) and (B’-B’’’’) Representative immunohistochemical staining demonstrates labeling of BrdU+ cells in the ipsilesional DG at 24 hours and 1 week post TBI respectively. Yellow dash lines outline the SGZ; yellow arrows point to BrdU+ cells. Scale bar = 60 μm. Two-way ANOVA, post-hoc within-group analysis, * denotes P ⩽ .05, error bars = SEM.
TBI alone bilaterally increased proliferation while binge alcohol alone increased proliferation in the ipsilesional GCL of the dentate gyrus at 1 week post TBI
Previous studies have reported that the proliferative response after TBI peaks at 1 week post-TBI.35 -37 In the current study, at 1 week post-TBI, many more BrdU+ cells were evident throughout all groups (Figure 2B, B’-B’’’’). There was a statistically significant Injury (TBI) × Treatment (alcohol) interaction for ipsilesional F(1,24) = 9.04; P < .05 and contralesional F(1,24) = 5.89; P = .024 BrdU+ cell counts. Pre-planned multiple comparisons showed that the mean number of proliferating cells in the GCL of the Vehicle/TBI group was higher than that of the Vehicle/Sham group for both ipsilesional (F(1,24) = 15.29; P < .005) and contralesional (F(1,24) = 11.71; P < .005) hemispheres, indicating that TBI alone induced an approximately 2 fold increase in cell proliferation in the GCL.
The mean BrdU+ cell count in the Alcohol/TBI group was not significantly different than that of the Vehicle/TBI group in the contralesional hemisphere (F(1,24) = 2.97; P = .099) but was very close to being significantly different in the ipsilesional hemisphere (F(1,24) = 3.92; P = .061) (Figure 2B, B’’’’). However, the number of proliferating cells of the Alcohol/Sham group was 2 fold higher than that of the Vehicle/Sham group in the ipsilesional (F(1,24) = 5.12; P = .034) hemisphere (Figure 2B, B’’), indicating that alcohol alone stimulated proliferation in the GCL at 1 week after the last binge.
Binge alcohol decreased BrdU+ cell number in the GCL of the dentate gyrus at 6 weeks post-TBI
We found that there was no significant main effect of Injury (TBI) or Alcohol treatment on the BrdU+ cell count of the contralesional GCL at 6 weeks post-TBI (Figure 3A). Multiple comparisons showed that the mean BrdU+ cell count was not significantly different among comparison groups. In the ipsilesional GCL, while there was no statistically significant effects of Injury or Injury x Treatment interaction, there was a significant overall difference between Vehicle and Alcohol groups (F(1,27) = 4.91; P = .036). This indicated that TBI had no effect on BrdU+ cell number in the GCL while binge alcohol still had a significant effect in decreasing the number of BrdU+ cells in the ipsilesional GCL at 6 weeks post-binge (Figure 3B-D).

At 6 weeks after injury, binge alcohol had an overall effect on decreasing GCL BrdU+ cell number: (A) plot of un-transformed BrdU+ cell count in the ipsi and contra-lesional GCL at 6 weeks post TBI, (B and C) representative immunohistochemical staining demonstrates labeling of BrdU+ cells in the ipsilesional DG at 6 weeks post TBI, and (D) interaction plot of square-root transformed 6 week data showing overall binge alcohol effect. Yellow dash lines outline the SGZ; yellow arrows point to BrdU+ cells. Scale bar = 60 μm. Two-way ANOVA, post-hoc within-group analysis, * denotes P ⩽ .05, error bars = SEM.
Binge alcohol altered the distribution of BrdU+ cells in dentate gyrus at 1 week and 6 weeks post-TBI
In adult animals, DG granule cells are being continuously generated from neural precursor cells located in the SGZ, and these cells express markers as seen during development. 38 Doublecortin (DCX) is a microtubule-associated protein and has become a reliable marker for neuronal differentiation. 39 At 1 week post TBI, BrdU+/DCX+ double labeled cells were seen populating the GCL of the dentate gyrus (Figure 4B). Also at 1 week, many BrdU+ cells within the GCL were expressing Iba1, a microglial marker (Figure 4E). At this timepoint, we did not see GCL BrdU+ cells with mature neuronal markers such as NeuN or Calbindin (Figure 4C, D); however, we only qualitatively examined the Vehicle TBI group. In contrast, at 6 weeks post-TBI, a number of GCL BrdU+ cells expressed NeuN, Calbindin, and Prox1 (Figure 5A-C) and very few expressed Iba1 (Figure 5D); as before, we only qualitatively examined the Vehicle TBI group. Of particular importance is Prox1, a transcription factor expressed during DG granule cell development that persists in adult granule cells. 38 At both 1 week and 6 week timepoints, we did not see many BrdU+ cells co-label with astrocyte markers (GFAP and S100B). Additionally, the distribution of BrdU+ cells in the GCL and part of the dentate hilus were measured at both 1 week and 6 weeks post injury (Figure 6A-D). At 1 week post injury, we detected a significant main effect of TBI on the number of BrdU+ cells in the 2 most distant layers from the SGZ, the Hilus 2 (ipsilesional F(1,25) = 10.27, P = .0037; contralesional F(1,25) = 4.47, P = .0446) and far GCL2 (ipsilesional F(1,25) = 15.31, P = .0006; contralesional F(1,25) = 5.71, P = .0247) as defined in the methods section. At 6 weeks post TBI, there was a significant main effect of alcohol on the percent of BrdU+ cells (defined as BrdU+ cell number in each zone divided by total BrdU+ cell number in the DG) in the contralesional Hilus 2 (F(1,22) = 6.67, P = .0170). Additionally, there was a significant main effect of alcohol on the number of BrdU+ cells in the ipsilesional Hilus1 (F(1,22) = 5.70, P = .026). Furthermore, there was a significant main effect of TBI on the percent of BrdU+ cells in ipsilesional GCL2 (F(1,22) = 6.71, P = .0167). Multiple comparisons showed that the percentage of BrdU+ cells was significantly higher in that of Alcohol TBI compared to Vehicle TBI groups in the contralesional Hilus 2. This also corresponds with a significant lower percentage of BrdU+ cells in Alcohol TBI compared to Vehicle TBI groups in the contralesional Hilus1. This indicates that in TBI groups at 6 week post TBI, binge alcohol shifts the distribution of newly generated cells to the more distal regions of the hilus, an area of ectopic granular cells as shown with BrdU+/Prox1+ double labeling in (Figure 6I-J), reported to be disruptive to hippocampal related tasks. 40

Representative immunofluorescence staining of BrdU+ (red) cells with proliferative, neuronal and glial markers from the ipsilesional DG of a vehicle treated rat at 1 week post TBI. BrdU+ cells co-labeled with Ki67 a proliferative marker (A) and DCX, a marker for new neurons (B), while it did not co-labeled with markers of matured neurons such as NeuN (C) and calbindin (D). BrdU+ cells in the GCL expressed Iba1, a microglial marker (E), while no co-labeling with the astrocyte marker GFAP could be seen (F).

Representative immunofluorescence of the ipsilesional DG from a vehicle treated rat showing BrdU+ cells within the GCL at 6 weeks post TBI expressing makers of matured neurons but very few expressed glial markers. BrdU+ cells (red nuclei) can be seen co-labeled with Prox1 (A), NeuN (B) and calbindin (C), which are markers of matured neurons in the GCL. Very few BrdU+ cells are seen co-labeled with the microglial maker Iba1 (D) and none can be seen co-labeled with the matured astrocyte marker S100B (E).
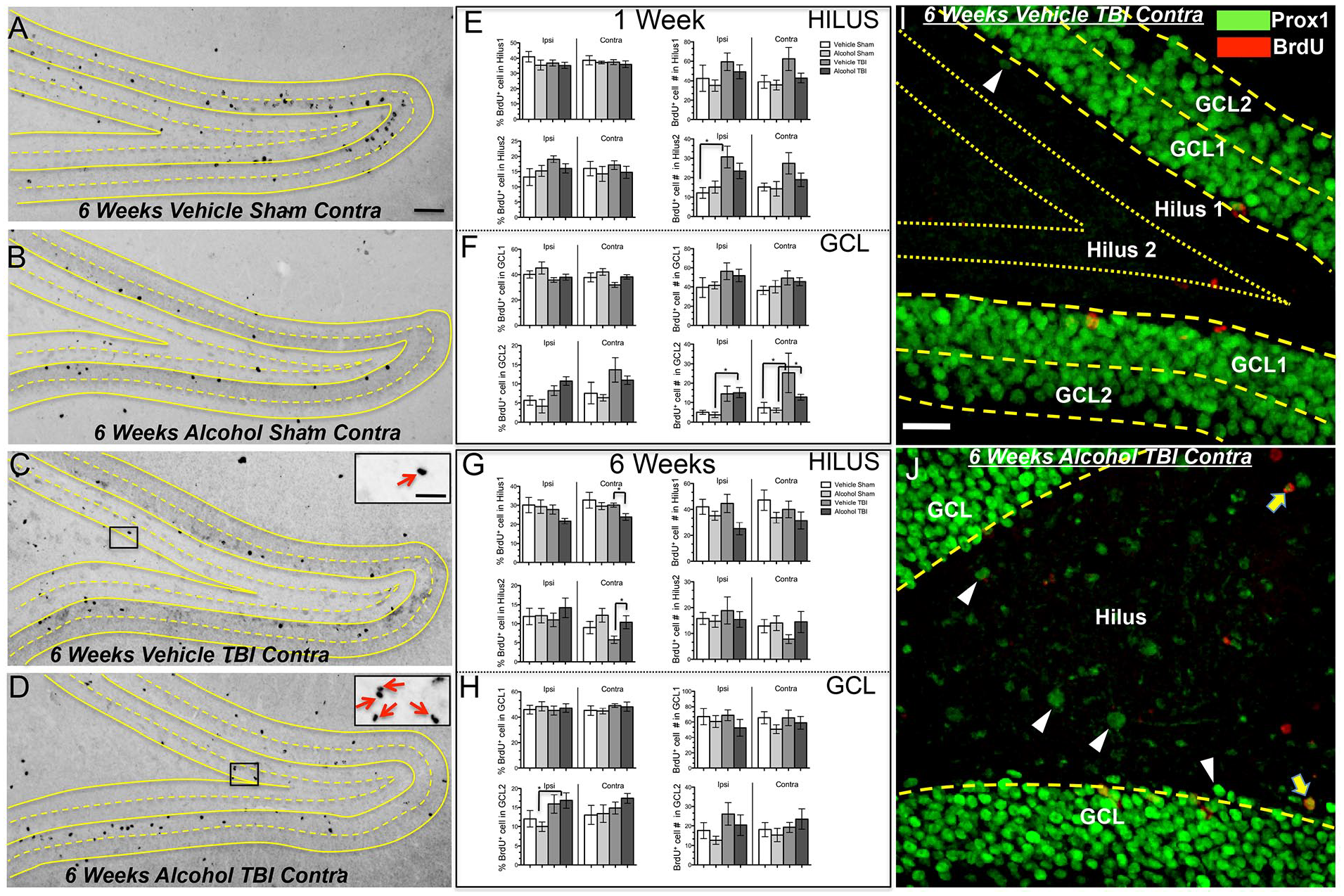
Binge alcohol and TBI altered the distribution of newly generated cells within different layers of the dentate gyrus, (A-D) BrdU DAB IHC of the contralesional DG from the 6 week time point. Inserts in C and D are a magnified view of the boxed area, and red arrows point to BrdU+ cells in the Hilus 2 zone, (E-H) Plots of BrdU+ cell number and percent of total BrdU+ cells over 2 coronal brain sections per rat encompassing the dorsal hippocampus in different layers of the DG at 1 week and 6 weeks post-TBI. (I, J) Representative Prox1 (green) and BrdU (red) immunofluorescence of contralesional dentate gyrus at the 6 weeks time point. (I) schematic of hilar ectopic granule cell quantification method. The GCL was sub-divided into 2 zones, GCL1 and GCL2. The dentate hilar area adjacent to the GCL was also sub-divided into the Hilus 1 and Hilus 2. Each zone was approximately 30 μm in thickness. Immunolabeled sections were processed for DAB and BrdU+ cells in each zone were manually counted. Scale bars: A = 60 μm, insert = 30 μm, I = 30 μm, and (J) alcohol TBI group showed more Prox1+ cells in the ectopic hilar region. White arrowheads point to single-labeled hilar Prox1+ cells, and yellow arrows point to double-labeled cells (hilar subdivisions not shown). Two-way ANOVA, post-hoc Dunn’s multiple comparison, * denotes P ⩽ .05, Error bars = SEM.
Discussion
Our results show that a single episode of binge alcohol (3 consecutive daily doses) before a TBI resulted in a reduction in DG proliferation when measured at 24 hours post-injury. Although TBI did not have a significant effect on the number of proliferating cells in the DG when measured at 6 weeks post injury, binge alcohol overall significantly reduced the number of BrdU+ cells. Surprisingly, binge alcohol alone increased DG proliferation in the short-term when measured at 24 hours and 1 week after the last binge episode. Furthermore, binge alcohol prior to TBI led to an increase in the number of BrdU+ cells in the hilar region of the dentate gyrus. The staining pattern of these cells suggests they are mismigrated granule cells (hEGCs), which may play a role in increased susceptibility to seizures and negatively contribute to hippocampal function.
TBI alone increased proliferation in the dentate GCL
In our study, cell proliferation in the DG increased post-TBI when measured at 24 hours and 1 week post injury, as in agreement with past literature.41,42 Experimental manipulations that resulted in increased hippocampal neurogenesis such as exercise or environmental enrichment lead to improvements in learning, memory and other hippocampal-related tasks. 38 Additionally, blocking hippocampal proliferation resulted in worse cognitive recovery post-TBI. 23 Furthermore, the DG proliferative response to TBI in our study appeared to peak after 1 week and is similar to other studies.41,43 Interestingly, qualitative examination of BrdU staining of vehicle/sham groups (Figures 2A’, 2B’, 3B) indicated that in control animals, there was incorporation of additional new cells into the GCL during this time. This appears to be possible due to a several reasons such as the cumulative BrdU labeling strategy, the high sensitivity of the immunohistochemistry method and the heterogeneity of the proliferative pool of the DG. 38 However, the cell proliferation effect of TBI at early time points did not extend to the 6 weeks post-injury time point. In fact, the mean BrdU+ cell count was lower in the vehicle TBI group as compared to vehicle sham group, bilaterally. This indicates that the elevated cell count as seen early on at 24 hours and 1 week did not persist, and these new cells could have either migrated away from the GCL or undergone cell death as seen in another study. 44
Additionally, we saw that many BrdU+ cells co-expressed the microglial marker Iba1 at 1 week post-TBI (Figure 4E), but not many of these double-labeled BrdU+/Iba1+ cells were seen in the dentate GCL at 6 weeks. Microglia are highly motile cells and therefore could have migrated away from the GCL during the time between 1 week and 6 weeks post-TBI. Microglia play a major role in the inflammatory response in the secondary phase of TBI, thought to be both beneficial and detrimental to CNS recovery.45,46 Pertinent to this study, neuroinflammation can stimulate neurogenesis,47,48 yet, excessive microglial activation and inflammatory cytokines released by microglia such as IL-1b may cause the neurogenic microenvironment of the DG to be non-supportive for the survival and maturation of newborn cells.49,50
Binge alcohol alone increased short-term proliferation in the dentate GCL but had the opposite effect in the long-term
We found that binge alcohol alone elicited a bilateral proliferative response in the DG, as alcohol only treated animals exhibited up to a 4 fold increase in BrdU+ cells at 24 hours compared to controls. While certain proliferation is thought to be beneficial after brain injuries, stem cells within the dentate SGZ can only undergo a finite number of cell divisions, 51 and the increase in proliferation as seen in the binge alcohol alone group could deplete the regenerative capacity of the hippocampus in the long run. Our finding is in contrast with some previous studies that used different rodent models of alcohol intoxication. In these other studies, alcohol caused significant depression in SVZ and SGZ proliferation as examined at various time points (5 hours and up to 41 days) post alcohol exposure.52 -54 However, several studies found an increase in SVZ and SGZ proliferation after alcohol cessation.52,55,56 Specifically, an increase in proliferation in the SVZ at 3 days after abstinence from chronic alcohol was reported. 55 Following binge alcohol, up to a 4 fold increase in DG proliferation at 7 days was detected. 56 While the majority of BrdU+ proliferating cells detected at 2 days post-binge alcohol expressed markers for microglia, 57 later time points showed BrdU+ cells expressed markers of immature neurons. 56 Although the mechanism is unclear, aberrant neurogenesis following alcohol withdrawal has been attributed to an increase in brain hyperexcitability. 58
The BrdU+ cell count at 6 weeks post-binge alcohol revealed that overall, regardless of whether animals received TBI or not, binge alcohol treatment resulted in less BrdU+ cells in the dentate GCL, demonstrating that only a 3 day binge episode had long-term effects in the hippocampus. Although not investigated in this study, the long-term effects of binge alcohol on the DG could be due to a number of reasons, including the change in expression of important trophic factors, such as BDNF, GDNF and EGF, as reported in studies using various binge administration protocols.59,60 Another factor may be due to alcohol’s effect on the epigenetic regulation. 61 These possible mechanisms are currently under investigation in our lab.
Binge alcohol combined with TBI decreased TBI-induced proliferation in the dentate GCL
We found that animals receiving binge alcohol had up to a 4 fold decrease in proliferating cells in the dentate GCL at 24 hours post-TBI. At later time points, the number of proliferating cells in the GCL of animals in the vehicle TBI and alcohol TBI groups were not significantly different. However, at the 1 week time point, the mean number of BrdU+ cells in the ipsilesional dentate GCL of animals in the alcohol TBI group was 40% lower than that of the animals in the vehicle TBI group. Our previous study also reported a similar significant effect of alcohol but in a different proliferative location, that is, the SVZ. 19 At both the 24 hour and 1 week time points, alcohol significantly decreased the TBI induced SVZ cell proliferative response. In adult rats, the SVZ produces new neurons that migrate toward cortical lesion sites post-injury. Therefore, the negative effect of binge alcohol on SVZ neurogenesis could be the underlying cellular basis for the reduced recovery in skilled forelimb use as we have reported.14,19 In regards to the current study, proliferation may contribute to functional recovery post-TBI by incorporation of new neurons into areas of the hippocampus, 62 and the reduction of the DG proliferative response associated with binge alcohol as shown here could worsen TBI associated cognitive deficits.
Binge alcohol combined with TBI altered the distribution of BrdU+ cells in the dentate gyrus
We found that binge alcohol significantly altered the distribution of BrdU+ cells in the DG at 6 weeks after TBI. The shift in the distribution of newly generated cells (BrdU+) was consistent with mismigration of granule cells into the hilus, known as hilar ectopic granule cells (hEGCs), which rarely occur under normal circumstances. 63 HEGCs express immunohistochemical markers that are found in granule cells such as the homeobox protein PROX1 and calcium-binding protein calbindin, with PROX1 thought to be the best marker for these cells. 63 We saw an elevated number of PROX1+ cells in the hilar region of binge alcohol TBI animals, but very few in vehicle TBI animals (Figure 6I, J). Elevated numbers of hEGCs have been detected under conditions such as experimentally induced seizure, 40 BDNF infusion, 64 or in transgenic BAX (a critical regulator of programmed cell death) null mouse lines. 65 Furthermore, one group reported elevated numbers of ectopically located DCX+ cells (putative hEGCs) in the hilus of mice exposed to TBI as juveniles and subsequently treated with binge alcohol as adults. 66 Interestingly, hEGCs have intrinsic properties and axonal projections to the hippocampal CA3 area similar to that of normal granule cells located in the GCL layer.40,67,68 Projection axons originating from hEGCs are similar to mossy fibers and innervate pyramidal cells of CA3 forming large boutons, 69 however, hEGCs have higher spontaneous firing activity than normal granule cells located in the GCL.40,65 This interesting property of hEGCs has been shown in a computational model to disrupt circuitry functions involved in pattern separation and completion, 70 specific processes that underlie higher cognitive functions that depend on the neural circuitry between the DG and CA3. 70 While our study quantified the number of BrdU+ cells within different layers in relation to the anatomical location of the SGZ, it is possible some of the cells counted could have non-neuronal identities. Therefore, it will be important in future studies exploring the relationships between binge alcohol, hEGCs and TBI to utilize double-labeled immunohistological methods.
In conclusion, our results show that TBI in the forelimb motor cortex robustly stimulated DG proliferation short-term in the adult male rat and that binge alcohol given prior to TBI dampened this reactive proliferative process. Furthermore, binge alcohol had a broad effect in reducing the number of newly generated cells in the DG at the 6 week time point, and led to an increased distribution of BrdU+ cells in the ectopic hilar DG area. These putative hEGCs are known to be detrimental to hippocampal dependent functions. Important to public health, a better understanding of the post-injury proliferative process could lead to better therapy to improve recovery, and reduce co-morbidities following TBI complicated by alcohol intoxication.
Footnotes
Author Contributions
Conceptualization: STT, ICV, S-YT, GLK; Methodology: STT, ICV, S-YT, GLK; Investigation: STT, S-YT, JYW, KH, NSA, JPG; Formal Analysis: STT, TEO; Writing – Original Draft: STT, GLK; Writing – Review & Editing: STT, GLK, S-YT, NSA; Visualization: STT; Supervision: GLK; Funding Acquisition: STT, GLK.
Significance Statement
Binge alcohol drinking is the most common mode of excessive alcohol consumption and patients presenting at the hospital for traumatic brain injury (TBI) are often under alcohol intoxication. This study provides evidence that a binge pattern of alcohol administration prior to TBI reduced injury-induced hippocampal proliferation in the short term, and suggests an increase in aberrant hilar ectopic granule cells in the long term. These changes in the proliferative capacity of the brain have important implications for recovery from TBI for many patients suffering with these injuries.
Declaration of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding:
We thank the Office of Academic Affiliations VA Advanced Fellowship in Polytrauma/Traumatic Brain Injury Rehabilitation; Department of Veterans Affairs; NIH/NIAAA T32 AA01352 and NIH/NIAAA-R21 AA020951 for generous support.