
Abstract
Coalition to Cure CHD2 (CCC) is a patient advocacy group dedicated to improving the lives of those affected by CHD2-related disorders (CHD2-RD) by increasing education, building community, and accelerating research to uncover a cure. CHD2 is a chromatin remodeler that was identified in 2013 as being a genetic cause for developmental and epileptic encephalopathies. Pathogenic changes in CHD2 can cause treatment-resistant epilepsy, intellectual and developmental delays, and autism, and some individuals experience neurodevelopmental regression. There are currently no targeted therapies available for CHD2-related disorders. Haploinsufficiency of CHD2 is a causative mechanism of disease for individuals with pathogenic variants (primarily truncating) in CHD2. Recently, identification of individuals with deletion of nearby gene CHASERR, a regulator of CHD2 gene expression, has established dosage sensitivity in CHD2 and solidified the CHASERR gene as a potential therapeutic target for CHD2 levels. Through collaboration with our community and our scientific advisory board, CCC has created a Roadmap to Cure CHD2 as our guide toward a targeted cure that can benefit our community, with steps including (1) identifying and defining patients, (2) developing models of CHD2, (3) studying models of CHD2, (4) testing therapies, (5) involving patients, and (6) reaching a cure. Despite some of the challenges inherent in CHD2 research including establishing animal and cellular models that recapitulate the CHD2 clinical phenotype, identifying measurable outcomes and reliable biomarkers, or testing emerging therapeutic approaches, CCC continues to engage with our community to support ongoing research that aligns with our priorities. CCC sees new and exciting opportunities for additional research that can move our community toward our common goal of a cure that will improve the lives of individuals and their families now and in the future.
Plain language summary
Coalition to Cure CHD2 (CCC) is a nonprofit founded in October 2020 to fund research towards a cure for individuals with CHD2-related disorders. The CHD2 gene was discovered as a genetic cause for epilepsy in 2013. Individuals with CHD2 typically experience seizures that can be resistant to treatment, intellectual disability, delayed development, autism, and other symptoms. The nearby CHASERR gene has been found to regulate CHD2 and is a possible therapeutic target. Individuals with a deletion of CHASERR have been identified - these individuals have too much CHD2 and more severe symptoms.
CCC has created a Roadmap to Cure CHD2 as a guide for their journey towards a targeted cure for CHD2-related disorders. The steps in the roadmap include: (1) identify and define patients, (2) develop models of CHD2, (3) study models of CHD2, (4) test therapies, (5) involve patients, (6) reach a cure.
CCC has worked with CHD2 families to identify family-level priorities for therapeutic development (e.g. seizures, behavior, etc), to capture the impact of disease through qualitative research, and to collect patient health data and tissue samples for scientific analysis.
The development of CHD2 models, mouse models in particular, has been challenging as the mice do not develop seizures. Additional models are underway including frogs, zebrafish, and patient-derived cells. These models have provided crucial insight into the biology of CHD2 but scientific questions remain unanswered.
A variety of therapeutic approaches have been proposed including novel treatments that directly target CHD2 biology as well as the repurposing of existing FDA-approved compounds. Establishing measurable outcomes, including biomarkers, and finding treatments that can reach the brain will be important.
By continuing to follow this roadmap, the CCC believes that one day there will be a cure for CHD2-related disorders.
Keywords
Introduction
Coalition to Cure CHD2 (CCC), founded in 2020, is a nonprofit organization dedicated to improving the lives of those affected by CHD2-related disorders by increasing education, building community, and accelerating research to uncover a cure. The CHD2 gene was identified as a genetic cause of early onset epileptic encephalopathy in 2013, and the most common features of CHD2-RD include seizures (96%), intellectual disability/developmental delay (95%), and autism spectrum disorder (56%) with other notable features including photosensitivity.1,2 Patients typically experience multiple seizure types of a refractory nature including myoclonic, absence, myoclonic-atonic, eyelid myoclonia, and tonic-clonic seizures. 3 Although the understanding of CHD2 has come a long way since its discovery as an epilepsy gene 10 years ago, further important scientific research is required for CCC to achieve its goal of a cure and to improve the lives of individuals with CHD2-RD.
CHD2 is a DNA-binding protein involved in chromatin remodeling and a member of the CHD (chromodomain helicase DNA-binding) family of genes. 3 Chromatin remodeling involves the modification and control of transcription, regulation, and expression of genes. A large proportion of the identified pathogenic (disease-causing) variants in CHD2-RD are predicted to cause premature truncation (nonsense, frameshift, splice variants) of CHD2; full gene deletions that lead to haploinsufficiency have also been reported. Pathogenic variants are almost exclusively de novo (not present in either parent). 1
Although CHD2 was recently implicated as a dosage-sensitive gene though large-scale population studies, CCC is unaware of any known CHD2-only duplications. 4 The recent discovery of three individuals with de novo deletions in CHASERR, a long noncoding RNA (lncRNA) located immediately upstream of CHD2, has added to what was previously understood about the nature of CHD2-RD. Notably, these newly identified patients with CHASERR deletions exhibit a far more profound phenotype than even the most severely impacted CHD2 patients. Features unique to individuals with CHASERR deletions include hypomyelination of the brain, cortical atrophy, and complete lack of the developmental milestones achieved by individuals with CHD2. Research using patient-derived cell lines from individuals with deletions of CHASERR has revealed overexpression of both CHD2 mRNA and protein, establishing CHD2 as a dosage-sensitive gene. 5
While the full impact of loss of CHASERR is unknown, it is believed that excess CHD2 protein drives the more severe phenotype as human deletions covering both CHASERR and CHD2 resemble CHD2 haploinsufficiency. CHASERR may also have roles beyond regulating CHD2 6 ; similar to other neurodevelopmental genes, it has been implicated in several cancers.7–9 In mouse models, CHASERR’s mouse equivalent, Chaserr, negatively regulates CHD2 levels in cis, pointing to CHASERR as a potential therapeutic target to restore CHD2 levels in patients with CHD2 haploinsufficiency. 10
Figure 1 compares the phenotypes of CHD2 and CHASERR-related disorders. 5 While CHD2 and CHASERR are distinct conditions with unique phenotypes, both disorders meet the technical definition of developmental and epileptic encephalopathies. 11

Comparison of CHD2 and CHASERR Haploinsufficiency.
Creating a cure roadmap
To help guide our journey to cure CHD2-RD, CCC created a Roadmap to Cure CHD2 (Figure 2) that maps out the steps that we, as a community, need to take to improve the lives of those living with CHD2-RD. Through a research roadmap, we can compartmentalize the current landscape of CHD2 research into the key milestones to drive us to our goal, which is a cure. We can identify gaps and work to fill these gaps. A cure is likely to be a novel therapeutic approach that restores CHD2 to normal levels, but there may be other approaches that would help some of the symptoms of the disorder and improve quality of life. Increasing research and knowledge of CHD2 and CHD2-RD not only propels us along the roadmap to a cure but also helps our community understand the impact of CHD2 and how their lives could be impacted in the future, both with and without a cure. One of the biggest areas of distress to patients and their caregivers facing a rare disease like CHD2-RD is the lack of information and uncertainty. We hope this roadmap will not only lead to a cure but also guide our community with better information. Through a disciplined approach, we hope to define the clinical trajectory into both measurable deliverables and quantifiable risk factors and then shepherd our community toward the finish line. We are laser focused on our goal, and it is important that we learn from other patient-advocacy groups and find ways we can work together toward a common goal.
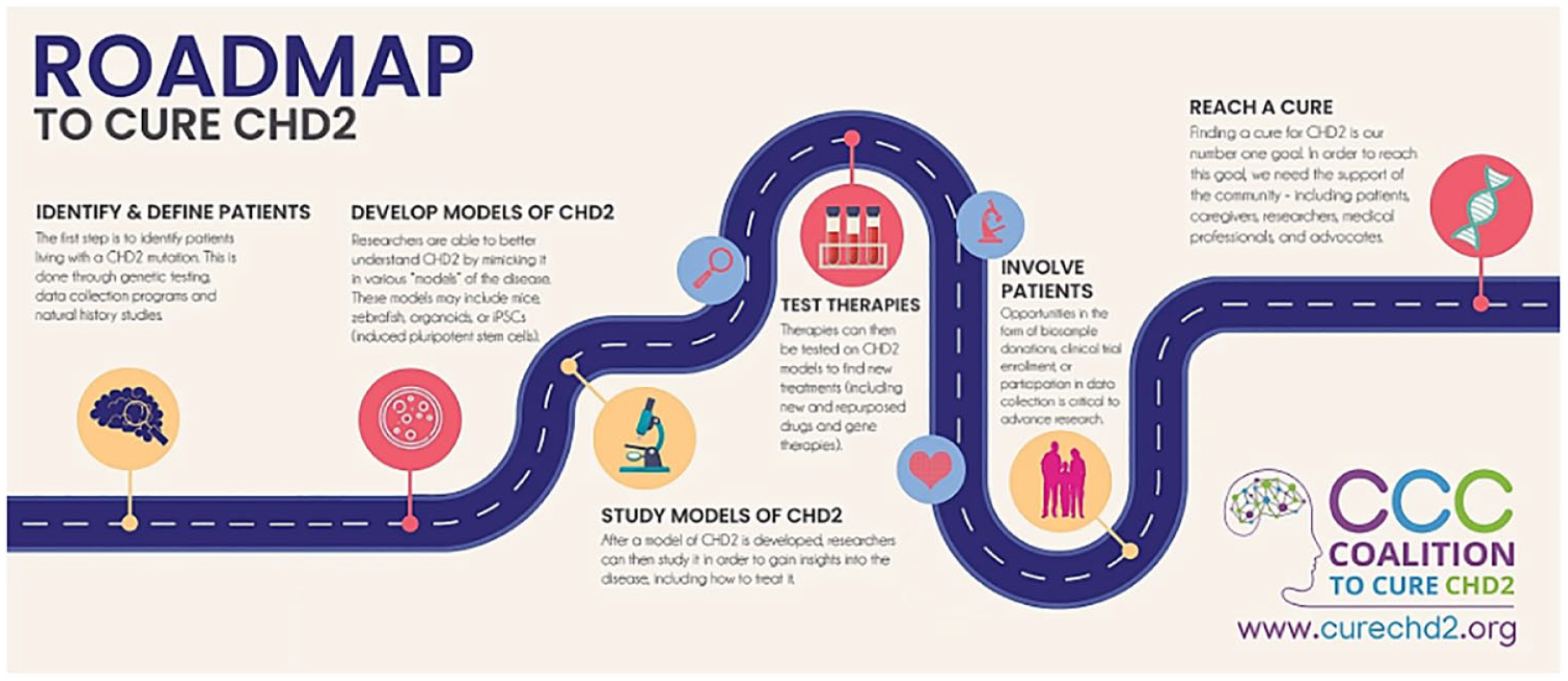
CCC’s roadmap to cure CHD2.
Research priorities
When we think about our “Roadmap to Cure CHD2,” we first need to think about what we expect at the end of the road, or as Stephen Covey stated in his seminal book “The Seven Habits of Highly Effective People” to “Begin With the End in Mind.” In other words, what is a cure for the CHD2 community? Understanding the symptoms and quality of life challenges help us target our efforts on projects that are aligned with our patient community. To understand the community’s challenges and the areas where families would like a therapeutic to make a difference, CCC has undertaken several data collection efforts. The first was a poll, administered through the private CHD2 Facebook group for caregivers or support figures for those with CHD2-RD. The CHD2 Facebook group is open to any family member with a CHD2 diagnosis after approval by an administrator of the group. In a Facebook post, members were first asked to email their top three research priorities in a completely open-ended format. These initial responses were then converted into the basis of a poll. Caregivers could vote for their top three research priorities from the options sourced from the email responses or add their own if an area were missing. There were 75 participants in the final poll. The top eight most important community research priorities are shown in Figure 3. While social media online data collection has its limitations, it was an easy way for CCC to get an initial pulse of what is most important to our community.

Top eight CHD2-RD community research priorities.
Control of seizures was the biggest priority in the survey. Many CHD2 patients do not achieve seizure control despite trying many different medications; those that do achieve seizure control often find it is not permanent. Some families noted that they felt like they are playing a game of “seizure roulette” causing a great deal of strain including medicine side-effects, issues with school activities, and caregiver burnout. An improvement in seizure control is a requirement of any therapy developed. The second priority was behavior. CHD2-RD can cause autism spectrum disorder (ASD) and attention deficit hyperactivity disorder, and a subset of individuals exhibit severe aggression. Some therapeutics can positively impact seizure activity but have negative behavioral side effects, and since improvement in both areas are priorities, it is important to keep this balance in mind for future therapeutics. The third priority is addressing regression, which can include changes in cognition, impact on academic and learning skills, and self-care abilities with or without worsening seizures. The regression experienced by some patients also creates a sense of urgency to find treatments that may preserve neurological function and/or to prevent it from happening to other patients. Hopefully, a cure that biologically restores CHD2 to its more natural levels will create measurable improvements in many of these manifestations that matter to patients and their families. Other priorities included improving intellectual disability, greater visibility on phenotypic severity, medication side effects, and improving speech.
The second data collection effort revolves around the first conceptual disease model for CHD2. The FDA recommends the use of conceptual disease models for patient-focused drug development as they provide a framework for capturing family experiences and priorities to inform selection of outcome measures of interest for therapeutic development.12,13 In 2022, CCC co-presented a poster on a “draft conceptual disease model” for CHD2-RD 14 at the American Epilepsy Society meeting that contained results from a triangulated methodological approach: a literature review, three family interviews, and survey data from a digital natural history study that was conducted in 2019, led by Dr. Anne Berg, Research Professor at Ann & Robert H. Lurie Children’s Hospital of Chicago and the Northwestern University Feinberg School of Medicine, and Gerry Nesbitt, MBA, who created the software platform, CLIRINX, that housed the project. The goal of the natural history study was to better understand the parent perspective regarding seizures and comorbidities associated with CHD2-related disorders. There were 62 participants in total who completed online surveys on the digital platform. The median age of symptom onset was 2 years old. Individuals had difficulties across all five domains of the aberrant behavior checklist of hyperactivity, irritability, social withdrawal, speech, and stereotypies. Thirty-five percent of participants had raw scores that would permit inclusion into ASD trials.
To expand on the draft conceptual disease model, CCC undertook a rigorous qualitative approach led by Dr. Christina SanInocencio, a qualitative researcher and board member of CCC. Preliminary findings from interviews of 15 CHD2 caregivers were reported at CCC’s inaugural family and research conference in June 2023 and a final publication is planned for submission for late 2024.
Identify and engage patients
CHD2 is a rare disorder with an unknown prevalence. In a report of nearly 10,000 individuals referred for genetic testing using a comprehensive epilepsy panel, 0.25% of individuals had a CHD2 pathogenic variant identified. 15 In the United States, CHD2 is included on common epilepsy and autism/intellectual disability panels offered by major clinical testing companies (see Figure 4), but as exome or genome sequencing is increasingly becoming the recommended first-line genetic test for individuals with unexplained seizures and/or intellectual disability, gene content of genetic panels becomes less important. A smaller percentage of patients have deletions that include CHD2 (and sometimes also CHASERR) that are detected on chromosomal microarray testing. Of note, CHD2 is 1 of only 8 genes that are covered by all the Epilepsy and Autism/Intellectual Panels from four major US laboratories (Figure 4).

CHD2 and CHASERR coverage on genetic panels in the United States.
Another tool for CHD2 diagnosis is methylation signature (or “episignature”) testing. CHD2 haploinsufficiency is one of several genetic conditions with a distinct episignature16–18; clinical episignature testing can be used to resolve variants of uncertain significance (VUS). If an individual with a VUS has the CHD2 episignature, the CHD2 variant is likely pathogenic; conversely if the episignature is not present, the variant is likely benign. Some families with VUS appear resistant to methylation follow-up because of the fear of losing a diagnosis that seems to fit, which is disappointing because they likely will not qualify for any of the treatments CCC is working so hard to develop without resolving these variants. We have also encountered families with a strong medical background that simply did not know that this Episign test existed. CCC has already held one webinar on methylation and plans to continue patient education efforts in the future. A methylation signature for CHASERR is still being evaluated.
Genetic testing for adults is arguably behind pediatric genetic testing for a variety of factors including lack of access to genetic testing and inconclusive prior testing done under older protocols, but we are seeing a shift in the age distribution of new cases coming to CCC as adult genetic testing becomes more pervasive. Last year, CCC learned of a patient in their early 40s who received genetic testing for the first time. This patient is believed to be the oldest known patient with a CHD2 diagnosis. An ongoing study of affected adults by Dr. Danielle Andrade of the University of Toronto (currently unpublished) will eventually lead to a published paper that presents new information about the features of CHD2 across the lifespan. We have also learned of a child who was diagnosed at 9 months due to generalized hypotonia, well before the average onset of seizures of 24 months. As access to genetic testing continues to gain traction, we expect the phenotypic spectrum of CHD2 to continue to expand.
Identifying new patients with CHASERR haploinsufficiency is far more challenging because not only is CHASERR not offered on the genome panels mentioned in Figure 4, but also that CHASERR is not covered on any of the genetic panels offered on their websites. Currently, patients have only been identified through genome sequencing rather than through smaller gene panels or exome sequencing that are more commonly used in clinical practice. To highlight the difficulty of finding patients, there was almost 2 years between the first CHASERR case and the second case and a similar gap between cases two and three. As Alu-mediated nonallelic homologous recombination is the causal mechanism of the three known cases, reanalysis of chromosomal microarray data may yield additional cases provided there is coverage of CHASERR. 5 As CHASERR symptoms may manifest shortly after birth, it is prime to benefit from the expanded use of genome sequencing, especially in neonates and in children with Neonatal intensive care unit (NICU) admissions under 1 year old, due to numerous studies citing benefits to these approaches in a clinical setting.19–24 As the methods for genome sequencing become more established 25 and complementary methods, such as RNASeq are added, 26 the testing yield should improve, and the diagnostic odyssey should shorten for patients.
The next step after a diagnosis is to engage patients and encourage them to become part of our community and participate in current and future research. In other words, we need patients to find CCC. This is done through referrals by genetic testing companies (e.g., GeneDX, Invitae), engagement of healthcare providers who order genetic testing (neurologists, geneticists, genetic counselors, other physicians, etc.), increasing awareness of CCC at professional conferences (e.g., AES), and search engine optimization and organic searches, which lead families to our website, www.curechd2.org. Once a patient/family member navigates to the CCC website, they are asked to register their loved one as a confirmed case to count them as a unique patient. Their input also adds to a global map of cases that is displayed on the CCC website (see Figure 5). We also participate in the Google nonprofit program which allows us to buy advertisements on keywords (such as “CHD2”) and promotes CCC website to potential visitors. Additionally, CCC maintains a social media presence on Facebook, Instagram, X, and LinkedIn. Further, Facebook has proven to be an important computer-mediated channel for connection, social support, and information seeking for CHD2 families, and the “CHD2 Support and Research” Facebook support page demonstrates the high user activity of our community with 850 current members as of 5/7/2024.

World Map highlighting location of people with CHD2 in CCCs contact registry.
This CHD2 patient map allows us to visualize areas across the globe where a diagnosis of CHD2 is not being made or where CCC needs to expand its reach to involve these communities. It also allows us to see cases registered on a local basis and recognize gaps with connecting with patients. For example, we know that there is a neurologist at Boston Children’s Hospital that has seen quite a few patients, but our patient registry map for New England is sparsely populated. We hope to fill in these gaps with improved outreach. Potential industry partners who are considering investing in therapeutic development in a rare disorder may view registries as a way of evaluating engagement of the disorder’s community, pinpointing geographical hot spots that would be good locations for clinical research and aiding communication with individual patients.
We have multiple projects to get identified patients engaged with active clinical research projects including natural history studies and the collection of outcome measures. Developing and validating outcome measures for CHD2-RD are essential for clinical trials. We are currently using the RARE-X platform hosted by Global genes which are a research data collection platform. Caregivers or individuals with specific disorders complete validated questionnaires on a broad range of health-related subjects including quality of life data. This ensures the whole phenotype is assessed. These surveys can be repeated over time to begin collecting longitudinal data which may help to define regressions. Currently, there isn’t a mechanism to collect prescribed medication information in RARE-X. Some of the RARE-X outcome measures could be used in clinical trials. RARE-X allows patient/caregiver ownership of their own data; they can choose who is able to see their data and can withdraw at any time. Caregiver-reported data from RARE-X needs to be collated with physician-collected data. CCC has enrolled patients living in the USA in the Ciitizen platform. Ciitizen collects, summarizes, and provides participants digital access to their full medical record. Researchers then can apply for access for those who have consented to review a particular disorder. The medical records are de-identified. To allow quantitative analysis of these medical records an analysis method called Human Phenotype Ontology is used. This is a standardized set of phenotypic terms which are used by computational analysis to produce data. The Ciitizen platform has the advantage of being clinician-collected objective data with data over time, but it is retrospective and biased toward phenotype aspects assessed by the clinician rather than the whole phenotype. For example, a neurologist may not report gastrointestinal symptoms. Both the RARE-X method and Ciitizen method need to be collated against in-person prospective physician assessments to triangulate the data and give us greater objective depth on a smaller group of patients. This in-depth work has begun in Dr. Heather Mefford’s Lab at St. Jude Children’s Hospital.
Develop models of CHD2 and CHASERR
Models of CHD2 are essential to ensure we can understand the basic science that leads to pathology and test potential therapeutics. Disease models include human cell models (cells from patients or cells engineered to have a CHD2 variant) or model organisms such as mouse, zebrafish, fly, or frog. Ideally a model of CHD2 exhibits key features of CHD2-RD; the most measurable phenotype may be seizures, EEG readouts (electroencephalogram), or electrophysiological abnormalities in cells. There are several induced CHD2 pluripotent stem cells (iPSC) in both human and CRISPR (Clustered Regularly Interspaced Short Palindromic Repeat) cells induced around the world. There is a current NIH grant to expand a line of iPSCs into cortical organoids to model the cell type-specific cellular and molecular mechanisms underlying the pathophysiology of CHD2 mutations. 27
There have been several iterations of a CHD2 mouse model. The first mouse model targeted the 27th intron of CHD2 resulting in a protein that lacked the DNA binding domain. The phenotype of the mice included growth defects, kidney abnormalities, and early lethality; however, there were no spontaneous seizures. 28 A review of CHD2 noted that this model may have had some CHD2 gain of function, which is an interesting observation in hindsight given the learnings from CHASERR and CHD2 overexpression. 29 The Hunt Lab of University of California, Irvine created a newer CHD2 mouse model that targeted the 3rd exon of CHD2 and showed deficits in neuron proliferation and a shift in neuronal excitability that included divergent changes in excitatory and inhibitory synaptic function. 30 This model also did not create clinical seizures and did not appear to have a 50% CHD2 protein reduction relative to controls in protein quantification. 30 The Ulitsky Lab created another mouse model of CHD2 (again based on the third exon of CHD2) that also instituted a two nucleotide frameshift. 10 This mouse model, which was characterized by Dr. Moran Rubenstein at Tel Aviv University in part through a grant from CCC, did have some measurable phenotypes and unusual EEG activity, but also did not have clinical seizures (manuscript pending with preliminary data presented at 2023 CHD2 Family Conference). One possible scenario for a better CHD2 model is to use CRISPR to delete exons further down the protein or to model known human pathogenic CHD2 mutations. This approach to recapitulate the human mutations was used in a recent rat model for SYNGAP1 31 , a different genetic form of epilepsy, and presented at their 2022 Family Conference. The SYNGAP1 model, based on deletion of exons 8–12, did produce measurable seizure activity on EEG in heterozygous Syngap1 rats. 31 Even though CHD2 and SYNGAP1 are distinct diseases with their own biology, finding an animal model with measurable seizures is a major goal for CHD2 and their success should not be ignored.
Other models of CHD2 exist including a zebrafish model from University of California, San Francisco (UCSF) by the Baraban Lab. 32 Also in zebrafish, E2I2-MO-injected larvae exhibited prominent morphological and behavioral alterations including pericardial edema, microcephaly, body curvature, absent swim bladder, stunted growth, and epileptiform discharges. 33 Willsey Lab at UCSF has recently explored loss of CHD2 in Xenopus tropicalis (diploid frogs). In Xenopus, for reasons we do not fully know, there is a stronger phenotype seen in morpholinos 34 than in CRISPR editing (unpublished research funded by CCC), and further research is needed to explore this difference in greater detail. The phenotype of spontaneous seizures across animal models of many different DEEs (developmental and epileptic encephalopathy) (not limited to CHD2) appears to be far less prevalent than anticipated, the reasons for which remain unclear, but may suggest species-specific sensitivities to DEE gene function that are particularly marked in humans.
Looking outside of animal models, there are other opportunities to expand our understanding of CHD2. One opportunity is to look for patterns on EEGs of a cohort of CHD2 patients to identify an EEG “biomarker.” Another opportunity is to take blood, urine, or other biosamples from CHD2 patients to look for other biomarkers of CHD2 found in these fluids. CCC is currently involved in a precompetitive biomarker study looking at metabolites and proteins through our membership in COMBINEDBrain (short for the Consortium for Outcome Measures and Biomarkers for Neurodevelopmental Disorders), which is a nonprofit consortium dedicated to speeding the path to clinical treatments for patients with rare neurological disorders. Finally, CHD2 has a proven methylation signature in blood, 18 but there is an opportunity to look for signatures in other cell types.
Developing models of CHASERR haploinsufficiency is far more challenging due to the inherent challenges of working with lncRNAs with one of the major issues being sequence variability. 35 Even as one of the most evolutionarily conserved lncRNAs, 36 CHASERR is not immune to these challenges. The Model Organisms Core of the Undiagnosed Diseases Network, which creates and studies animal models in zebrafish, fruit flies, and worms, did not believe they could create a CHASERR model with actionable insights. The Ulitsky Lab created a CHASERR mouse model with a strong phenotype including an almost two-fold increase in CHD2 production. 10 However, this mouse line has early lethality, which is not abnormal for genes associated with hypomyelination, such as POLR3B. 37 A conditional knockout model may solve the early lethality issues similar to the new mouse model recently developed for 4H Leukodystrophy. 38
Instead of using a CHASERR knock-out approach that theoretically causes total loss of function (but may be prone to alternative transcription start sites) another opportunity is to shift to an approach that uses CRISPR to target very specific sequences of CHASERR that seem important, and then to study changes to those sequences. lncLOOM is a tool to elucidate the most conserved sequences of lncRNAs, and it identified several well conserved sequences of CHASERR mostly around the last exon of CHASERR and closer to the start of CHD2. 39 We believe work on the conserved sequences of CHASERR has been ongoing and that results will be published sometime in 2024. A secondary opportunity is to use bioinformatics approaches to look at other sequences of CHASERR that lack sequence conservation but appear to have the greatest potential biological impact based on their amino acid structure.
Finally, we believe there is a greater opportunity to expand work on Xenopus models for both CHD2 and CHASERR as these frog models are relatively low-cost and allow for high-throughput drug screening. 40 Our understanding is that both underexpression and overexpression of CHD2 in Xenopus exhibit a strong phenotype, although these experimental results have been achieved through protein manipulation as opposed to true in vivo models of pathogenic mutations.
Previous research in Xenopus has shown that CHD2 localizes to microtubules, suggesting that CHD2 may play a role in regulating tubulin in addition to regulating histones similar to other chromatin remodelers. 34 Interestingly, the human CHASERR phenotype leads to significant issues with the corpus callosum, a phenotype that has been referred to as the classic ciliopathy41,42 and both hypoplasia of the corpus callosum and ventriculomegaly (observed in two CHASERR patient MRIs) 5 have been associated with known ciliopathies. 43 Changes in microtubules may not be driving the phenotype in either the CHD2 or CHASERR patients and the MRI changes seen are not exclusive to ciliopathies, but we believe these initial findings warrant further investigation and could provide insights as to whether tubulin biology is dosage-dependent. Xenopus is an ideal model for evaluating this biology.44,45
Study models of CHD2
It is important for CCC to understand the basic science of CHD2 and how this leads to pathology. It was initially unknown why pathological variants affecting CHD2 would result in epilepsy or neurodevelopmental phenotypes. CHD2 is one of the nine chromodomain helicase DNA-binding family proteins responsible for remodeling chromatin which controls the three-dimensional architecture of the genome and gene expression. 10 The majority of pathological variants in CHD2 lead to truncation, while pathogenic missense variants tend to occur in the highly conserved DNA-binding or helicase domains, which likely hinders the ability of CHD2 to remodel chromatin. 1
Wild type and haploinsufficient CHD2 human embryonic stem cell models have been compared to determine the mechanisms by which CHD2 regulates human brain development. It has been identified that during cortical interneuron (hcIN) differentiation CHD2 has genome-wide binding profiles. 46 CHD2 has a role in promoting gene expression during hcIN development. CHD2 has direct targets and mechanisms by which CHD2 prevents precocious hcIN differentiation, and this is one mechanism where pathogenic CHD2 variants can cause developmental disorders. 46 It was also demonstrated that CHD2 haploinsufficiency can cause increased expression of many neuronal genes which can lead to autism and other neurodevelopmental disorders. 46
In vivo work in Xenopus frog models has shown that CHD2 localizes to microtubules of the mitotic spindle. 34 Pathological variants in CHD2 cause disruption in localization at the microtubule with cell cycle stalling which leads to DNA damage and cell death. 33 Tubulin biology is quite elaborate,34,47 and it is unknown the degree to which microtubule defects are driving disease pathology versus transcriptional changes. 34 Xenopus could be an ideal organism to isolate the impact of microtubule defects due to the ability to use Xenopus cell-free cytoplasmic egg extract system, where spindles can form in the absence of nuclei.34,48
For both CHASERR and CHD2, there appears to be an opportunity to examine cell-type specificity and to look at cell types beyond neurons. Young found that CHD2 was widely expressed in oligodendrocytes, almost to the same extent as neurons, but admittedly did not study this cell type in their mouse model of CHD2 and pointed at both oligodendrocytes and astrocytes as a potential for further study. 30 This approach is especially important because lncRNA expression can be more cell-type specific.49,50
Finally, protein quantification of CHD2 is an important issue that needs to be addressed in the future. CHD2 is lowly expressed in certain cell types, and it can be difficult to find good controls. With some of the mouse models of CHD2, you see irregular results including CHD2+/– models that do not appear to achieve 50% protein reduction 30 and CHD2–/– models that still show measurable protein on Western blot. 10 To have the best understanding of CHD2, we need to work with protein experts to sharpen our methods to measure CHD2 and create scientific protocols on how to measure CHD2.
Therapies
Targeting pathological variants for CHD2-RD is difficult for a multitude of reasons, but CCC and our invested researchers see an opportunity for deep thinking and novel approaches. Traditional adeno-associated viruses (AAVs) vectors for gene therapies have the capacity to introduce up to approximately 4.7 kb of single-stranded DNA 51 ; the CHD2 protein is 1828 amino acids long and the entire CHD2 coding sequence exceeds 5 kb. In addition to gene-size issues, the cis-acting feedback loop between CHD and CHASERR 10 would likely introduce additional challenges to therapy via gene-replacement.
One of the most promising approaches for CHD2-RD is using antisense oligonucleotides (ASOs) which are small single-stranded nucleotides that target a specific RNA to modify protein expression. 52 For CHD2, the aim would be to increase CHD2 expression to overcome haploinsufficiency. Special attention will need to be given to the balance of increase of CHD2 expression, given that overexpression of CHD2 could lead to a more severe phenotype as observed in individuals with CHASERR deletions with increased CHD2 expression and also to be mindful that increasing CHD2 expression may also increase the expression of the pathological allele and could cause detrimental effects. Targeting the lncRNA CHASERR with an ASO is being explored. In mice, targeting Chaserr to reduce its expression leads to an increase of CHD2 messenger RNA (mRNA) and subsequent protein levels. 10 We are informed that there are also other ASO targets being explored that can increase CHD2 expression.
Another therapeutic avenue is utilizing microRNA (miRNA). These are small, noncoding RNA which fine-tune protein expression and are critical to brain development. Dysregulation of the miRNA system can lead to epilepsy. 53 Because miRNA can manipulate multiple RNA targets, they could become therapeutic by utilizing them to selectively upregulate individual transcripts, which could overcome haploinsufficiency. 53 Further research is required to determine if they can be manipulated for CHD2-RD. ASOs are established in clinical trials already and we wait to see when miRNAs will achieve the same to pave a way forward for this therapeutic approach.
Targeted drug repurposing screens in animal models are currently underway, and this approach has been successful in other rare diseases. The most famous repurposed drug in the epilepsy sphere is fenfluramine. 54 Originally developed as an appetite suppressant, it is now licensed for Dravet syndrome under a controlled access program to minimize risk. 54 In ADNP (activity dependent neuroprotector homeobox), another chromatin-remodeling related gene with neurological symptoms and the use of low-dose ketamine was found to produce several measurable improvements in behavior. 55 While these examples are nothing more than confidence boosters, small molecule screens can and should also be considered to find a potential small molecule disease modifying agent for CHD2-RD.
Another technique to be considered is stop codon read through therapy for nonsense variants. This is where a ribosome reads past a stop codon and continues translation rather than resulting in a truncated protein. 56 However, this can only be utilized for patients with nonsense variants of specific amino acids and is not of use to the whole CHD2-RD community, making it lower priority.
For CHASERR haploinsufficiency, potential therapies are complicated by the unknowns and complexity of CHASERR/CHD2 biology. From a size perspective, CHASERR appears amenable to AAV gene replacement therapy, but it is unknown whether a transgene could decrease CHD2 levels given the cis-acting nature of CHASERR. RNA Interference (RNAi) 57 could be a good tool to reduce CHD2 levels provided that the protein levels can be controlled in a way that does not cause CHD2 levels to drop too low. Another possibility is to target an activator toward the last exon of CHASERR as it contains the most conserved sequences in CHASERR, and these motifs may be driving the regulation of CHD2 expression levels. 39 CHASERR was presented to the n-Lorem Foundation but was not accepted due in part to the lack of a pharmacodynamic biomarker to measure CHD2.
Biomarkers are a valuable tool to independently demonstrate the efficacy of a treatment. In CHD2-RD this is essential as too much CHD2 (as in the case of the CHASERR pathological variant) also causes a phenotype. There is likely a therapeutic window which needs to be established and finding a clinically useful biomarker can help us to titrate a therapeutic to the most effective dosage. We also need to be mindful that for a potential therapeutic to reach the affected tissue in the body (i.e., brain), it is likely to be injected directly into the cerebrospinal fluid (CSF). Therefore, an effective biomarker either needs to be measurable within the CSF or the effect of the therapeutic needs to cross the blood–brain barrier to be read in plasma. DNA methylation is a strong biomarker candidate given the strong methylation signature with CHD2 haploinsufficiency. 18 A reversal or change of this signature after a therapeutic could help determine the efficacy of a therapeutic.
Conclusion
It is important to note that a research road map is not a straight road with twists and turns along the way but a multilane highway, where multiple projects are traveling alongside one another, and all can influence the other. The natural history work will inform the basic science work. The best example for this is our inaugural family meeting hosted by CCC in June 2023. Our community benefited from meeting others affected by CHD2-RD and learning from experts in the field who presented high-level clinical and scientific talks. Alongside this, the researchers also hugely benefited from meeting the community. This allowed them to deepen their understanding of CHD2-RD in the face of the first ever gathering of patients, understand the impact of CHD2-RD on individuals and their families, and influence future research directions. This continual crisscross of knowledge is important to keep our families informed to increase engagement and for researchers to have a fuller understanding of the condition.
Collaboration is the key to success—not only among our researchers and patients but also across other rare diseases and their associated patient advocacy groups (PAGs). We can learn a huge amount from each other’s successes and failures and gain understanding of each other’s basic science, and how each disorder may overlap not only in symptoms but also in their disease mechanisms. We have begun reaching out to other rare genetic disorders that are involved in chromatin and transcription. Koolen De–Vries Syndrome Foundation (KANSL1 gene) is a PAG pursuing an ASO through a regulatory lncRNA similar to CHASERR 58 and we believe that Kleefstra syndrome (EHMT1) and ADNP also may have similar regulatory lncRNAs. Interestingly, CHD2, KANSL1, EHMT1, and ADNP all have distinct methylation signatures 16 and are captured in the EpiSign Complete test.
Our top priorities that need addressing to lead to a cure that meets the community’s priorities are developing an animal model which can be used for therapeutic testing; developing a biomarker to titrate and measure response; and further therapeutic development to treat CHD2-RD. In the short-term, we hope to have the drug repurposing results and natural history study work ready for publication. We hope the natural history work will help to develop clinical guidelines to help our community in the short term. To advance our roadmap forward, we are holding an in-person scientific meeting in mid-2024 for CHD2 researchers to meet, share knowledge and flesh out the most important scientific priorities and to encourage collaboration on these projects.
CCC continues to build and inform our community. We have regular updates with our passionate and knowledgeable researchers. We have more researchers joining our community. We continue to raise awareness of CHD2-RD and learn from other neurodevelopmental communities. We continue to fundraise and hold webinars and other events to keep our community up to date. This is all in the hope of propelling the multilane highway across the finish line to a successful therapeutic for CHD2-RD, including those caused by CHASERR.