
Abstract
Background:
The International Rare Diseases Research Consortium (IRDiRC) is an international initiative that aims to use research to facilitate rapid diagnosis and treatment of rare diseases.
Objective:
IRDiRC launched the Chrysalis Task Force to identify key financial and nonfinancial factors that make rare disease research and development attractive to companies.
Methods:
The Chrysalis Task Force was comprised of thought leaders from companies, patient advocacy groups, regulatory agencies, and research funders. The Task Force created a survey that was distributed to companies of different sizes with varied investment portfolios and interests in rare disease research. Based on the survey results, the Task Force then conducted targeted interviews.
Results:
The survey and interview respondents identified several factors that make rare disease research and development attractive (e.g. a good understanding of the underlying biology) as well as barriers (e.g. absence of an advocacy organization representing the affected community’s needs). The concept of Return On Investment allowed the exploration of factors that were weighed differently by survey and interview respondents, depending on a number of intrinsic and extrinsic issues.
Conclusions:
The Chrysalis Task Force identified factors attributable to rare disease research and development that may be of interest to and actionable by funders, academic researchers, patients and their families, companies, regulators, and payers in the medium term to short term. By addressing the identified challenges, involved parties may seek solutions to significantly advance the research and development of treatments for rare diseases.
Plain language summary
The International Rare Diseases Research Consortium (IRDiRC) is an international initiative that aims to speed the diagnosis and treatment of rare diseases through research. The IRDiRC Chrysalis Task Force, comprised of thought leaders from companies, patient advocacy groups, regulatory agencies, and research funders, identified key factors that make rare disease research and development attractive to companies. The Task Force distributed a survey to companies with varied investment portfolios and interests in rare disease research, followed by in-depth interviews based on the survey results. The survey and interview respondents identified both attractive factors and barriers to rare disease research and development. The concept of Return On Investment was used to frame discussion of factors that companies weighed differently, depending on a number of issues that were a function of both the company itself and outside factors. The identified challenges can be addressed by funders, academic researchers, patients and their families, companies, regulators, and payers, which hopefully will lead to significant advances in the research and development of treatments for rare diseases.
Background
The International Rare Diseases Research Consortium (IRDiRC) was established as an international initiative to facilitate the rapid diagnosis and treatment of rare diseases through research. 1 Corporate interest and success in rare disease therapeutics lag behind patient needs and demand. 2 Although the reasons for this mismatch have been addressed at the continental level, a truly global approach is lacking, and previous studies have only focused on specific aspects of the problem.3–5 To address this gap, IRDiRC launched the Chrysalis Task Force, which aimed to identify key financial and nonfinancial factors that make rare disease research and development attractive to companies from a company perspective.
Methods
The selection of the topic for the Chrysalis Task Force was made after a year-long iterative and competitive process within the IRDiRC Consortium Assembly, which represents the IRDiRC membership. Task Force co-chairs identified three objectives, namely, to (1) identify key criteria that determine the attractiveness of rare disease research to industry, (2) identify gaps in the current funding landscape to attract industry in rare disease research, and (3) identify other nonfinancial barriers of industry in relation to rare disease research. Task force members were selected by the co-chairs of the Chrysalis Task Force and the IRDiRC Scientific Secretariat via an open-application format that was widely advertised on social media, paying attention to diversity in terms of geographic location, gender, and professional background. Task force members represented thought leaders from companies, patient advocacy groups, regulatory agencies, and research funders.
Survey questions were drafted by the IRDiRC Scientific Secretariat based on the Task Force objectives, which were then revised by Task Force members. A pilot survey was sent to six companies, and minor revisions were made to produce the final version that was sent to 70 companies identified by the Task Force as potential respondents. Particular attention to the geographic diversity of the respondents, as well as the specific corporate interests of the entities, their size, and percent of their portfolio dedicated to rare diseases, was made in the selection process. One set of reminders was sent to companies that did not respond to the initial inquiry. Consent to participate was obtained in the online survey. Only one entry was allowed per company. The survey questions are available in the Supplemental Material. Survey responses were used to identify key issues for more in-depth interviews, which were conducted by members of the Task Force using Microsoft Teams. The topics for the interviews were based on open-ended responses to questions that required clarification or suggested the need for further discussion, particularly on complex topics. Notes were taken by all interviewers and collated by the IRDiRC Scientific Secretariat. Results of the surveys and interviews were analyzed by the Task Force and synthesized by broad category (summarized in Table 1), discussed below. Because of the descriptive nature of the survey, only descriptive statistics were used.
Attractive features and potential barriers for company involvement in rare disease.

HCP: healthcare personnel; IP, intellectual property; R&D, Research and Development.
Results and discussion
The survey received responses from 38 companies out of the 70 that were distributed to a group of companies that ranged across the spectrum in terms of size and interests, and in-depth interviews were conducted with survey respondents from 19 companies. In examining the respondent pool, it was evident that the goal of representativeness in company size was not achieved, although 16 of the 38 companies have multicountry representation (e.g. multinational work sites and scope), suggesting that the goal for international representation was met (Supplemental Table 1).
Most of the respondents to the survey were private, for-profit companies, but public companies were also highly represented (Figure 1). Most companies employed more than 500 people (Supplemental Figure 1). Most companies were focused on pharmaceuticals, but some were also involved in other therapies like gene- and cell-based therapies (Figure 2). A plurality of companies only allocated a small amount of resources to non-rare indications (Supplemental Figure 2). The vast majority of companies were involved in the discovery through postmarketing phases of therapy development (Supplemental Figure 3). Respondents represented a wide range of leadership functional domains (Supplemental Table 2).
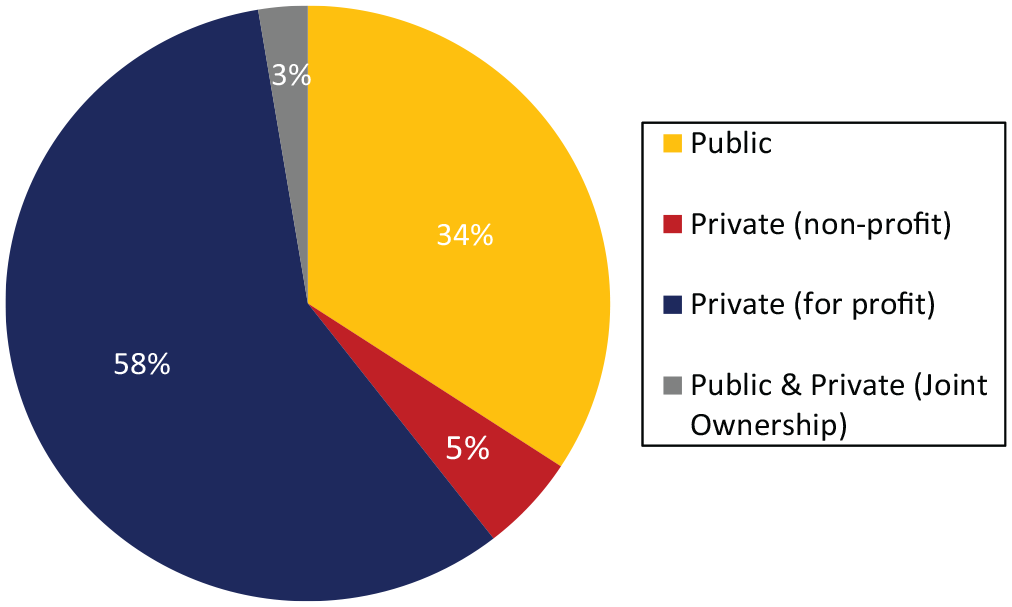
Company type for those responding to the survey.

Company focus for those responding to the survey. Colors indicate company size (numbers of employees).
For new drug development in rare disease, companies prioritized the assessment of patient unmet need, nonexistent current treatment for the indication, the availability of preliminary scientific data, and the competitiveness of the landscape (Figure 3). Potential global market size, expected development cost/good Return On Investment (ROI) followed in importance, and potential regulatory incentives, while the expected time to marketing authorization, an indication attractive to investors, and marketing incentives were considered the lowest priority (Figure 3).

Order of importance for new drug development. The number of respondents is shown in all graphs. Ratings provided by respondents are shown in the color legend.
When evaluating available scientific data to support involvement in rare diseases, companies prioritized the existence of a reliable animal or cellular model, what was known of the safety profile, the mechanism of action of the therapy (if known), and the existence of natural history data for the disease (Figure 4). A lower priority was assigned to the existence of established or regulatory-accepted end points to measure the effect of the proposed therapy and patient-reported disease outcome data (Figure 4).

Order of importance when evaluating available scientific data. The number of respondents is shown in all graphs. Ratings provided by respondents are shown in the color legend.
When evaluating potential market size, respondents assigned the highest priority to the presence of a reimbursement scheme, prevalence of patients in high-income countries, estimated numbers meeting a predetermined threshold, and opportunities to expand an indication (Figure 5). A lower priority was assigned to whether the patient community was well identified and well organized (Figure 5).

Order of importance when evaluating potential market size. The number of respondents is shown in all graphs. Ratings provided by respondents are shown in the color legend.
In terms of access and reimbursement, companies prioritized duration of market exclusivity and opportunities for premium pricing with a somewhat lower priority for authorization for compensated access programs, while tax credits were considered a lower priority (Figure 6).
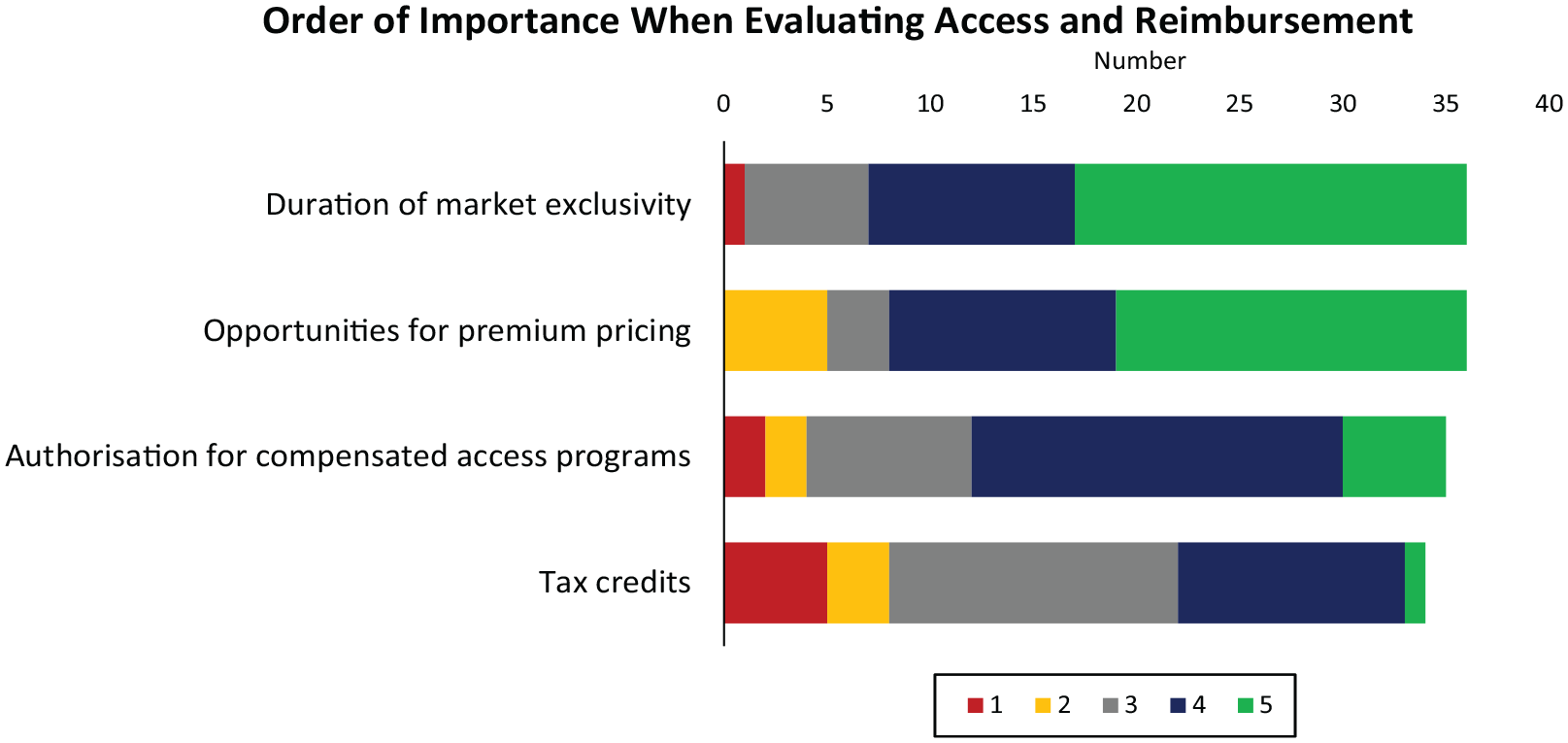
Order of importance when evaluating access and reimbursement. The number of respondents is shown in all graphs. Ratings provided by respondents are shown in the color legend.
In terms of landscape competitiveness, companies prioritized the existence of an approved therapy for the target disease, existence of emerging therapies at the clinical stage and track record of the related competitor, and number of approved therapies for symptoms of the targeted disease (Figure 7). A lower priority was given to market knowledge and customer reach, whereas the geographic footprint of competitors was considered the lowest priority (Figure 7).

Order of importance for when evaluating landscape competitiveness. The number of respondents is shown in all graphs. Ratings provided by respondents are shown in the color legend.
Priorities for collaboration with various stakeholders in the development of a drug or device portfolio were also ranked, with academia and research institutions, regulators (providing scientific advice), payers and health technology assessment (HTA) (national competent authorities), and patient advocacy groups were identified as high priority stakeholders (Figure 8). Intermediate priorities were assigned to contract research organizations and public clinical research centers. Lower priorities were assigned to Contract Development Manufacturing Organizations, venture capitalists and investors, and public or philanthropy funders (Figure 8). Challenges to effective industry and academia collaboration included factors such as complexity of alliance management, and differences in standards of research results (in terms of reproducibility, target validity, safety, delivery on time, etc.) (Figure 9). Quality of data and misaligned intellectual property (IP) timing or lack of IP protection were ranked intermediate, with lack of proper visibility into emerging (academic) science and conflicts of interest ranking last (Figure 9).

Order of importance for stakeholder collaboration when developing a drug or device portfolio. The number of respondents is shown in all graphs. Ratings provided by respondents are shown in the color legend.

Order of importance for challenges to effective collaboration between industry and academia. The number of respondents is shown in all graphs. Ratings provided by respondents are shown in the color legend.
Respondents were asked to identify the three main factors that would encourage companies to initiate new rare disease projects. Factors leading the list included the opportunity to be the first or best in the market, followed by accelerated market authorization, the existence of a patient registry to facilitate recruitment for a clinical trial, market exclusivity, and the possibility to closely collaborate with academia and patients to access preliminary scientific results and natural history studies (Figure 10).

List of main factors that would encourage companies to initiate new rare disease projects (participants were asked to rank their top three selections). The number of respondents is shown in all graphs.
Commercial viability
Investment decisions for any company involve analyzing ROI and risk-reward. This means estimating future cash outflows (investments) and inflows (returns) and, just as importantly, evaluating the likelihood of those cash flows (risk). The greater and the more certain the net cash inflow, the safer and more likely the investment. When evaluating investments in rare disease products, pharmaceutical companies face problems with both the scale and the certainty of cash flows. By definition, rare disease patient populations are small, so returns (cash inflows) are limited, while investment requirements (cash outflows) and barriers to development (risks) are potentially large.
To determine investment requirements, companies evaluate existing capabilities. To determine risk, they measure their comfort level and past experience in developing similar products. Companies identify potential barriers to development as part of their risk assessments, such as pathways to approval and access. It is very difficult for even large organizations to start from scratch: experience in clinical development, regulatory interactions, and commercialization in a particular disease or therapeutic area de-risks drug discovery and development. Smaller companies may accept greater risk but are more likely to partner with academic investigators (some with public or foundation funding) to mitigate that risk, followed by partnering with or acquisition by larger companies (i.e. bringing a drug to market might not be the goal). Likewise, a good understanding of the genetic cause of the disease, the biological mechanisms leading to pathology, clinical progression, and the existence of reliable models of disease and potential biomarkers that could be used in early clinical studies are other important de-risking factors.
Viability in the clinic and experience with regulators are also significant considerations. Many rare genetic diseases, while very serious and possibly life-threatening, can progress very slowly, making clinical development lengthy and very difficult from a sponsor, investigator, and participant perspective. Experience with regulators, regulatory exclusivity, and incentives reduce risk by increasing the probability of a ROI.
Even after clinical development, a small population size coupled with possible diagnostic challenges is a critical issue characteristic of rare diseases. Approaches to improve diagnosis (e.g. disease awareness) can be a financial burden that is separate from clinical development, with an additional, potentially negative, impact on ROI.
Finally, commercialization experience in a particular therapeutic area is important. Competing for internal resources to develop a new therapeutic area can be difficult, especially in an organization with many profitable focus areas outside of rare diseases. Market development, launch strategies, and physician education programs require significant effort, and without a firm commitment to rare diseases, it can be problematic to compete for resources. This is particularly problematic when the patient population is small and possibly heterogeneous, aggregate demand is low, risks are high, and returns are limited.
Probability of regulatory success
As noted above, the outlook for a pathway to regulatory approvals can have a major impact on company interest in rare diseases. Respondents noted that, of all the constituent groups involved in rare disease drug development, communication and collaboration with regulatory agencies around the world was the most critical. This was based on a range of concerns which, in aggregate, pose a high degree of risk in seeking approvals for rare disease products.
The lack of regulatory precedent for most rare diseases contributes to uncertainty and introduces a gap in communication and collaboration between companies and regulators concerning which evidence is needed and how to generate it. The shared lack of knowledge and availability of natural history for most rare diseases, the small number of patients involved, the potentially limited number of research participants, and the rapidly evolving knowledge of both diseases and potential treatments highlight the importance of this gap. In turn, this may lead to challenges in defining pivotal trial design (including which end points to study and study design) and evolving guidance about standards and manufacturing control.
Regulators also need to balance short-term patient needs and long-term outcomes, as gene-based treatments are increasingly used for rare diseases. These gaps have led to a limited consensus among global regulators on data standards and quality, or how natural history data may be used in drug development. Limited data sharing among rare disease stakeholders (such as academia, patient organizations, industry, and regulators) that could inform regulators and future product development exacerbate these challenges.
Major regulatory agencies offer opportunities to obtain advice on clinical study designs prior to trial initiation, including discussions around end points and what would be considered a clinically meaningful change. However, agencies typically do not commit to a defined path to approval at that stage and wait until trial data are presented to assess the benefit/risk of the product. This results in uncertainty and risk for companies developing trials and products that may be approved in one country but not in a different jurisdiction. Regulatory agencies have worked to resolve some of these issues in recent years, adding programs to qualify biomarkers, end points, and natural history models to inform disease fields broadly about best practices for trial design. However, the requirements for qualification may be unattainable for rare diseases with limited populations and no approved therapies.
Furthermore, although the International Council for Harmonisation guidelines describe the requirements for approval of pharmaceuticals for human use, there are differences in interpretation among regulatory agencies. Agencies also have different requirements for the qualification of biomarkers and end points, qualification for an orphan designation, and clinical trial design (e.g. requirements for studying different populations), making global drug development in rare diseases challenging. Respondents noted that there was even a lack of regulatory infrastructure to support drug repurposing in some cases.
Several respondents noted that increased regulatory incentives, such as accelerated marketing authority, market exclusivity, and faster regulatory processes, increase their interest in rare disease drug development, but this alone does not drive their decision to pursue an active rare disease drug development program. Despite the potential for financial rewards if the drug reaches the market, respondents indicated that there is still a heightened risk associated with low ROI for products that address small markets.
Overall, companies developing therapies for rare diseases perceive the different approaches of major regulatory agencies and limited early incentives for developing therapies for small populations as major challenges. This results in increased financial risk and uncertainty during drug development. Global concordance around the regulatory pathways for approval of rare disease therapies, including approaches to understanding rare disease natural history, acceptance of clinical trial designs, acceptance of end points, and other requirements for regulatory approvals, if achieved, could reduce inefficiencies in rare disease drug development. This would allow companies and regulators greater certainty around trial design, enable companies to run fewer clinical trials that are truly global, and allow companies to reach larger numbers of people living with rare diseases around the world. This would ultimately decrease the risks of developing rare disease therapies and encourage companies to work in the rare disease space while increasing the global reach of new therapies.
Payer/pricing models
Pricing and reimbursement considerations are typically factored into a company’s calculus early in rare disease drug development. Ideally, expected payer evidence requirements are typically integrated at the time of designing the clinical trials and when addressing regulatory hurdles. Recognizing that achieving reimbursement at a premium price is an important financial incentive for companies, payers are only willing to pay for the demonstrated value of the treatment in the context of their healthcare system. This latter commercial dynamic is all too often a secondary consideration during earlier stages of development when understanding the biology of disease is the priority for scientists. The financial reward for companies is realized only if the product is in demand and the value for society is assessed as worthy of payment. The value proposition for payers is substantiated by evidence (including the burden of disease, the efficacy and safety of the new therapy, and the added benefit compared to other therapeutic options), which has been developed for regulatory purposes in well-defined patient populations.
There are barriers related to pricing and reimbursement. Overall, the unpredictability of the reimbursement negotiations that may result in a price lower than expected has a negative impact on ROI. Variability across jurisdictions where HTA bodies unevenly apply value frameworks, and modifiers in their assessment leads to different reimbursement decisions. This may restrict the use to a subpopulation where the treatment is deemed most cost-effective rather than be used in the broader cohort accepted by regulators, essentially shrinking the size of the potential market. For companies, this is particularly damaging for prospects in small populations and rare diseases. Expectations for additional evidence after marketing approval may lead to financial commitments that can act as disincentives too if they are perceived as disproportionate. Based on methods developed for common diseases, HTA/payers expect mature data and high-quality evidence with limited uncertainties in support of premium prices. The many challenges in gathering large-scale evidence in rare diseases can result in rare disease treatments falling short of payer expectations of an unequivocal demonstration of significant benefit and high value for patients and society. Companies may resist creating new patient datasets when the probability of success is perceived as low. Pressure on prices also comes from other sources as payers mostly manage a fixed yearly drug budget, negotiations include important discounts, and prices erode over time with no adjustments to the cost of living outside the United States. Companies hedge the risk of additional evidence commitments and mandatory price discounts by requesting premium prices, and in return, payers may request new data to grant a price premium. Thus, even when the unmet need is high, price expectations might not be met. When appropriate, accepting surrogate end points and data from transnational registries can mitigate this problem. When other treatments are commercially available, they are used as comparators and may heavily influence the price point companies can expect. This is problematic when the available treatment is suboptimal and relatively inexpensive because the investments needed to demonstrate superiority in efficacy will be high.
An example where development incentives are totally lacking is in repurposing a medicine that no longer has market exclusivity. Developing a new indication for a rare disease still requires an important investment, but in this example, the price is set at the one from the existing commercialized formulation and is, therefore, nonnegotiable. As a result, no prospect of ROI exists for a new small or even large indication. In recognizing this challenge, the European Commission started a pilot program to support not-for-profit organizations and academia to gather or generate sufficient evidence on the use of an established medicine in a new indication with the view to having this new use formally authorized. 6 This pilot aims to stimulate drug repurposing but is not linked to financial incentives.
Collaborations with other involved parties
Respondents noted that collaborations with academia, research institutions, and regulators are crucial when developing their rare disease therapies/device portfolios. Collaborations with patient advocacy groups and payers also were important. Challenges and gaps inherent to collaborations with regulators and payers were previously outlined. Respondents noted that one of the main factors that would encourage them to initiate (new) rare disease(s) projects would be the possibility of collaborating closely with academia and patients to access preliminary scientific results and natural history studies (in other words, to align on business aspects that would advance rare disease drug development and confirm ROI). However, many operational impediments emerged as clear barriers to effective collaborations.
Academia was credited as a source of innovation, expertise, and ‘out-of-the-box’ thinking with a shared willingness and intent to collaborate with industry based on a mutual interest in science and the scientific process. Operational barriers to effective collaborations (cited by respondents) included disparate incentives between entities and inherent distrust of companies by academia. When academics develop a new approach, IP protection is frequently either not obtained at all or sought at the wrong time. Academics may believe that making an approach public will allow for cheap translation into a therapy when in fact, a lack of IP protection exposes a company to risk and disincentivizes drug development. Respondents also cited challenges related to contractual terms made by institutions with regard to IP for collaborative work. A lack of alignment on business deliverables with academic institutions was described (e.g. having different qualitative and quantitative standards for scientific rigor and validity, nonadherence to specified corporate timelines, and legal burden).
In addition, misalignment of focus and mandate was mentioned, wherein companies rely on metrics tied to ROI for sustainability, but academic metrics are placed elsewhere. Regarding publications, the tension between independence and transparency was repeatedly noted. Unique to rare diseases is the low number of academic and physician experts specializing in any given rare disease. Respondents described the challenges of having them collaborate in a meaningful way (alignment on clinical trial design, clinical trial execution, data sharing) without deference to individual visibility as ‘the expert’.
For most rare diseases, there is a lack of quantitative understanding of the disease burden. The impacts and progression of disease symptoms further complicate the ability to construct and conduct effective drug development, and accurately assess benefit–risk profiles. It is the benefit–risk calculus that ultimately drives value for the patient community and decision-making by regulators and payers. 7 Patient communities are critical collaborators in quantifying disease burden and unmet needs. Addressing the latter is the overwhelming reason many companies enter into rare diseases. Rare disease patient and advocacy communities have expertise in the ‘lived experience’, connectivity with the patient community, and relevance with all stakeholders in the rare disease ecosystem, and often build their capabilities to engage in the drug development and delivery continuum. Engagement by the industry with rare disease patients and advocacy communities during all stages of the therapy development process may greatly enhance the likelihood of success by de-risking the science, gaining access to tools and data resources, empowering participation in clinical research, and defining the objectives and end points that are most meaningful to patients. Frameworks have been developed for sponsors and patient groups to engage at every stage of drug development to build effective collaborations across the medicines development continuum, such as the Patient Group Engagement – Clinical Trials Transformation Initiative. 8
While respondents acknowledged the critical need to collaborate with patient advocacy groups in this research and commented on the richness and complexity of these relationships in advancing the rare disease ecosystem of care, access, and policy, they cited several barriers to effective collaborations specific to drug development. An obvious challenge is when a given rare disease patient community has not yet organized itself into an advocacy group with a shared longitudinal view and mission to advance drug development in collaboration with other stakeholders. If capabilities to engage in research and drug development are lacking by patient advocacy groups, then investments may be needed to build such capabilities, inclusive of resources such as time, capital, and people. Respondents reported that if there are several patient advocacy groups supporting a given rare disease community, then there may be competition for resources with trade-offs in building capabilities. In addition, when they do not work together, multiple groups may duplicate infrastructure (e.g. registries), creating fractured efforts that are less effective than a single combined effort. Respondents noted the potential risk to innovation if patient communities and organizations are unwilling to collaborate with companies coming into a therapeutic space where established treatments may already be on the market. Several respondents found it useful to perform a baseline assessment of the patient/advocacy landscape early in consideration of a rare disease research and development program to ensure a shared research and development agenda with the patient community moving forward.
Strengths and limitations
The survey and interview format used in this study provided valuable insights into the decision-making process of companies in the rare disease research space. Respondents represented a variety of countries and types of companies; however, it is important to note that the respondents may not be fully representative of all companies operating in this field. The candor of answers, particularly in the interviews, is also a strength of this work. As a result however, individual companies could not be identified because of this anonymity. Nonetheless, the general framework used here, which was developed by those experienced in the field, can serve as a useful starting point for discussions between stakeholders. The study did not explore the role of other funders such as angel investors, venture firms, and public–private partnerships but this is the focus of a new IRDiRC Task Force.
Conclusion
The unique perspectives and insights provided by the respondents identified several factors that make rare disease research and development attractive to companies, as well as barriers. The concept of ROI allowed the exploration of factors that were weighed differently by respondents, depending on a number of intrinsic and extrinsic issues. Clinical trial designs for rare diseases are evolving, as are regulatory practices. Innovative, public–private models are emerging, like the Bespoke Gene Therapy Consortium 9 and the Rare Disease Moonshot initiative, 10 which both seek to ease operational barriers to collaborations involving multiple interested parties, including companies, while enabling a shared goal of advancing gene therapies for ultra-rare diseases. Manufacturing costs for some genetic therapies are also likely to decrease over time. 11 As some investment indicators in disease-related R&D appear to be improving (e.g. Eroom’s Law), there is a reason for optimism in the long term. 12
The Chrysalis Task Force identified potentially actionable barriers that can be addressed by funders, academic researchers, patients and their families, companies, regulators, and payers. The identification of these barriers provides a path forward for the advancement of research and development of treatments for rare diseases.
Supplemental Material
sj-docx-1-trd-10.1177_26330040231188979 – Supplemental material for The IRDiRC Chrysalis Task Force: making rare disease research attractive to companies
Supplemental material, sj-docx-1-trd-10.1177_26330040231188979 for The IRDiRC Chrysalis Task Force: making rare disease research attractive to companies by Katherine L. Beaverson, Daria Julkowska, Mary Catherine V. Letinturier, Annemieke Aartsma-Rus, Jennifer Austin, Juan Bueren, Simon Frost, Misako Hamamura, Jane Larkindale, Greg LaRosa, Rita Magenheim, Annamaria Merico, Anna Maria Gerdina Pasmooij, Vinciane Pirard, Nicholas Ekow Thomford, Michihiko Wada, Durhane Wong-Rieger and Adam L. Hartman in Therapeutic Advances in Rare Disease
Footnotes
Acknowledgements
The authors gratefully acknowledge the important early contributions of former IRDiRC Scientific Secretariat members: Carla D’Angelo and Alexander Parry. This manuscript was prepared by the authors in their personal capacity. The views and opinions expressed here are those of the authors and do not necessarily reflect the views, opinions, or position of their employers or any subsidiary companies. This report does not represent the official view of the National Institute of Neurological Disorders and Stroke (NINDS), the National Institutes of Health (NIH) or any part of the US Federal Government. No official support or endorsement of this article by the NINDS or NIH is intended or should be inferred.
Declarations
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.