
Abstract
Background:
Maternal depressive symptoms are a common phenomenon during pregnancy and are related to negative outcomes for child development and health. Modifications in child DNA methylation are discussed as an underlying mechanism for the association between prenatal depressive symptoms and alterations in child outcomes. However, formerly reported genome-wide associations have yet to be replicated.
Methods:
In an epigenome-wide association study (EWAS), alterations of DNA methylation related to maternal prenatal depressive symptoms were investigated in buccal cell samples from 174 children (n = 52 exposed to prenatal depressive symptoms; 6-9 years old) of the German longitudinal study FRAMES-FRANCES. Whole blood samples from the independent, age-comparable ARIES subsample of the ARIES/ALSPAC study (n = 641; n = 159 exposed to prenatal depressive symptoms; 7-8 years old) were examined as a confirmation sample. Depressive symptoms were assessed with the Edinburgh Postnatal Depression Scale. DNA methylation was analyzed with the Infinium Human Methylation 450k BeadChip. Modifications in single CpGs, regions, and biological pathways were investigated. Results were adjusted for age and birth outcomes as well as postnatal and current maternal depressive symptoms. Analyses were performed for the whole sample as well as separated for sex.
Results:
The EWAS yielded no differentially methylated CpG or region as well as no accordance between samples withstanding correction for multiple testing. In pathway analyses, no overlapping functional domain was found to be enriched for either sample. A comparison of current and former findings suggests some overlapping methylation modifications from infancy to childhood. Results suggest that there might be sex-specific differential methylation, which should be further investigated in additional studies.
Conclusions:
The current, mainly nonsignificant, results challenge the assumption of consistent modifications of DNA methylation in children exposed to prenatal depressive symptoms. Despite the relatively small sample size used in this study, this lack of significant results may reflect diverse issues of environmental epigenetic studies, which need to be addressed in future research.
Introduction
The “Developmental Origins of Health and Disease” (DOHaD) hypothesis 1 suggests that environmental stimuli in the pre- and early postnatal period can lead to long-lasting alterations in an organism that influence development and health throughout life. One of these potential influencing stimuli are maternal prenatal depressive symptoms, which are a frequent phenomenon with an estimated prevalence between 10% and 25%. 2 Prenatal depressive symptoms are regarded as a risk factor for child development, which can manifest in preterm delivery, more difficult temperament, delayed cognitive development, or emotional and behavioral problems.3-5 Partially based on this evidence, the originally somatic DOHaD hypothesis was recently expanded to mental health.6,7 Epigenetic modifications of DNA methylation in children are discussed as an underlying biological mechanism for the association between maternal mental health in pregnancy and later child mental health.8,9 Environmental stimuli can modify DNA methylation of cytosine-guanine dinucleotides (CpGs), resulting in context-specific, sometimes contradicting effects, eg, attenuated or silenced gene expression 10 versus methylation-mediated transcription activation and several other developmental outcomes (eg, X-chromosome inactivation). 11 These outcomes based in adverse environmental stimuli can be found years after the exposure of the fetus. A review by Barker et al 12 reported several studies linking prenatal stress (such as prenatal depressive symptoms) to differential methylation in children and youth between 5 and 10 years of age and some other studies report associations to be persistent after up to 13 years. 13
Studies investigating DNA methylation in association with prenatal depression have reported epigenetic modifications in placental DNA,14,15 leading to an altered placental hormone sensitivity and transfer. Others found differences in the methylation of DNA extracted from newborns’ cord blood and child buccal or blood cells. 16 These alterations of infant and child DNA methylation are hypothesized to be associated with child phenotypic outcome, eg, neurodevelopment 17 and hypothalamic-pituitary-adrenal axis activity.18-21
Most of the studies regarding epigenetic modifications in association with prenatal depressive symptoms and phenotypic outcome have used a candidate-gene approach, investigating preselected genes which have previously been found to be involved in developmental processes or physiological mechanisms.19,22 Preselection enables the investigation of the influence on and the influence of specific established genes and pathways, but simultaneously prevents investigation of any additional genes that may help to identify new underlying mechanisms and relevant pathways. Using microarrays to conduct epigenome-wide association studies (EWASs) regarding genome-wide modifications of DNA methylation (for a list of EWAS regarding diverse prenatal risk-factors, see, eg, the work of the PACE [Pregnancy and Childhood Epigenetics] consortium) 23 yields the possibility for the selection and prioritization of new candidate loci, which then could be validated and examined in downstream analysis. 24
There have been some EWAS published investigating associations of prenatal depressive symptoms and DNA methylation in newborns or infants; however, the existing studies differ in sample size, risk operationalization, cells for DNA extraction, and statistical procedures for data analysis.25-29 Schroeder et al 25 conducted an EWAS with 201 newborns finding no differences in DNA methylation of umbilical cord whole blood samples dependent on maternal depression during pregnancy. Similar results were found by Wikenius et al 29 in the latest EWAS with 274 DNA saliva samples from 6 weeks and 12 months old infants. In contrast, Non et al 26 and Nemoda et al 27 reported modifications associated with prenatal depression in DNA extracted from umbilical cord blood when analyzing mixed leukocytes or T lymphocytes, respectively. Non et al 26 identified small differences in DNA methylation of 10 CpGs related to 9 genes in 13 children whose mothers were prenatally depressed compared with 23 children from healthy mothers. The EWAS by Nemoda et al 27 found methylation modifications at 145 CpGs of 139 genes in a sample of 44 newborns, mainly in the form of a hypomethylation in newborns from prenatally depressed women. Comparing the results of these EWASs,26,27 both reported predominantly small differences in DNA methylation, mostly in the form of hypomethylation. However, there is no concordance between the reported significantly differentially methylated CpGs. Furthermore, only 5 of 38 genes, which reached significance under a less stringent criterion in Non et al, 26 were also described by Nemoda et al. 27 In an EWAS by Viuff et al, 28 modifications were found in the DNA methylation of cord blood samples of 844 newborns depending on the timepoint of depression during pregnancy. Only depression in mid-pregnancy was associated with 2 differentially methylated CpGs. However, an attempt to replicate the significant findings in the Generation R Study with 1339 cord blood samples failed, despite using comparable methodology (with the exception of the operationalization of prenatal depression). 28 While these studies have focused mainly on differential methylation at single CpG sites, it is also possible to analyze differential methylated regions (DMRs), pooling together multiple sites of single-nucleotide methylation. The EWAS by Viuff et al additionally used this technique and identified 39 DMRs associated with prenatal depression, characterizing their effect by different timepoints during pregnancy. Yet, confirming these results using a replication cohort failed. 28 In another study, Liu et al 30 identified associations between prenatal depression and methylational differences in 2 distinct character DMRs associated with MEG3 and IGF2.
The small number of consistent results emphasizes the further need for validating EWAS findings in larger and independent cohorts using comparable methods. 31 In addition, there have been multiple studies which have found sex specificity of methylation19,32-34 suggesting that this aspect should also be taken into account. Furthermore, no EWAS has examined the stability of early found modifications over the course of childhood. Therefore, this study (1) investigated the association of prenatal depressive symptoms and epigenome-wide DNA methylation of primary school–aged children from a German longitudinal cohort and (2) compared the results with an independent, age-comparable British cohort using the same risk-operationalizing and strategies of data processing. (3) Results of modifications in newborn age reported by former studies were reevaluated in our study cohort in childhood. (4) All analyses were performed for the overall sample as well as separately for boys and girls, investigating sex-specific effects on modifications of DNA methylation.
Materials and Methods
Study design
The discovery sample consisted of a subsample of 192 mother-child dyads who provided child DNA samples for epigenetic analyses from the German longitudinal studies Franconian Maternal Health Evaluation Studies 35 (FRAMES, n = 1100, first data collection [t1]) and Franconian Cognition and Emotion Studies 36 (FRANCES, n = 245, second data collection of same cohort [t2]). For this study, exclusion criteria were as follows: (1) nonwhite ethnicity (n = 4), (2) multiple pregnancy (n = 0), (3) antidepressant medication intake during pregnancy (n = 3), and (4) incomplete data of depressive symptoms (n = 4). Furthermore, 7 dyads had to be excluded after sample quality control. Exclusions resulted in a discovery sample of n = 174 mother-child dyads (88 boys, 86 girls). At birth (t1), mothers (48.9% primiparous) had a mean age of 32.7 years (SD = 4.6, range: 19.9-44.3). At the timepoint of DNA extraction (t2), children were between 6 and 9 years old (M = 7.6, SD = 0.6, range: 6.0-9.3). The study was approved by the Local Ethics Committee of the Medical Faculty of the University of Erlangen-Nürnberg and conducted in accordance with the Declaration of Helsinki. All participants gave informed consent.
The confirmation sample was drawn from the participants of the Accessible Resource for Integrated Epigenomic Studies 37 (ARIES) subsample (n = 1018 whole blood samples at age of 7 years, t2), which is a part of the British Avalon Longitudinal Study of Parents and Children38,39 (ALSPAC) cohort (15 454 pregnancies, resulting in 14 901 children alive at 1 year of age, t1) and will be further on called ARIES/ALSPAC. Please note that the study website contains details of all the data that are available through a fully searchable data dictionary and variable search tool (http://www.bristol.ac.uk/alspac/researchers/our-data/). Exclusion criteria were transferred from the discovery to the confirmation sample: (1) nonwhite ethnicity (n = 27), unknown ethnicity (n = 5); (2) multiple pregnancy (n = 4); (3) prenatal antidepressant medication intake (n = 4); (4) incomplete or missing depressive symptom data (n = 115). Further dyads had to be excluded after sample quality control (n = 97) and genotype quality control (n = 115). Duplicates (n = 10) were also removed. Exclusions resulted in a confirmation sample of n = 641 mother-child dyads (320 boys, 321 girls). At birth, mothers (45% primiparous) were 29.9 years old (SD = 4.4, range: 18.0-42.0). At DNA extraction, children had a mean age of 7.4 years (SD = 0.1, range: 7.1-8.8). Informed consent for the use of data collected via questionnaires and clinics was obtained from participants following the recommendations of the ALSPAC Ethics and Law Committee at the time. Consent for biological samples has been collected in accordance with the Human Tissue Act from 2004. Ethical approval for this study was obtained from the ARIES/ALSPAC Ethics and Law Committee and the Local Research Ethics Committees (http://www.bristol.ac.uk/alspac/researchers/research-ethics/). Figure 1 summarizes the study design.

Schematic overview of the study design. Different timepoints important for this study: T1 (third trimester), birth, and T2. After quality control n = 174 FRAMES/FRANCES and n = 641 ARIES/ALSPAC samples were available for analysis. Of those mother-child dyads, 24.8% of the mothers showed prenatal depressive symptoms for the ARIES/ALSPAC sample, whereas 29.9% showed prenatal depressive symptoms for the FRAMES/FRANCES sample. For further insight, sample tissue and age (of both mother and child) are given. ARIES/ALSPAC indicates Accessible Resource for Integrated Epigenomic Studies/Avalon Longitudinal Study of Parents and Children; FRAMES/FRANCES, Franconian Maternal Health Evaluation Studies/Franconian Cognition and Emotion Studies.
Measures
Maternal depressive symptoms
Maternal depressive symptoms in the discovery and confirmation cohort were measured using the Edinburgh Postnatal Depression Scale (EPDS), 40 a self-rating scale assessing severity and frequency of depressive symptoms by 10 items with a 4-point Likert-type scale. The scale is validated for the prenatal and postnatal period, 41 with scores ⩾10 being interpreted as present depressive symptoms. In FRANCES, mothers rated their depressive symptoms during the third trimester (t1), 2 days postpartum and when the child was in primary school (t2). Comparable timepoints were used in ARIES/ALSPAC for assessing maternal depressive symptoms: third trimester (32nd gestation week), 8 weeks postpartum, and when child was in primary school.
Further child and family characteristics
Pregnancy characteristics, ie, gestational age (weeks), birth weight (g), and maternal age at birth (years), were registered immediately after delivery. To ensure comparability of both cohorts, children’s phenotypic outcome in terms of psychopathology and intelligence in primary school age was compared. In both cohorts, psychopathology was assessed using the 25-item Strength and Difficulties Questionnaire (SDQ) 42 completed by a parent. Subscales “Emotional Problems” and “Conduct Problems” were used as index for child’s psychopathology with higher scores representing more problems (range: 0-10), respectively. Children’s intelligence quotient (M = 100, SD = 15) was assessed using different methods (FRANCES: Intelligence and Development Scales [IDS] 43 ; ARIES/ALSPAC: Wechsler Intelligence Scale for Children [WISC-III]). 44
DNA methylation
DNA samples were obtained from buccal cells extracted with an OmniSwab (Whatman, Maidstone, UK) cotton stick from the inner cheek. The QIAamp DNA Mini Kit (Qiagen, Hilden, Germany) was used for DNA extraction. Genome-wide DNA methylation was analyzed by the Helmholtz-Zentrum München (Germany) with the Infinium Human Methylation 450K BeadChip (Illumina, San Diego, CA, USA) as described elsewhere. 45 For a more detailed description, please see Stonawski et al. 19 Within the ARIES cohort, DNA samples were extracted from peripheral blood. In accordance with FRANCES, DNA methylation was analyzed with the Infinium Human Methylation 450K BeadChip, with more detailed information described in Relton et al. 37
Epigenetic analyses
To provide methodological comparability between FRANCES and ARIES/ALSPAC, the preprocessing strategy for the methylation data of both cohorts was paralleled. Epigenetic analyses, including quality control and preprocessing, were performed with R (FRANCES: version 3.5.2; ARIES/ALSPAC: version 3.2.0) using the package meffil (version 1.1.0) as described more detailed in Min et al. 46 Quality control parameter thresholds were paralleled between cohorts using the provided ARIES/ALSPAC user guide. Samples had to be excluded after quality control regarding (1) age mismatch (estimated after Horvath), 47 (2) sex mismatches or outliers, (3) ratio of methylated/unmethylated signal, (4) dye bias, (5) detection P value, and (6) low bead numbers. For a detailed description of quality control parameters, see Supplement S2. Together with the excluded samples given by the study design exclusion criteria described earlier, this resulted in n = 174 samples for the FRANCES discovery sample and n = 641 samples for the ARIES/ALSPAC confirmation sample.
Samples were normalized using functional normalization (FN) 48 implemented in meffil. The optimal number of principal components used for normalization was estimated by scree plots of the control matrix provided by meffil (FRANCES: nPCs = 15 and ARIES/ALSPAC: nPCs = 10). In addition, bisulfite-conversion plate (“plate”) and beadchip (“slide”) were included as batch variables, as well as row and column of the microarray and, in case of non sex-specific analyses, sex of the participants. Estimated methylation level of each CpG was assessed using β-values representing the ratio of methylated signal and overall signal. 49 Adjustment after linear regression was performed with Independent Surrogate Variable Analysis (ISVA). 50 As single-nucleotide polymorphisms (SNPs) at a target CpG site yield the possibility of confounding the measurement of the methylation level by increasing within-tissue variation, SNPs containing CpG sites were removed before performing FN. To assess which CpGs are associated with an SNP, the extended Illumina 450k annotation by Price et al 51 was used (Gene Expression Omnibus [GEO] accession number: GSE40279), resulting in a removal of 4.3% of all CpG probes. The ARIES/ALSPAC samples have been normalized for cell-type composition using the Houseman et al 52 method to a reference data set from Reinius et al 53 (GEO accession number: GSE35069). No adjustment for cell-type composition has been applied for the FRANCES sample, due to missing reference data sets for buccal cells. Normalization reports were generated with meffil as described by Min et al, 46 evaluating associations between potential batch variables with control probes as well as normalized data. This approach assists in the identification of batches and technical artifacts which are not resolved using FN and might therefore be problematic.
Statistical analyses and covariates
Epigenome-wide analyses of differentially methylated CpGs associated with exposure to prenatal depressive symptoms were run across normalized beta values using the EWAS pipeline implemented in meffil. To control other factors potentially influencing methylation, the following covariates were included: gestational age, birth weight, EPDS postpartum, current EPDS, age (children at t2 and mother at time of birth), and—in case of nonsex-specific analyses—sex. Prenatal, postpartum, and current EPDS were used as categorical variable with the described threshold. For the investigation of sex-specific effects, separate EWAS analyses were subsequently performed for boys and girls. To correct for multiple testing, P values were adjusted by false discovery rate (FDR) procedure. 54 Adjusted P values were labeled as q values, with q < 5% indicating a significant finding. For assessment of replication, a threshold of P < 10−3 was set as equilibrium between correcting for multiple testing and comparing the samples in an exploratory fashion. A CpG has been defined as replicated, if it showed a P value of P < 10−3 in both samples and the directions of the observed effect (hyper/hypomethylation) have been congruent.
Up to now, other working groups identified 272 differentially methylated CpGs associated with prenatal depression: Non et al 26 : 42 CpGs, q < .01; Nemoda et al 27 : 145 CpGs, q < .05; Viuff et al 28 : 68 CpGs, q < .05. Using an exploratory procedure to identify overlaps between former studies and the current one, all CpGs which were found to be P < .05 (not corrected for multiple testing) in both FRANCES and ARIES/ALSPAC sample were compared with the previous published findings.
Analysis of DMRs was performed using the R package seqlm (version 0.1.0) 55 using standard parameters. Functional gene clustering was performed with an online tool provided by the Database for Annotation, Visualization and Integrated Discovery (DAVID, version 6.8).56,57 The CpGs with a P value of <10−3 have been considered for analysis, identifying annotated genes using the Illumina HumanMethylation450 v1.2 Manifest File (as implemented in meffil). The classification stringency used for clustering was set to medium (as recommended) and contained the following parameters: similarity overlap = 3, similarity threshold = 0.5, initial group membership = 3, and multiple linkage threshold = 0.5. To focus on potentially biological meaningful clusters, only clusters with an enrichment score >1.50 have been taken into account.
Results
Sample characteristics
Comparing the FRANCES sample with the ARIES/ALSPAC sample, no differences were found for child sex—χ2(1) = 0.02, P = .88, ϕ = .01. At t2, children of the FRANCES sample were older than ARIES/ALSPAC children—FRANCES: M = 7.60, SD = 0.60; ARIES/ALSPAC: M = 7.40, SD = 0.10; t(177.1) = 3.1, P = .003, d = 0.69. Mothers in the FRANCES sample were about 3 years older at birth compared with those of the ARIES/ALSPAC sample—FRANCES: M = 32.70, SD = 4.60; ARIES/ALSPAC: M = 29.90, SD = 4.40; t(813) = 7.50, P < .001, d = 0.63. Mothers did not differ in depressive symptoms in pregnancy, t(255.1) = 1.1, P = .27, d = 0.11, or at t2, t(813) = 1.3, P = .20, d = 0.10. At postpartum, mothers in the FRANCES sample reported fewer depressive symptoms than mothers in the ARIES/ALSPAC sample, FRANCES: M = 4.40, SD = 4.70; ARIES/ALSPAC: M = 5.60, SD = 4.40; t(813) = 3.2, P = .002, d = 0.27, probably due to the discrepant assessment points (FRANCES 2 days postpartum vs ARIES/ALSPAC 8 weeks postpartum). Regarding pregnancy characteristics, in the FRANCES sample, pregnancy was shorter than in the ARIES/ALSPAC sample—FRANCES: M = 39.30, SD = 1.50; ARIES/ALSPAC: M = 39.60, SD = 1.50; t(813) = 2.13, P = .03, d = 0.20. There were no differences in birth weight, t(813) = 0.78, P = .44, d = 0.07. Relevant sample characteristics of mothers and children are summarized in Table 1. Further sample characteristics, which were not part of the following analyses, can be found in Table S1 in the supplement.
Sample characteristics and group differences between exposed and nonexposed children/mothers in the FRANCES and ARIES/ALSPAC sample.

Sample size varied due to missing values in FRANCES between n = 170 and n = 174 and in ARIES/ALSPAC between n = 608 and n = 641. Continuous variables are expressed as mean (SD) and tested with independent t tests (df varied according to different sample sizes between df = 170 and df = 639), with d corrected for unequal sample sizes 58 as measure of effect size. The t scores are displayed as absolute values. Categorical variables are expressed as No. (%) and tested with chi-square tests (df varies according to the number of categories of the tested variable between df = 1 and df = 3), with ϕ coefficient used as measure of effect size.
Prenatal EPDS score <10 interpreted as nonexposed, ⩾10 as exposed.
At time of DNA sampling.
df adjusted for unequal variances based on Levene.
P < .05. **P < .01.
EWAS analysis of the FRANCES sample showed no differentially methylated CpG based on exposure to prenatal depressive symptoms
Based on the exposure to prenatal depressive symptoms, a comparison of both groups (exposed to prenatal depressive symptoms vs no exposition) in the FRANCES sample was performed using an EWAS with an ISVA model. 50 There were 46 833 CpGs that yielded significance under P < .05 for the whole sample, 41 363 for boys and 52 560 for girls. Correction for multiple testing via Benjamini-Hochberg procedure 54 yielded no significant differentially methylated CpG. This holds true for the whole sample (all q values >.99) as well as for sex-specific analyses (boys: q values ranging from q = .85 to q = .99; girls: all q values >.99). The QQ plots for the given EWAS models are shown in Figure 2; the Manhattan plots could be found in Supplementary Table S3. The 10 lowest observed P values for the EWAS are given in Table 2. Results of the same analysis for the ARIES/ALSPAC sample are presented in Supplementary Table S4.

QQ plots (ISVA) for the FRANCES sample. (A) Whole sample and (B, C) sex-specific analyses. FRANCES indicates Franconian Cognition and Emotion Studies; ISVA, Independent Surrogate Variable Analysis.
The CpGs with the lowest observed P value for the FRANCES cohort’s subsample: the whole sample and separated by sex.

Overlapping CpGs between subsets are highlighted in bold.
Abbreviations: CpGs, cytosine-guanine dinucleotides; FRANCES, Franconian Cognition and Emotion Studies.
Comparison of both EWAS results (FRANCES vs ARIES/ALSPAC) revealed one accordant CpG based on exposure to prenatal depressive symptoms
Using a threshold of P < 10−3 for the whole sample, 227 CpGs have been found for FRANCES with P values ranging from P = 2.67 × 10−6 to P = 9.99 × 10−4 and 357 CpGs for ARIES/ALSPAC, with P values ranging from P = 4.24 × 10−6 to P = 9.98 × 10−4. For the boys, 301 CpGs could be found for the FRANCES sample with P values ranging from P = 6.52 × 10−6 to P = 9.98 × 10−4, and 338 CpGs for the ARIES/ALSPAC sample with P values ranging from P = 2.73 × 10−6 to P = 9.99 × 10−4. In the group of girls, 288 CpGs could be found for FRANCES with P values ranging from P = 1.39 × 10−5 to P = 9.96 × 10−4 and 434 CpGs for ARIES/ALSPAC with P values between P = 5.27 × 10−8 and P = 9.98 × 10−4.
Analyzing overlapping CpGs for all subsets, no overlaps were found for the whole subsamples and girls. However, for boys, 1 CpG could be found in both samples (cg08741921, FRANCES: P = 5.16 × 10−4, ARIES/ALSPAC: P = 5.56 × 10−4, no associated gene), which therefore represents the only CpG meeting the beforehand set definition of replication.
DMR analysis of the FRANCES sample could not identify any significant DMR
No significant DMRs could be found using the given samples and exposition groups. This is true for the whole sample as well as for the sex-specific analyses. The 5 segments with the lowest FDR correction found via seqlm 55 and their associated genes are shown in Table 3. The results for the same analysis for the ARIES/ALSPAC sample are shown in Supplement S5. No overlapping segments or associated genes could have been identified between the FRANCES and the ARIES/ALSPAC cohorts’ subsamples.
The 5 segments with the lowest FDR as given by seqlm, including chromosomal location and UCSC RefSeq Gene Name, for the whole FRANCES sample as well as for boys and girls separately.

Abbreviations: FDR, false discovery rate; FRANCES, Franconian Cognition and Emotion Studies.
Comparison of the present primary school age results with previous EWAS results from newborns identified 4 common CpGs being associated with depressive symptoms
We compared the differentially methylated CpGs obtained from FRANCES and ARIES/ALSPAC with former published EWAS results regarding prenatal exposure to maternal depressive symptoms. Using an exploratory threshold of P < .05, 4 overlapping CpGs were identified between FRANCES, ARIES/ALSPAC, and former results (Nemoda et al 27 : 3 CpGs; Viuff et al 28 : 1 CpG) as shown in Table 4.
Comparison of FRANCES and ARIES/ALSPAC EWAS results with differentially methylated CpGs obtained in former studies: overlapping CpGs with P < 0.05 in both samples.

Abbreviations: ARIES/ALSPAC, Accessible Resource for Integrated Epigenomic Studies/Avalon Longitudinal Study of Parents and Children; CpGs, cytosine-guanine dinucleotides; EWAS, epigenome-wide association study; FRANCES, Franconian Cognition and Emotion Studies.
Pathway analysis of FRANCES and ARIES/ALSPAC
DAVID functional clustering was used to check for overrepresentation of pathways among differentially methylated CpGs with P < 10−3. There were 158 unique genes associated with the FRANCES subsample which gave 22 clusters using DAVID standard settings. No cluster remained when applying the cut-off enrichment score >1.50. A total of 262 unique genes associated with the ARIES/ALSPAC sample yielded 49 clusters—after filtering for enrichment score, one cluster remained, which was annotated with protein phosphorylation, signal transduction, and cell-cell junction (enrichment score: 2.02).
In sex-specific analyses, 209 unique genes associated with the male subset of the FRANCES cohort yielded 38 clusters, with one cluster showing an enrichment score >1.50 (annotated terms: nucleotide binding/ATP binding, enrichment score: 1.96). There were 227 genes associated with the ARIES/ALSPAC boys aggregated into 30 clusters, with 2 of them showing an enrichment score >1.50: (1) cell junction/postsynaptic processes (enrichment score: 1.79) and (2) membrane and transmembrane components (enrichment score: 1.70). For girls, 198 unique genes in the FRANCES sample gave 32 clusters, with no cluster being enriched. Out of 302 unique genes from the girls of the ARIES/ALSPAC sample, 47 clusters could be formed, with 3 of them reaching the beforehand defined threshold of enrichment: (1) Pleckstrin homology-like domain (enrichment score: 2.20), (2) membrane-attached proteins (enrichment score: 1.67), and (3) adrenergic signaling/insulin secretion/aldosterone synthesis/estrogen pathway/digestion (enrichment score: 1.53). As the CpGs of FRANCES and ARIES/ALSPAC did show only one overlap in all CpGs with P < 10−3, a clustering analysis for those overlapping CpGs was not conducted. A summary of the found clusters with their associated genes can be found in Table 5. A more detailed description of each functional term within the cluster can be found in Supplementary Table S6.
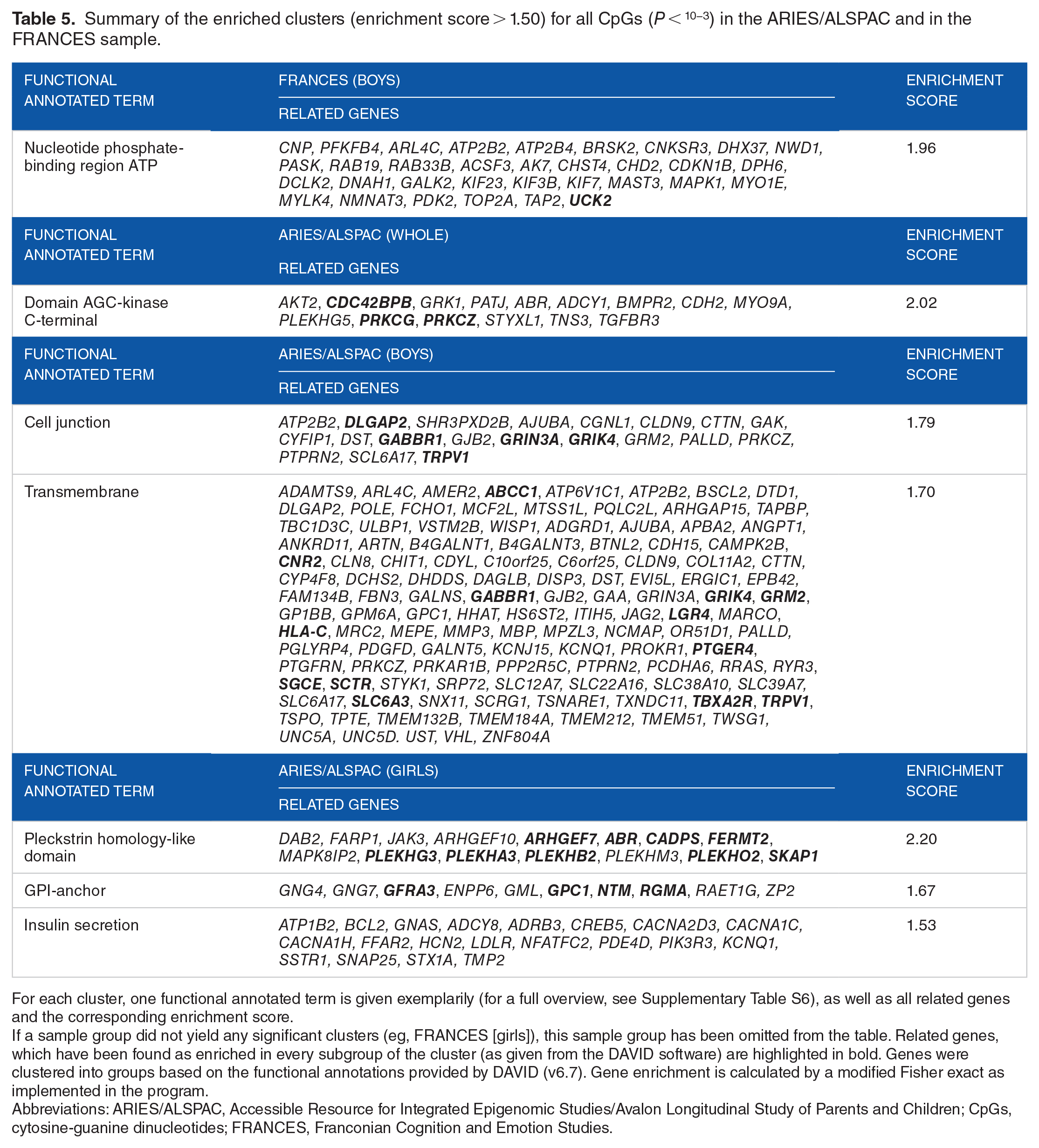
For each cluster, one functional annotated term is given exemplarily (for a full overview, see Supplementary Table S6), as well as all related genes and the corresponding enrichment score.
If a sample group did not yield any significant clusters (eg, FRANCES [girls]), this sample group has been omitted from the table. Related genes, which have been found as enriched in every subgroup of the cluster (as given from the DAVID software) are highlighted in bold. Genes were clustered into groups based on the functional annotations provided by DAVID (v6.7). Gene enrichment is calculated by a modified Fisher exact as implemented in the program.
Abbreviations: ARIES/ALSPAC, Accessible Resource for Integrated Epigenomic Studies/Avalon Longitudinal Study of Parents and Children; CpGs, cytosine-guanine dinucleotides; FRANCES, Franconian Cognition and Emotion Studies.
Discussion
This study was performed to identify and validate CpGs and genes implicated in DNA methylation modifications in school-aged children associated with prenatal exposure to maternal depressive symptoms by investigating 2 independent cohorts from Germany and Great Britain. In our discovery sample FRANCES, we could not identify any differentially methylated CpG withstanding correction for multiple testing—in the whole sample or in sex-specific analyses. A comparison of both samples, FRANCES and ARIES/ALSPAC, using all CpGs with P < 10−3, showed only one overlapping CpG for boys. However, this CpG did not survive FDR correction. Looking at the data on a lower level of resolution, analyses of DMRs also did not yield significant findings.
In summary, analyses of single CpGs as well as regional analyses regarding subsamples from 2 independent cohorts with comparable child age did not result in significant or consistent findings of modifications in DNA methylation associated with prenatal depressive symptoms. This is in line with former studies which were also not able to identify modifications in child DNA methylation associated with maternal depressive symptoms throughout pregnancy 25 or specifically in late pregnancy as investigated in this study. 28 Regarding earlier findings from studies using a candidate gene approach, many of the reported findings would also not withstand corrections for multiple testing, 16 which might challenge the assumption of existing effects on child DNA methylation in general. On the basis of the current results, one may argue that prenatal exposure to maternal depressive symptoms is not associated with DNA methylation in childhood. Alternatively, modifications might be too small for identification and replication in the current samples. Small effects and effect sizes are discussed as a common finding in environmental epigenetic studies resulting in debates regarding their biological relevance. 59 However, there are existing findings showing functional consequences of small methylation changes. 59 It is hypothesized that psychological phenomena such as depressive symptoms or subjective stress, which were investigated here, might result in smaller DNA modifications than other prenatal adversities, eg, smoking or dietary habits, which would more severely affect the fetus. 12
Evaluating the stability of previously described DNA modifications from birth to childhood by comparing the current results with former study findings resulted in 4 overlapping CpGs with a P value of <.05 in both FRANCES and ARIES/ALSPAC. One overlapping CpG (cg07051728, SULF1) was reported by Viuff et al 28 and has not been described in the context of prenatal depression in any other study. The other 3 CpGs were also reported by Nemoda et al 27 ; however, they used an exploratory approach without a correction for multiple testing. Of these 3 CpGs, 1 CpG was found for the whole sample (cg17889623, KIF21B), whereas the other 2 were only found for the subset of girls (cg01431057, S100A8; cg15086439, EDARADD).
Referring to the enrichment analyses in Nemoda et al, 27 S100A8 is associated with leukocyte quantity, migration, and necrosis. Leukocytes are a prominent nonspecific inflammatory marker, and their associations with psychiatric disorders have been extensively studied in the past (eg, depressive symptoms).60,61 Furthermore, S100A8 was found to be expressed differently in prefrontal cortices of mice with depression-like behavior after chronic unpredictable stress 62 as well as in postmortem brains of individuals with schizophrenia. 63 These findings support S100A8 as a potential candidate for further research. KIF21B has been recently described as involved in methylation mechanisms in other fields of research (ie, tumor research 64 or toxicological mechanisms), 65 but it has not yet been described in the context of depression in humans. However, Morikawa et al 66 found KIF21B-knockout mice lacking in fear extinction, which might suggest some kind of involvement of KIF21B in neurocognitive mechanims. Similarily, EDARADD has not yet been described as associated with depressive symptoms; however, it has been reported as a marker of age in pediatric populations, with methylation levels declining over the course of childhood and youth. 67 As FRANCES and ARIES/ALSPAC are not perfectly age-homogeneous, the methylation differences for this CpG/gene might be interpreted as an artifact of age divergence.
The identified overlapping CpGs between this study and findings from Nemoda et al 27 suggest there may be some stable modifications; however, most of the findings from newborn or infant age26-28 have not been seen in childhood. Low agreement between results might be explained by low stability of methylation modifications during development, as a recent study found significant epigenetic remodeling processes taking place during the first 5 years of life. 68 Longitudinal changes from birth to the age of 2 years were also reported for DNA methylation of genes implicated in important biological functions including immunity and inflammation. 69 Supporting this, in an investigation of the stability of DNA methylation modifications associated with attention-deficit hyperactivity disorder trajectories in the ARIES/ALSPAC cohort, modifications at birth were not seen at 7 years of age. 70 In contrast, Ladd-Acosta et al 71 reported a high concordance between modifications of DNA methylation associated with prenatal smoking in infancy and childhood. Thus, there is still need for longitudinal studies examining the occurrence and maintenance of DNA methylation after birth to validate findings of DNA modifications in children exposed to prenatal risks, especially mental health adversities.
A lack of consistent epigenetic findings has been described by Ryan et al 31 for prenatal mental well-being and they suggest these discrepancies could be due to methodological differences, small underpowered cohorts, divergent covariates, or the time frames investigated. As the methylation effects due to psychological phenomena are estimated to be small, divergent settings in analyses could already change results distinctly. So, besides instability of DNA methylation modifications mentioned earlier, methodological differences regarding epigenetic analyses as well as assessment of prenatal depression might contribute to the diverging results between the present and former studies. While differences between the FRANCES and ARIES/ALSPAC cohort have been addressed via study design, it has to be kept in mind that DNA was extracted from different tissues for both cohorts (buccal cells in FRANCES vs whole blood samples in ARIES/ALSPAC). Nonreplication might be explained by tissue specificity of DNA methylation. 72 It has been reported that—due to their matching ectodermal origin in embryonal development—buccal cells might yield better representability of brain mechanisms, but methylation results from buccal cells might not be comparable with whole blood samples (ie, leukocytes are of mesodermal origin). 73 Further-more, taking into account that ARIES/ALSPAC was started in the early 1990s and FRANCES about 15 years later, there might be cohort effects with different behavioral patterns or dietary habits in pregnancy which might have resulted in different DNA methylation patterns.74,75 So, more research is needed, eg, in the form of large consortia, using a longitudinal, prospective design with comparable methodology to clarify these issues.
Regarding the confirmation sample, ARIES/ALSPAC (n = 641), one differentially methylated CpG for girls could be identified (cg11251378, PLEKHA3, MIR548N, P = 5.27 × 10−8, q = .02). However, this CpG did not show a significant differential methylation in the discovery sample FRANCES (P = .676, q = .99), so it should be interpreted cautiously. This identified CpG of the PLEKHA3 gene has been found to be associated with state-dependent and region-specific transcriptome changes in a rodent model of depression and antidepressant effects; however, these effects did not hold up after corrections for multiple testing. 76 MIR548N has been identified as differentially methylated in a twin study investigating early-onset major depression and its associations with DNA methylation. 77 In addition, a study by Kim et al 78 investigating somatic deletions in a population of patients with schizophrenia and unaffected controls found that MIR548N activity interfered with the prefrontal cortex cells of patients with schizophrenia. This might imply some unspecific function of MIR548N in cellular diversity in psychiatric populations which should be monitored in future research.
Besides analyzing individual sites and regions, functional relevance of the modifications was investigated by pathway analyses. No matching enriched cluster was found between samples, which leads to 2 possible conclusions: concordant functional sections in both samples do not show enough enrichment in this sample or there are different epigenetic mechanisms at hand. Looking at the analyzed functional annotated terms per sample, there was one cluster enriched for the FRANCES sample with one gene appearing in every functional annotated subpart of this cluster—UCK2. While this gene has been shown to play a role in, eg, cancer mechanisms, 79 it is yet to be reported in the context of depressive symptoms. In addition, the most enriched cluster for the girls subsample in the ARIES/ALSPAC sample is associated with the previously mentioned significant differentially methylated PLEKHA3 gene, which further supports the conclusion that these functional related genes (and their associated CpGs) are strong candidates for further research. This seems especially true when looking at the results for the other genes reported for this cluster (see Table 5). Nearly all genes out of this list have been previously reported to play a part in mechanisms of cognitive/psychiatric disorders. An overview of these potential candidate genes is given in Table 6. However, it is important to note that these candidate genes can only be investigated for girls, as none of the genes/pathways have been found to be differentially methylated or enriched for the subsamples of boys.
Overview of potential candidate genes for girls derived from EWAS/pathway analyses.

Given are the gene symbols as well as the associated cognitive/psychiatric disorders and the researched species.
Abbreviation: EWAS, epigenome-wide association study.
Last but not least, in this study, epigenome-wide analyses were performed for the first time separately for boys and girls to evaluate sex-specific methylation modifications. Several studies have investigated prenatal environmental risk factors, eg, stress or depression, on child development; these studies have reported sex-specific effects on both phenotypic outcome, eg, psychopathology or neurobiological measures,85,86 and DNA methylation.19,22,32 Variations in placental DNA methylation have been discussed as a mechanism which leads to altered placental functioning between male and female fetuses.87,88 In addition, sex-specific alterations of infant DNA methylation might result in diverging patterns of gene expression in response to the in utero environment. In this study, overlapping methylation sites were identified in boys and girls; however, no CpG or region reached significance. Despite nonsignificance, in FRANCES, modified CpGs in boys reached larger effects than in girls, which is in adherence to former results.19,22 It is hypothesized that boys may be more affected by prenatal adversities than girls, partially due to diverging brain development between the sexes. 89 In contrast, 3 of the 4 CpGs found in both this study (primary school–aged children) and the former studies (newborns/infants) were identified only in girls at primary school age and only in the ARIES/ALSPAC results. The only differentially methylated CpG was also found only in girls. This is in line with other studies hypothesizing stronger effects of prenatal stimuli in girls.32,33,90 In summary, despite having mostly nonsignificant findings and not being able to answer whether boys or girls are more strongly affected by prenatal exposure to depression, current results underline the hypothesis that there are sex-specific methylation patterns. This finding may help to expand the understanding of mechanisms underlying sex differences, eg, in later psychopathology. However, this is the first EWAS addressing sex-specific methylation; to best evaluate these findings, much more research is needed regarding general sex-specific methylation and specifically how it is associated with prenatal depressive symptoms.
Limitations
Age has been reported to have a possible impact on DNA methylation in samples of children.67,68 In FRANCES, children exposed to prenatal depressive symptoms were significantly older than nonexposed children, which can be attributed to an extended contacting process with the affected families. Furthermore, children in the FRANCES sample were slightly younger than children of the ARIES/ALSPAC sample at DNA extraction (M = 7.4 vs M = 7.6 years). Child age was statistically controlled for during the preprocessing of DNA methylation and has been added as a potential covariate for further analyses; however, an impact of age cannot be definitively ruled out.
Regarding assessment of maternal depressive symptoms, an EPDS score of 10 or higher in the third trimester was interpreted as exposure to prenatal depressive symptoms. This threshold is used in several studies but indicates only a potential minor clinical depression. Besides the self-rating questionnaire, no other objective measure for validation was used and mothers exposed to pharmacologically treatment have been explicitly excluded from further analysis. Both of these points could potentially be involved in the methylation patterns we reported. In addition, postnatal and current depressive symptoms have been added as a covariate in all analyses which included prenatal depressive symptoms. However, an influence of postnatal or current depression cannot be ruled out definitely. Furthermore, depressive symptoms were assessed just once in pregnancy; analyses regarding the prenatal timing of risk influence were not possible. Moreover, while controlling for maternal psychopathology, this study did not consider other postnatal factors which should be included in future longitudinal studies. Controlling for critical life events during childhood is essential, especially as there is evidence for an effect of childhood adversity on DNA methylation. 91 In addition, later parent mood disorders, mother-child interaction style, and child maltreatment might mediate effects of prenatal depressive symptoms on methylation.
Regarding epigenetic analyses, there are some further limitations. It is known that DNA methylation changes, especially in the gene promoter region, can influence gene expression; however, not all promoter regions of every gene are included on the 450k BeadChip. Therefore, investigation of important gene regulation sites might not have been possible for all candidate or potentially modified genes. Modifications in DNA methylation are assumed to result in altered gene expression; however, gene expression data, which would be necessary for validating this assumption, were not available. Furthermore, there was no analysis regarding the association of differently methylated CpGs with target variables (eg. developmental, behavioral, and emotional or neurobiological markers) to identify their potential functional relevance. In addition, tissue for DNA extraction differed between both cohorts. Due to tissue specificity of DNA methylation, 72 conclusions from peripheral tissues upon brain processes and functioning as well as comparisons between both samples should be drawn cautiously. This is especially true, as no cell-type adjustment for the FRANCES sample has been done, which could alter the comparability between both samples even further. Furthermore, there are other findings which include novel biomarkers into the analysis of prenatal depressive symptoms and epigenome-wide changes, eg, epigenetic gestational age. 92 Incorporation of such biomarkers might be valuable for further research, as well as assessing cell types which can be present in both blood and buccal cells. Last but not least, this study focused on modifications in DNA methylation, but did not take into account other potential methylation relevant mechanisms. For example, histone modifications have been shown to interact with DNA methylation. 93 Including histone modification data into further analysis could enrich the interpretation possibilities. Furthermore, interaction effects regarding DNA methylation have been reported for genetic and environmental factors, 94 which were not considered in the current analyses. Including these aspects would strengthen future research.
Conclusions
This study could not identify modifications in child DNA methylation in primary school age related to intrauterine exposure to prenatal depressive symptoms. However, results suggest some stable modifications in methylation from newborn and infant age to childhood. Furthermore, the current findings support the hypothesis that there are sex-specific methylation processes. More large, prospective longitudinal studies are needed to clarify whether altered DNA methylation in children is associated with prenatal depressive symptoms while assessing for sex-specific differences. In addition, integrating genetic information, epigenetic mechanisms, and phenotypic data to evaluate the functional relevance of genetic changes would help to expand the current understanding of prenatal influences on postnatal outcomes.
Supplemental Material
Supplement_S1_Revised_xyz35773a9d23438 – Supplemental material for Genome-Wide DNA Methylation Patterns in Children Exposed to Nonpharmacologically Treated Prenatal Depressive Symptoms: Results From 2 Independent Cohorts
Supplemental material, Supplement_S1_Revised_xyz35773a9d23438 for Genome-Wide DNA Methylation Patterns in Children Exposed to Nonpharmacologically Treated Prenatal Depressive Symptoms: Results From 2 Independent Cohorts by Valeska Stonawski, Jakob Roetner, Tamme W Goecke, Peter A Fasching, Matthias W Beckmann, Johannes Kornhuber, Oliver Kratz, Gunther H Moll, Anna Eichler, Hartmut Heinrich and Stefan Frey in Epigenetics Insights
Supplemental Material
Supplement_S2_xyz340136898738d – Supplemental material for Genome-Wide DNA Methylation Patterns in Children Exposed to Nonpharmacologically Treated Prenatal Depressive Symptoms: Results From 2 Independent Cohorts
Supplemental material, Supplement_S2_xyz340136898738d for Genome-Wide DNA Methylation Patterns in Children Exposed to Nonpharmacologically Treated Prenatal Depressive Symptoms: Results From 2 Independent Cohorts by Valeska Stonawski, Jakob Roetner, Tamme W Goecke, Peter A Fasching, Matthias W Beckmann, Johannes Kornhuber, Oliver Kratz, Gunther H Moll, Anna Eichler, Hartmut Heinrich and Stefan Frey in Epigenetics Insights
Supplemental Material
Supplement_S3_xyz34013e5c2fea3 – Supplemental material for Genome-Wide DNA Methylation Patterns in Children Exposed to Nonpharmacologically Treated Prenatal Depressive Symptoms: Results From 2 Independent Cohorts
Supplemental material, Supplement_S3_xyz34013e5c2fea3 for Genome-Wide DNA Methylation Patterns in Children Exposed to Nonpharmacologically Treated Prenatal Depressive Symptoms: Results From 2 Independent Cohorts by Valeska Stonawski, Jakob Roetner, Tamme W Goecke, Peter A Fasching, Matthias W Beckmann, Johannes Kornhuber, Oliver Kratz, Gunther H Moll, Anna Eichler, Hartmut Heinrich and Stefan Frey in Epigenetics Insights
Supplemental Material
Supplement_S4_xyz34013d8a10197 – Supplemental material for Genome-Wide DNA Methylation Patterns in Children Exposed to Nonpharmacologically Treated Prenatal Depressive Symptoms: Results From 2 Independent Cohorts
Supplemental material, Supplement_S4_xyz34013d8a10197 for Genome-Wide DNA Methylation Patterns in Children Exposed to Nonpharmacologically Treated Prenatal Depressive Symptoms: Results From 2 Independent Cohorts by Valeska Stonawski, Jakob Roetner, Tamme W Goecke, Peter A Fasching, Matthias W Beckmann, Johannes Kornhuber, Oliver Kratz, Gunther H Moll, Anna Eichler, Hartmut Heinrich and Stefan Frey in Epigenetics Insights
Supplemental Material
Supplement_S5_xyz34013cb77c00b – Supplemental material for Genome-Wide DNA Methylation Patterns in Children Exposed to Nonpharmacologically Treated Prenatal Depressive Symptoms: Results From 2 Independent Cohorts
Supplemental material, Supplement_S5_xyz34013cb77c00b for Genome-Wide DNA Methylation Patterns in Children Exposed to Nonpharmacologically Treated Prenatal Depressive Symptoms: Results From 2 Independent Cohorts by Valeska Stonawski, Jakob Roetner, Tamme W Goecke, Peter A Fasching, Matthias W Beckmann, Johannes Kornhuber, Oliver Kratz, Gunther H Moll, Anna Eichler, Hartmut Heinrich and Stefan Frey in Epigenetics Insights
Supplemental Material
Supplement_S6_xyz3401382b311ea_1 – Supplemental material for Genome-Wide DNA Methylation Patterns in Children Exposed to Nonpharmacologically Treated Prenatal Depressive Symptoms: Results From 2 Independent Cohorts
Supplemental material, Supplement_S6_xyz3401382b311ea_1 for Genome-Wide DNA Methylation Patterns in Children Exposed to Nonpharmacologically Treated Prenatal Depressive Symptoms: Results From 2 Independent Cohorts by Valeska Stonawski, Jakob Roetner, Tamme W Goecke, Peter A Fasching, Matthias W Beckmann, Johannes Kornhuber, Oliver Kratz, Gunther H Moll, Anna Eichler, Hartmut Heinrich and Stefan Frey in Epigenetics Insights
Footnotes
Acknowledgements
The authors thank all families, who participated in FRAMES-FRANCES, as well as all colleagues and student assistants, who contributed to this study. Moreover, we are extremely grateful to all the families who took part in the ARIES/ALSPAC study, the midwives for their help in recruiting them, and the whole ARIES/ALSPAC team, which includes interviewers, computer and laboratory technicians, clerical workers, research scientists, volunteers, managers, receptionists, and nurses. In addition, the authors thank Dr Matthew Suderman (University of Bristol) for his technical support and general help with interpretation of our epigenetic findings. The present work was performed in partial fulfillment of the requirements for obtaining the degree Dr. rer. biol. hum. for Jakob Roetner.
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This project was specifically funded by the Universitätsbund Erlangen-Nürnberg e.V., Germany (grant to A.E.). The Staedtler-Stifung (Nürnberg, Germany) covered the costs for the DNA methylation analysis of FRANCES data (grant to G.H.M.). The U.K. Medical Research Council and Wellcome (grant ref. 102215/2/13/2) and the University of Bristol provide core support for ALSPAC. Methylation data of the children gathered in ARIES have been funded through various sources: The BBSRC (grant ref. BBI025751/1 and BB/I025263/1), funding for the MRC Integrative Epidemiology Unit (MC_UU_12013/1, MC_UU_12013/2, and MC_UU_12013/8), through the National Institute of Child and Human Developmental Grant (grant ref. R01HD068437), the NIH (5RO1AI121226-02), and through the CONTAMED EU collaborative Project (grant ref. 212502). This publication is the work of the authors and Jakob Roetner will serve as guarantor for the contents of this paper.
Declaration of Conflicting Interests:
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Hartmut Heinrich works at the neuroCare Group, Munich, and is Research Fellow at the Research Institute Brainclinics, Nijmegen.
Author Contributions
VS, JR, HH and SF analyzed the data and/or interpreted the results. TG, PF, MB and JK initiated and designed the FRAMES project. TG and PF supervised the clinical data acquisition in FRAMES. OK, GM, AE and HH were responsible for the study design and the data acquisition of FRANCES. VS, JR and SF wrote the manuscript. All authors reviewed the manuscript.
Access to Underlying Research Material
If you want to access underlying research material (eg, data, samples, or models) please contact ![]()
Supplemental Material
Supplementary material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.