
Abstract
Background:
How epigenetic modifications of DNA are associated with gestational age at birth is not fully understood. We investigated potential effects of differential paternal DNA methylation (DNAm) on offspring gestational age at birth by conducting an epigenome-wide search for cytosine-phosphate-guanine (CpG) sites.
Methods:
Study participants in this study consist of male cohort members or partners of the F1-generation of the Isle of Wight Birth Cohort (IoWBC). DNAm levels in peripheral blood from F1-fathers (n = 92) collected around pregnancy of their spouses were analyzed using the Illumina 450K array. A 5-step statistical analysis was performed. First, a training-testing screening approach was applied to select CpG sites that are potentially associated with gestational age at birth. Second, functional enrichment analysis was employed to identify biological processes. Third, by centralizing on biologically informative genes, Cox proportional hazards models were used to assess the hazard ratios of individual paternal CpGs on gestational age adjusting for confounders. Fourth, to assess the validity of our results, we compared our CpG-gestational age correlations within a Born into Life Study in Sweden (n = 15). Finally, we investigated the correlation between the detected CpGs and differential gene expression in F2 cord blood in the IoWBC.
Results:
Analysis of DNAm of fathers collected around their partner’s pregnancy identified 216 CpG sites significantly associated with gestational age at birth. Functional enrichment pathways analyses of the annotated genes revealed 2 biological pathways significantly related to cell-cell membrane adhesion molecules. Differential methylation of 9 cell membrane adhesion pathway-related CpGs were significantly associated with gestational age at birth after adjustment for confounders. The replication sample showed correlation coefficients of 2 pathway-related CpGs with gestational age at birth within 95% confidence intervals of correlation coefficients in IoWBC. Finally, CpG sites of protocadherin (PCDH) gene clusters were associated with gene expression of PCDH in F2 cord blood.
Conclusions:
Our findings suggest that differential paternal DNAm may affect gestational age at birth through cell-cell membrane adhesion molecules. The results are novel but require future replication in a larger cohort.
Background
Several studies have reported that preterm birth (<37 weeks of gestational age) is a leading cause of infant mortality and morbidity.1-5 In the United States, the proportion of preterm children in 2016 was 9.85%. 6 Identified risk factors related to gestational age at preterm birth only explain part of its variability. Attempts to explain the nongenetic factors that contribute to gestational age at birth have focused on prenatal maternal exposures to environmental risk factors, such as maternal smoking, 7 intrauterine infection, 8 neighborhood poverty,9,10 ambient air pollution exposure, 11 maternal education, 12 availability of healthy food to the mother, 13 perceived maternal stress 14 maternal age, 15 maternal obesity,16,17 maternal diabetes, 18 maternal hypertension, 19 maternal asthma, 20 and pre-eclampsia. 21 Paternal factors are less well investigated. 22
Despite insight into genetic and epigenetic factors that may contribute to gestational age at birth,23,24 the spotlight is on the contribution from maternal DNA methylation (DNAm) 25 and infants’ DNAm. 26 Findings of paternal genetics and epigenetics explaining gestational age at birth are rare and controversial.27,28 A study on twins’ offspring in the Netherlands suggested that there is a correlation between the gestational age of the first-born offspring whose mothers were monozygotic female twins and non-twin sisters. No correlation was found in gestational age at birth among offspring whose parents were monozygotic male twins, non-twin brothers, or brother-sister. This study also reported that maternal genetic factors contributed only 34% of heritability of gestational age at birth. No paternal heritability was found for gestational age at birth. 27 However, novel findings provided some clues that differential paternal epigenetics may contribute to preterm deliveries22,29 by affecting placental gene expression. Wang et al 28 in a horse-donkey model with hybrid offspring found that paternally expressed genes predominate in the development of placenta. The reciprocal hybrid offspring of horse and donkey, the mule has a donkey as father and horse as mother, and the hinny which has a donkey as mother and horse as father, showed physiological variations in placentas that might be attributable to imprinted genes. Imprinted genes expressed in a parent-of-origin-specific pattern were regulated by epigenetic modification which completely or partly silenced either the paternal or maternal origin allele. 30 They identified multiple nucleotide sites that were only different between horse and donkey, but not polymorphic in both species. Horse-origin allele expression levels were quantified among previously identified nucleotide sites in placental tissue among mule and hinny which carried same genotype. The results showed that more genes with paternal origin than genes with maternal origin were expressed in the placenta. Besides, variation in parental allele-specific methylation status was corresponding to the allelic expression bias. These paternally-origin bias expressed genes were considered as candidate imprinting genes. Generally, a placental-specific expression bias of maternal imprinted (paternally expressed) genes was critical for the normal development of placenta. 31
To investigate whether differential paternal DNAm plays a role in gestational age at birth, we examined epigenome-wide DNAm in peripheral paternal blood collected around the time of the pregnancy of their female partners. We used data from the Isle of Wight Birth Cohort (IoWBC) 32 that includes the grand-parental (F0), the parental (F1), and a grandchild generation (F2). In the F1 generation, we have paternal DNAm data and information on gestational age at birth when F1-mothers were pregnant with F2. We tested DNAm in the Swedish “Born into Life” study for agreements with our results.
Methods
Discovery cohort
The IoWBC was established in 1989 in the United Kingdom to observe natural history and to examine risk factors of asthma and other allergic conditions. 32 Pregnant F0 women were enrolled at the time of delivery in 1989/1990. Newborns (F1) were followed to date (samples analyzed until 2018). Between 2011 and 2018, part of their F1 daughters and spouses or female partners of F1 sons became pregnant. Peripheral blood samples of the F1-mother and their partners were collected during pregnancy. Questionnaires were completed during pregnancy to gather information about behavior and medical conditions.
Ethics approval was achieved by the Isle of Wight Local Research Ethics Committee before recruiting participants between January 1989 and February 1990. The investigation of the third generation was approved by the Isle of Wight, Portsmouth, and SE Hampshire Local Research Ethics Committee (Research Ethics Committee reference number: 09/H0504/129; December 4, 2009). The internal review board of the University of Memphis approved the project (FWA00006815, December 7, 2012). Permission was also granted for the collection of samples for genetic studies. Written informed consent forms were obtained from all participants before they were enrolled in the study.
Replication cohort: Born into Life study
The Swedish Born into Life study focuses on maternal factors and early biomarkers during pregnancy on child’s growth and health outcomes later in life. In this cohort, 92 pregnant women were recruited and have been followed up in 2010-2012. This study was described in a previous publication. 33 DNAm data were available for 15 fathers to replicate our results. Ethical approval was obtained from the Regional Ethical Review Board in Stockholm, Sweden, and written informed consents were obtained from fathers in the Born into Life study.
Variables
In the Isle of Wight study, gestational age estimates were based on the last menstrual period and/or by ultrasound measurement. Data on smoking during pregnancy were obtained from questionnaires administered at 12, 20, and 28 gestational weeks. Maternal smoking during pregnancy was assessed in each trimester and information on treatment for premature labor was ascertained during pregnancy. Maternal and paternal age at conception, race of father, education level of father, paternal smoking, maternal chronic disease during pregnancy, mode of delivery, gravida, parity, and number of unsuccessful pregnancy were collected and considered as confounding variables. In the Born into Life study, information of gestational age at birth, treatment for preterm labor, and form of delivery were obtained from Medical Birth Register. Maternal smoking during pregnancy, maternal smoking before pregnancy, fathers’ education levels, maternal chronic disease history, parity, and history of unsuccessful pregnancies were assessed in the first trimester from maternal reports to the midwife at the first antenatal visit. Maternal age at delivery and paternal age when the child was born were estimated by birth data of the child and birth data of the parents.
Biological sample collection and DNA extraction
In the Isle of Wight study, paternal peripheral blood samples were collected in EDTA tubes during their spouse‘s pregnancy. After the delivery of the infant and cutting of the umbilical cord, blood from umbilical cord was collected into PAXgene Bone Marrow RNA tubes (Qiagen, Valencia, CA, USA). For DNAm, blood samples were centrifuged at 3,000 r/min to separate blood cells from plasma and stored at −80°C. Later, DNA was extracted by using standard salting out procedure. 34
In the Born into Life study, paternal peripheral blood samples were ascertained before or during their partners’ pregnancies. DNAm was measured by the MethylationEPIC BeadChip, using the manufacturer’s standard protocol (Illumina, Inc., San Diego, CA, USA) at the Mutation Analysis Facility, Karolinska Institutet (http://www.maf.ki.se/).
DNAm analysis
One microgram of DNA from each subject was converted with sodium bisulfite by means of the EZ-96 DNAm kit (Zymo Research, Irvine, CA, USA). Genome-wide DNAm was measured using the Illumina Infinium Human Methylation450 beadchip (Illumina, Inc.). Methylation data were extracted from image data files using the methylation module of Genome Studio software. The methylation β-values (percent methylation) represent the proportion of methylated (M) over the sum of methylated and unmethylated (U) allele intensities (β = M/[M + U + c]), where c indicates a constant to prevent dividing by zero. 35
Methylation data pre-processing
The Bioconductor IMA (Illumina methylation analyzer) package 36 and the ComBat package 37 were applied to preprocess raw DNAm data, including removing background noise, adjusting for inter-array variation, performing peak correction, and removing batch effects. 37 During data preprocessing (1) cytosine-phosphate-guanine (CpGs) (cross-reactive probes) that were related to probes containing SNPs (single nucleotide polymorphisms) were removed from the list of all CpG sites if the minor allele frequency (MAF) of the probe SNPs based on European white population was larger than 5% (N = 89,678). This step was conducted, since probes containing SNPs that are at or near the targeted CpG sites may interfere with the measurement of DNAm. (2) CpGs located on the sex chromosomes were excluded. (3) CpGs with signals that were not distinguishable against background noise were taken out while keeping probes with detection P value of <1e−16. (4) Finally, CpGs with more than 5% missing probes were removed (sample call rate > 95%). Due to these procedures, the number of eligible CpG sites was reduced to 274,710 CpGs.
The estimateCellCounts() function from the Minfi package 38 was employed with an adult reference panel, 39 to generate cell proportions of the respective blood sample. Using cell-specific CpGs, we assessed 7 cell-types CD4T, CD8T, natural killer cells, B cells, monocytes, granulocytes, and eosinophils applying the estimatecellcounts() function in minfi with the default settings using the adult reference panel and the method by Houseman et al 40 and Jaffe and Irizarry. 41
In the Born into Life study, GenomeStudio Software was used to process the raw methylation intensities, and the detection P value for each CpG was used as a quality control measure of probe performance. The Bioconductor Illumina Minfi package 36 and the ComBat package 37 were used to remove background noise, adjust for inter-array variation, perform peak correction, and remove batch effects.
Gene expression–level measurement
In F2-offspring, total RNA was extracted from umbilical cord blood according to the manufacturer’s instructions. Total RNA yield and the absence of DNA contamination was measured by Qubit 2.0 fluorometer (Life Technologies, Grand Island, NY, USA). RNA quality was evaluated by a Bioanalyzer 2100 via RNA 6000 Nano Chips (Agilent Technologies, Santa Clara, CA, USA). RNA was reverse transcribed into cDNA by Agilent’s Single-Color Microarray-Based Gene Expression Analysis protocol version 6.0. Then cDNA was amplified, labeled, and hybridized on SurePrint G3 Human GE v2 8x60K Agilent Microarray slides (Agilent Technologies). Slides were scanned with an Agilent’s G2565AA Microarray Scanner System. Expression data were reported using Agilent’s Feature Extraction Software version 9.5. A subset of probes corresponding to the annotated genes was measured to examine the expression level.
Statistical analysis
Statistical and biological assessments followed the 5 steps (Figure 1). First, in the discovery cohort, the statistical analyses started with screening for significant CpG sites potentially associated with gestational age at birth by using training-testing screening (ttScreening) (v1.5) package. 42 The ttScreening approach has the ability to detect more true positives CpGs than traditional methods that controlled for multiple testing by false discovery rates (FDR), and Bonferroni-based methods. During simulation, sensitivity based on ttScreening is comparable with that from the FDR-based method, but ttScreening provides a higher specificity. Compared with Bonferroni-based method, the ttScreening method showed better sensitivity and comparable specificity. ttScreening uses 100 randomly selected training and testing sub-samples to estimate and test the effects of the primary variable. 42 The selection probabilities indicate how often CpGs gained statistical significance both in the 100 training and testing sub-samples. To account for skewed distributions of gestational age at birth with β values, the β values were logit-transformed. In the screening process, the purpose of which is the detection of associations, CpGs were the dependent and gestational age at birth was the independent variable. Potential confounding from the leukocyte cell composition was controlled in the process of ttScreening. The CpG sites which met the 50% cut-off in selection proportion were considered to be important. 42

Study flowchart of statistical and biological assessment in the course of the study.
Second, following the screening, the study concentrated on biological pathways to explain the function of the identified CpGs. Function enrichment analysis was conducted for the genes of the discovered CpGs obtained from the methylation label file (Infinium MethylationEPIC v1.0 B4 Manifest File). CpGs for which the gene was not documented in the manifest, the nearest gene names were identified using SNIPPER (https://csg.sph.umich.edu/boehnke/snipper/) 43 and the University of California Santa Cruz (UCSC) Genome Browser (https://genome.ucsc.edu/). 44 The chromosome number and map info of the CpGs were queried (using Human GRCh37/hg19) and the nearest gene to the site of the CpG was selected. Once all genes were identified, the list of genes was entered into ToppFUN (https://toppgene.cchmc.org/) 45 to identify biological pathways related to these genes. Significant pathways (adjusting for multiple testing P value of ⩽.05) were selected for the next steps.
Third, to test whether the biological pathway-related CpGs were associated with gestational age at birth adjusting for confounders, Cox proportional hazards models were applied. Hazard ratios (HRs) were estimated. In this step, to estimate the risk of CpGs for the duration of gestational age, the CpGs were used as independent variables and weeks of gestational age at birth as the dependent variable. We adjusted for the proportion of leukocyte cell-composition, maternal smoking during pregnancy, maternal age at conception, paternal age at conception, and paternal smoking. To minimize false-positive findings, the P values were adjusted for FDR. 46 To graphically demonstrate associations, we chose one CpG site with largest HR and plotted the Kaplan-Meier curve of the gestational age at birth versus DNAm. For the figure, DNAm was dichotomized into 2 groups based on their median.
Fourth, to replicate our results, Spearman’s correlation analyses were performed between DNAm levels of the pathway-related CpG sites and gestational age at birth from our cohort and from the replication dataset from Born into Life Study (n = 15). The 95% confidence intervals (CI) of the IoWBC Spearman’s correlation coefficients were obtained and investigated whether the correlation coefficients of the replication cohort were within the respective 95% CI. To better demonstrate the correlation between gestational age at birth and methylation level of pathway-related CpG sites in both IoWBC and Born into Life Study, graphs were plotted by using log-transformed gestational age at birth.
Finally, fifth, to explore the biological implications from pathway-related CpGs, correlations between methylation levels of specific CpGs from offspring and gene expression in offspring’s umbilical cord blood of corresponding genes were estimated in IoWBC. In addition, the correlations were also tested in a subgroup of participants who had available paternal DNAm data, transcript data, and DNAm data for offspring’s umbilical cord blood.
For all analyses, a P value of ⩽.05 was considered as statistically significant. Statistical analyses were performed by R 3.4.4. and SAS 9.4 (SAS Institute, Cary NC).
Results
Participants characteristics in IoWBC
A total of 96 F1-fathers were enrolled in the IoWBC until 2018, either as male F1-offspring or as partners of F1 female participants. Of these 96 fathers, DNAm was measured in 92 fathers around the period when their partners were pregnant. Fathers were predominantly white (98.90%) and between 17 and 37 years of age (Table 1). The gestational age at birth of their partners’ pregnancy ranged from 35 to 42 weeks; the mean gestational age at birth was 39.6 weeks. Only 3 (3.26%) F2-offspring were born preterm (<37 weeks).
Characteristics of the factors may be related to gestational age at birth in IoWBC.

In the case of yes versus no question, only the proportion of positive answers is tabulated.
Values represent mean ± SD.
Values represent n (%).
Replication cohort
A total number of 15 fathers with available DNAm and child’s gestational age information were included to test the discovery results in an external sample (Table 2).
Characteristics of the replication cohort (Born into Life study, n = 15).

In the case of yes versus no question, only the proportion of positive answers is tabulated.
From Medical Birth Register.
EWAS search for differential paternal DNAm associated with gestational age at birth in IoWBC
By using ttScreening, 216 CpG sites were identified to be potentially associated with gestational age at birth in 50% of the different samples (Additional file 1: Table S1). A total number of 297 annotated genes were identified corresponding to these 216 CpG sites by choosing 1 to 3 closest genes.
Biological functions enrichment analysis
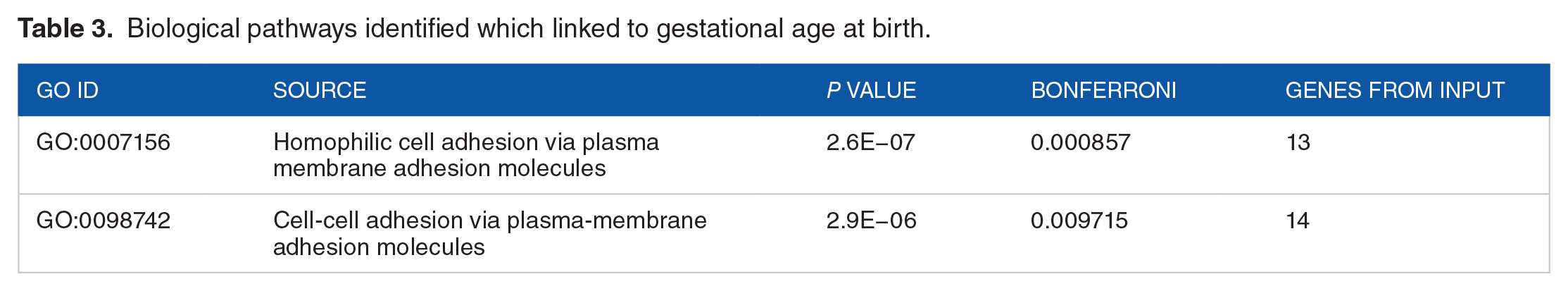
Applying ToppFUN, two statistically significant biological pathways were identified: homophilic cell adhesion (13 genes, GO:0007156, Table 3) and cell-cell adhesion (14 genes, GO:0098742, Table 3). Except for one gene 1L1RN in GO:0098742, the remaining 13 genes enriched in these two biological pathways were the same; 12 belonged to the protocadherin superfamily, and one belonged to the cadherin family (Additional file 1: Table S2). This step led us to focus on 9 pathway-related CpGs corresponding to these 14 genes in subsequent analyses.
Biological pathways identified which linked to gestational age at birth.
Survival analysis
DNAm levels of all 9 CpGs corresponding to 14 genes in the two biological pathways were significantly associated with gestational age at birth after adjustment of confounders (Table 4). The hazard ratio (HR) expresses the relative risk increase per percent change of DNAm. All CpG sites with the exception of cg18608017 and cg11408933 were less methylated when the gestational age at birth was longer. Accordingly, the Kaplan-Meier curve of cg11408933 showed that the higher the DNAm level, the longer the gestational age at birth (Figure 2). All 9 CpG sites remained significant after correction for multiple testing.
Hazard ratios of 9 biological pathway-related CpG sites after adjustment for confounders.
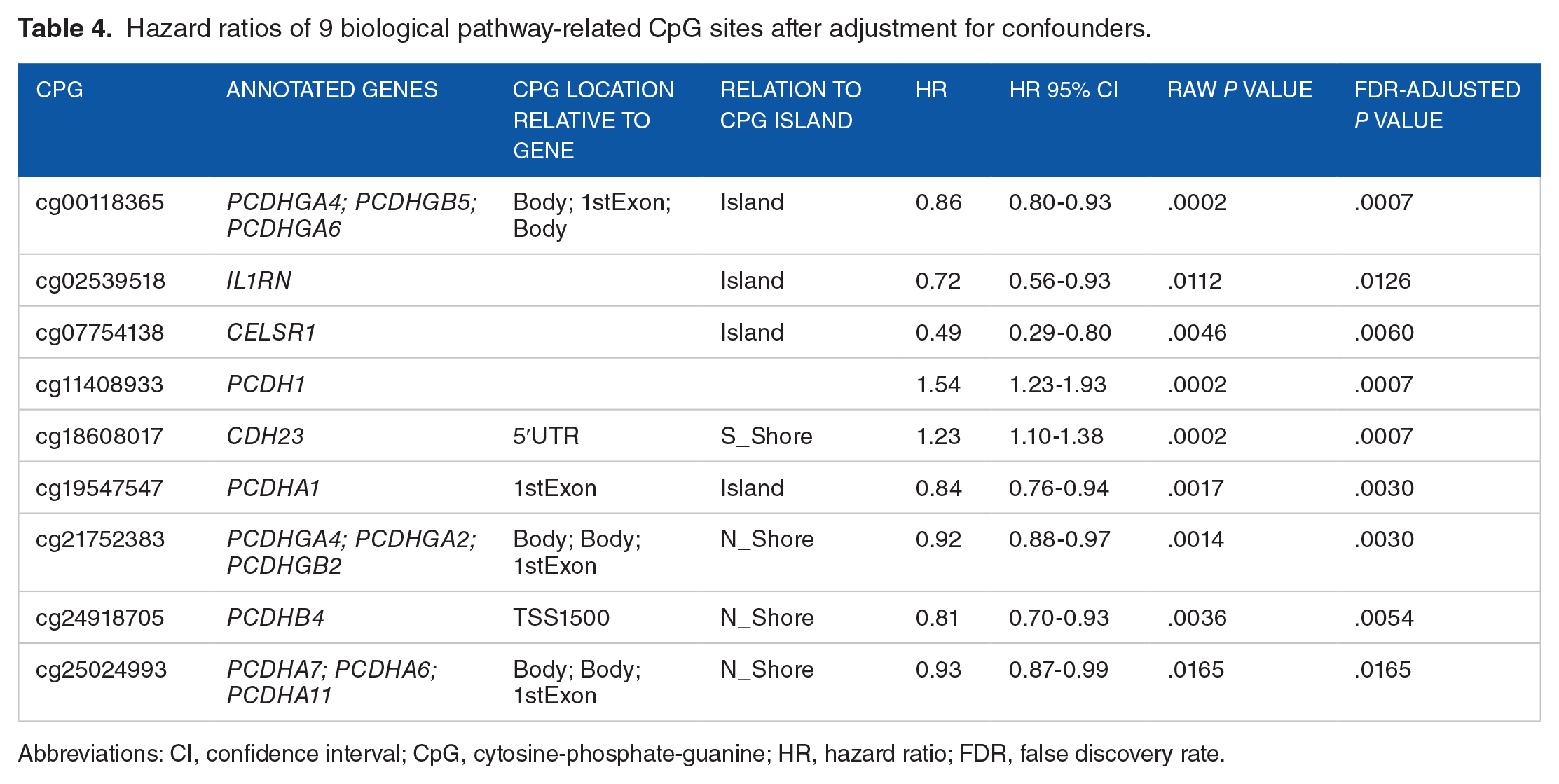
Abbreviations: CI, confidence interval; CpG, cytosine-phosphate-guanine; HR, hazard ratio; FDR, false discovery rate.

Kaplan-Meier curve showing gestational age at birth with DNA methylation levels of cg11408933 (PCDH1, protocadherin 1) dichotomized at the median. Group 1 of the CpG cg11408933 includes subjects with lower DNAm levels and group 2 subjects with higher DNAm levels.
Replication
The data of 8 out of the 9 pathway-related CpG sites were available in the Swedish Born into Life study. Of note, the gestational age at birth in the replication cohort did not vary much (Table 2). The Spearman’s coefficient of correlation between DNAm and gestational age at birth in the Born into Life Study of two (cg00118365; cg18608017) out of 8 pathway-related CpG were within 95% CI of Spearman’s coefficients of the IoWBC (Table 5, Figure 3; Supplemental Figure S1), suggesting replicable results. A third CpG (cg21752383) was just outside the lower confidence limit (Table 5), but showed a similar direction in the plot of its β-values with log-transformed gestational age.
Correlation coefficients for DNAm and gestational age at birth in IoWBC and Born into Life Study.

Abbreviations: CI, confidence interval; CpG, cytosine-phosphate-guanine; DNAm, DNA methylation; IoWBC, Isle of Wight Birth Cohort.
Bolded mark represents CpG sites which were got replicated in Born into Life study.
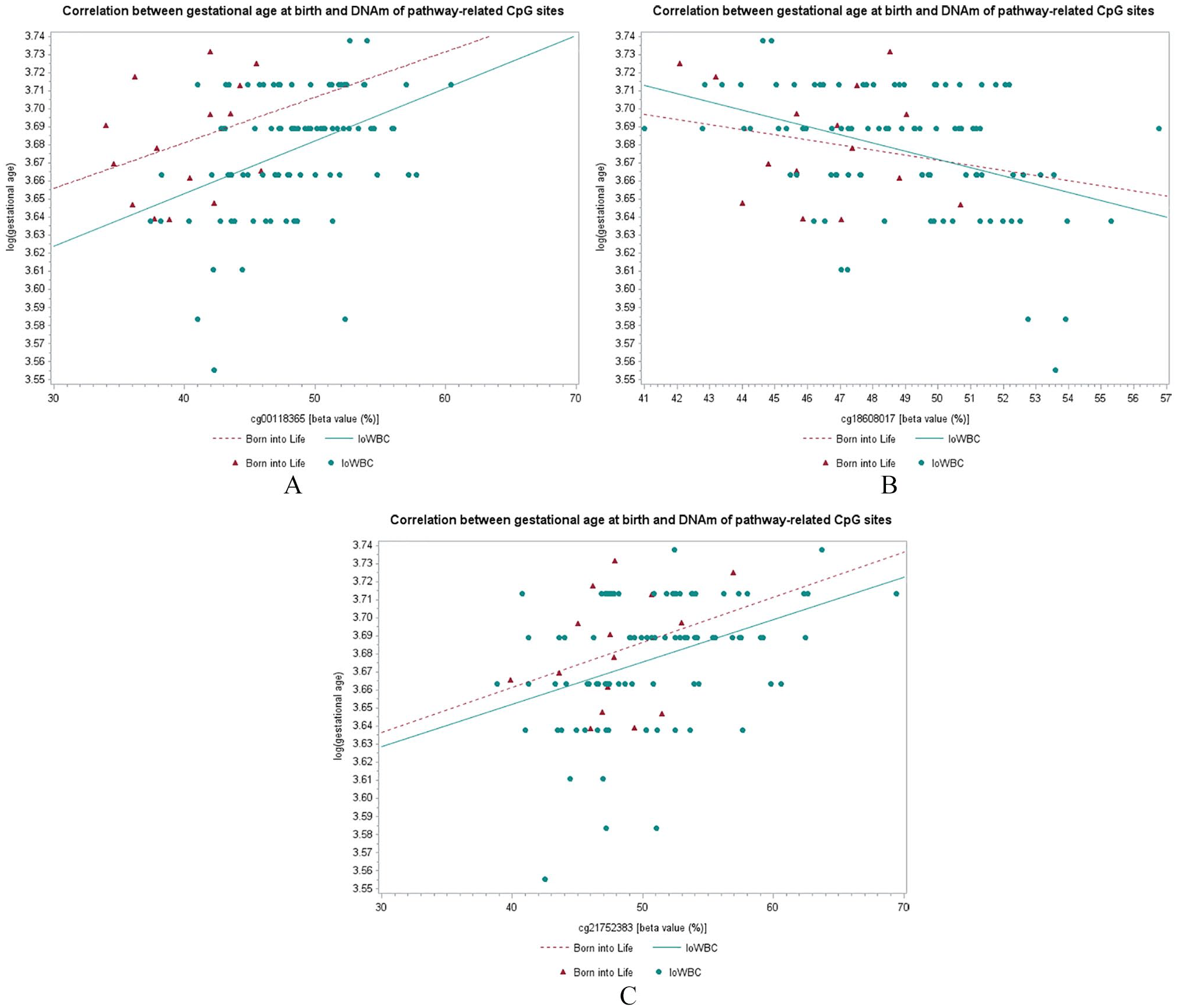
Correlation between gestational age at birth and DNAm of CpG sites of the genes that were related to the cell-cell membrane adhesion pathway. (A) cg00118365, (B) cg18608017, and (C) cg21752383.
Correlation with DNAm and gene expression in offspring’s umbilical cord blood
Considering complex overlapping genes in PCDH gamma cluster and PCDH alpha cluster (Figure 3), gene expression levels of 22 genes in PCDH gamma cluster and 15 genes in PCDH alpha cluster were included. Note, the association of DNAm and gene expression was tested in cord blood to determine whether the methylation may be associated with genes expression. Of the 9 candidate CpGs, correlations between 2 CpG-gene expression pairs, cg21752383-PCDHGB4 (rs = 0.2079, P = .0092) and cg25024993-PCDHA4 (rs = −0.1620, P = .0434), were statistically significant (Table 6, Additional file 1: Table S3). However, the correlation between these 2 correlations did not remain statistically significant in the subgroup of 47 participants who also have paternal DNAm data (Additional file 1: Table S4).
Significant correlation between DNA methylation levels of pathway-related CpGs and expression levels of annotated genes in offspring umbilical cord blood (IoWBC, n = 157).

Discussion
In samples of 92 fathers from the F1-generation of the IoWBC, paternal CpGs of genes in pathways related to cell-cell adhesion molecules were significantly associated with gestational age at birth. Comparing rank correlations, the results of 2 CpGs out of 9 were replicated in the Swedish Born into Life Study; a third CpG graphically showed a similar association. Expression levels of 2 pathway-related genes were found statistically correlated with DNAm of corresponding CpGs in umbilical cord blood of IoWBC. This study provided novel insights into that paternal DNAm may contribute to the variance among gestational age at birth of their partners’ pregnancy.
We employed a 2-step screening approach to identify CpG sites which are both statistically significant and biologically reasonable. ttScreening was employed as a selection approach to perform epigenome-wide association analysis. 42 A 50% cut-off of selection probability was sensitive to identify CpG sites, which were significantly associated with gestational age at birth. Moreover, the main purpose of our study was to identify CpG sites that are both statistically and biologically meaningful. Therefore, functional enrichment analysis was carried out as a second filter to select CpG sites before subsequent survival analysis.
How might the paternal epigenetic alteration contribute to gestational age at birth? One explanation is that paternal genome regulates normal placental development through imprinted genes.47-49 Wang 28 previously reported that a unique epigenetic signature of imprinted genes in the placenta contributes to variation in the intrauterine environment. Imprinted genes are those expressed in a parent-of-origin specific pattern that may cause one of the alleles of paternal or maternal origin to be epigenetically silenced. 30 Imprinted genes are functionally haploid, which increases susceptibility to mutations and epimutation. 31 In the placenta, more maternal-origin genes were inactivated by epigenetic modification than paternal-origin genes. 28 Variability in methylation status in paternal-origin genes might contribute more to gestational age at birth through affecting placenta development and nutrient transfer. Therefore, the pathway-related genes identified in this work can be treated as candidate imprinted genes which especially require further investigation in human placenta. However, previous studies rarely focused on paternal factors and the placenta. Our study provides a suggestion for future investigations to explain the epigenetic paternal role of gestational age at birth.
Our results are further supported by the exciting findings that the 2 identified biological pathways were both linked to cell-cell adhesion. As part of the 2 identified biological pathways, the annotated genes in the protocadherin (PCDH) gene played a crucial role in fetal development, especially in neural circuit formation, 50 and were reported to be associated with gestational age at birth. 51 Higher methylation levels of CpGs in PCDH clusters were linked with decreasing risks of having shorter gestational age at birth (Table 4). The DNAm sites cg21752383 and cg25024993, both close or within PCDH clusters, were positively associated with PCDHGB4 expression level and negatively associated with PCDHA4 expression level, respectively (Table 6). And cg21752383 seems to have a similar effect on gestational age in both cohort: the IoWBC and the Born into Life Study (Figure 3).
Pretreatment of Interleukin 1 receptor antagonist (IL1RN), one of the major anti-inflammatory cytokines, has been shown to prevent interleukin-1-induced preterm labor in animal models. 52 A significantly reduced expressed level of IL1RN in cervicovaginal fluid was considered as a biomarker for spontaneous preterm labor. 53 Besides, a previous study showed that fetuses who carried IL1RN risk alleles to be at increased risk for preterm premature rupture of membranes, subsequent preterm delivery, 54 and differrential gestational age at birth. 55 Meanwhile, maternal carriage of risk alleles of IL1RN did not show any impact on the risk of preterm deliveries, 54 suggesting that the fetal effect of IL1RN risk alleles comes from paternal side. Accordingly, we found that higher methylation of IL1RN from paternal side was associated with shortening gestational age at birth (Table 4). One locus in IL1RN was found to be associated with gestational age at birth in infants.
Several limitations in this study should be amended in future investigations. The sample size of the paternal sample, the variation of gestational age at birth, and the number of preterm births (n = 5 of 92) were limited in this investigation. As few studies have ever focused on the father’s effect on gestational age at birth, the sample size of the replication cohort also is small with a sample size of n = 15 and showed little variation in gestational age at birth. Furthermore, gene expression levels from placental samples were not available to assess the potential effect of paternal DNAm on gestational age at birth by affecting placental gene expression. However, DNAm level and gene expression levels were available in umbilical cord blood. By testing the correlation between DNAm and gene expression levels in umbilical cord blood, additional support for our biological implications was provided. Furthermore, information on methylation linked to genetic polymorphism could not be tested. Hence, we cannot be sure that a differential gestational age at birth was determined by paternal methylation or due to inheritance of genetic polymorphisms to the offspring. Finally, the study population did not include other races than white Europeans. Hence, future studies with larger sample sizes, wider ranges of gestational ages at birth, more racially diverse populations, and multiple tissue samples are warranted to test the proposed association between paternal DNAm and gestational age at birth.
Conclusions
Our results suggest that paternal epigenetics may be associated with gestational age at birth of their partners’ pregnancy, which may offer clues for novel preventive strategies for short gestational age at birth. Paternal DNAm seems to associate gestational age at birth through 2 biological pathways; both focus on cell-cell adhesion molecules. Additional studies are needed to investigate the potential generational impacts of paternal DNAm on offspring.
Supplemental Material
Supplementary_Tables_S1-S4 – Supplemental material for Paternal DNA Methylation May Be Associated With Gestational Age at Birth
Supplemental material, Supplementary_Tables_S1-S4 for Paternal DNA Methylation May Be Associated With Gestational Age at Birth by Rui Luo, Nandini Mukherjee, Su Chen, Yu Jiang, S Hasan Arshad, John W Holloway, Anna Hedman, Olena Gruzieva, Ellika Andolf, Goran Pershagen, Catarina Almqvist and Wilfried JJ Karmaus in Epigenetics Insights
Footnotes
Acknowledgements
The authors gratefully acknowledge study participants and appreciate the hard work of the Isle of Wight research team and Born into Life Study team.
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by funding from the National Institute of Allergy and Infectious Diseases (R01 AI091905) and the National Heart, Lung, and Blood Institute (R01 HL132321) to W.K. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health, USA.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
RL conducted the statistical analyses, interpreted the data, and drafted the manuscript. WJJK, NM, SC, and YJ contributed to conceptualization and methodology. HA was responsible for data collection. JWH conducted DNAm measurements. AH, OG, EA, GP, and CA contributed to the replication using the “Born into Life Study.” All authors contributed to manuscript drafting and final proofreading.
Abbreviations
CI: confidence intervals; CpG: cytosine-phosphate-guanine; DNAm: DNA methylation; FDR: Adjusted p value with Benjamini-Hochberg method; HR: hazard ratios; IMA: Illumina methylation analyzer; IoWBC: Isle of Wight Birth Cohort; MAF: Minor Allele Frequency; SD: standard deviation; SNPs: single nucleotide polymorphisms; ttScreening: training-testing screening; UCSC: University of California Santa Cruz; UTR: Untranslated region.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.