
Abstract
Hemoglobinopathies are genetic disorders that lead to abnormal structure of the hemoglobin molecule. Sickle cell disease, the most common inherited blood disorder, is characterized by defective oxygen transport. Almost every part of the eye can be affected by sickle cell disease; however, proliferative sickle cell retinopathy is the primary cause of vision loss, either from vitreous hemorrhage or retinal detachment. Here we review the various manifestations of hemoglobinopathies on the eyes of children and adolescents, with a specific focus on sickle cell disease and its different phenotypes. Newer, more sensitive ophthalmological imaging modalities, including ultra-widefield fluorescein angiography, spectral-domain optical coherence tomography, and optical coherence tomography angiography, are available. These sensitive modalities allow for a more thorough examination of the retinal periphery where sickle cell retinopathy is often present. Utilization of such modalities will help with the early detection of the disease in children, which provide a better understanding of the pathogenesis of the disease and guide future screening and treatment regimens.
Introduction
Hemoglobinopathies are caused by genetic mutations that lead to major changes in the hemoglobin molecule structure. This causes dysfunctions related to changes in shape, oxygen-carrying capacity, or ability to clump together, leading to obstruction of the vascular system. 1 Hemoglobinopathies typically fall into two main groups: thalassemia syndromes and structural hemoglobin variants. Alpha (α)- and β-thalassemia are the main types of thalassemia; the main structural hemoglobin variants are HbS, HbE, and HbC. There are many subtypes and combined types in each group.2,3 Sickle cell disease (SCD), the most common inherited blood disorder, is a disorder of chronic hemolysis, vascular injury, and tissue ischemia affecting multiple organ systems.
In SCD, erythrocytes undergo rapid yet reversible shape changes because of deoxygenation. Intracellular polymerization of HbS molecule alters the normal flexible biconcave shape into an elongated rigid form. Sickled erythrocytes adhere to the endothelium of blood vessels, leading to vasospasms, vasoconstriction, and inflammation.4,5 Endothelial adhesion is also significantly affected by alterations in erythrocyte hydration. 6 Because of their increased viscosity, these sickled red blood cells (RBCs) sludge in the circulatory system causing obstruction of the microvasculature, 7 leading to a broad range of acute and chronic complications from oxygen deficiency in various organs and tissues. Vaso-occlusive crisis can be precipitated by multiple factors including cold weather-induced vasospasm, hypoxia, infection, dehydration, acidosis, and alcohol intoxication. 8
Although individuals with homozygous hemoglobin S disease (HbSS) present with more severe systemic morbidities than those with HbSC, individuals with HbSC show an increased risk of developing retinal disease.9–11 SCD can affect every vascular bed in the eye; however, the most significant changes occur in the retina. These changes can be classified into proliferative sickle retinopathy (PSR) and non-proliferative retinal changes. 12 PSR can ultimately lead to visual loss through ischemia, vitreous hemorrhage, or retinal detachment. Therefore, early screening to detect neovascularization can prevent the consequences of PSR. 13 Application of newer, more sensitive imaging modalities to screen the pediatric population will play a major role in early detection of sickle retinopathy in children and adolescents.
Here we review the various ophthalmic manifestations of hemoglobinopathies in the pediatric population as well as the latest technologies to detect disease.
Ophthalmological manifestations of sickle cell disease
Anterior segment involvement
Comma-shaped vessels, usually found in the inferior bulbar conjunctiva, also known as “conjunctival sign,” are a common presentation.14,15 These conjunctival changes vary with the oxygenation status and are best seen under high magnification. They are more commonly observed in HbSS than in HbSc. 16 Hyphema of the anterior chamber is another possible complication due to SCD. Hyphema can lead to clogging of the trabecular meshwork subsequently leading to elevated intraocular pressure (IOP) even with relatively small amount of intracameral blood. 17 Numerous publications have reported risk of retinal artery occlusion even with modest IOP elevations.18,19 Studies have suggested surgical intervention for hyphemas of any size with IOP more than 24 mmHg for more than 24 hours in individuals with sickle cell trait or disease; 20 however, there is little scientific evidence supporting surgical interventions in these case scenarios. 21 The decision for medical and surgical management of these patients should be made on a case-by-case basis, until more evidence is readily available.
Orbital involvement
Orbital involvement is a less common manifestation of SCD, yet it is critical to recognize the potential risk of loss of vision. Although vaso-occlusive crises often affect the bone marrow of vertebrae and long bones, they may involve the orbital walls in children where the marrow content is higher.22–24 Presentation may be severe pain, lid edema, proptosis, ophthalmoplegia as well as diplopia.22–25 Orbital bone infarction can lead to an orbital hematoma, which in turn may lead to orbital compartment syndrome. 26 Both osteomyelitis and orbital cellulitis may present similarly, and thus confirmation of the diagnosis of orbital bone infarction is of great importance.27,28 The lacrimal glands can be affected, presenting as either bilateral or unilateral pain and swelling. 29
Retinal involvement
Retinopathy is the most serious and vision threatening manifestation of SCD. Sickle cell retinopathy can either be non-proliferative (NPSR) or proliferative sickle retinopathy (PSR). Described clinical findings found in NPSR include salmon-patch hemorrhages (Figure 1), iridescent spots, and black sunbursts, all of which result from peripheral arterial occlusion.30,31 These lesions are not typically associated with vision loss as they occur in the periphery. Other findings include angioid streaks, sickle disk sign, retinal depression sign, posterior vascular tortuosity, and retinoschisis.32,33 Peripheral neovascularization seen in PSR can lead to serious visual loss due to its sequelae as vitreous hemorrhage and retinal detachment. The most widely used scale by which retinal changes are graded is the Goldberg classification. 34

A widefield fundus photograph showing multiple salmon-patch hemorrhages (white arrows) as a result of peripheral arteriolar occlusion in a patient with sickle cell disease.
Goldberg described the natural history of untreated PSR and the sequence of retinal changes. Stage I consists of peripheral arteriolar occlusions, and stage II is characterized by peripheral arteriovenous anastomosis (Figure 2). Stage III is defined by the classic “sea-fan” neovascularization that takes place at the border of perfused and non-perfused retina (Figure 3). Auto-infarction occurs spontaneously in 20–60% of sea fans as a result of chronic, recurrent vaso-occlusion within the sea fans themselves. 35 Stage IV is characterized by vitreous hemorrhage, which is seen when the sea fan bleeds, and stage V is defined by tractional retinal detachment resulting from peripheral fibrovascular membranes.34,35 This classification scheme was later updated by Penman, where he more strictly defined proliferative retinopathy as requiring the presence of neovascularization (Goldberg Stage III–V). 36

Ultra-widefield fluorescein angiography showing large areas of peripheral non-perfusion (yellow asterisks), arteriolar occlusion (red arrow), and arteriovenous anastomosis (blue arrow) in a patient with sickle cell retinopathy.
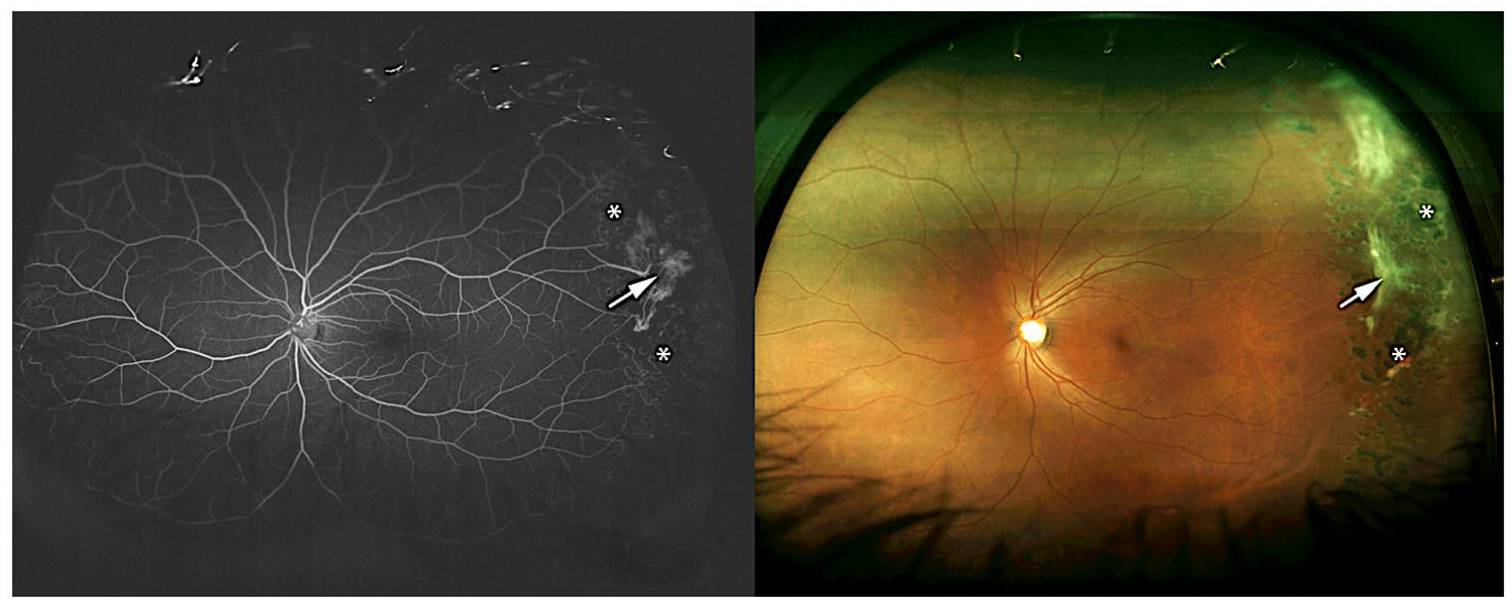
Ultra-widefield fluorescein angiography (left) and widefield fundus photography showing an area of sea-fan neovascularization (white arrow) surrounded by retinal laser photocoagulation marks (asterisks) in a patient with sickle cell retinopathy (Goldman stage III).
While clinical exam findings are essential in detecting retinopathy signs and preventing visual loss, newer imaging modalities have made earlier detection and screening for retinopathy more possible.
Ophthalmic imaging techniques in sickle cell disease
Recent advances in retinal imaging modalities, including ultra-widefield fluorescein angiography (UWFA), spectral-domain optical coherence tomography (SD-OCT), and optical coherence tomography angiography (OCT-A), have revealed significant retinal findings in asymptomatic sickle cell patients.37–42 PSR presence has been significantly linked to some of the silent findings. 41
Ultra-widefield fluorescein angiography
Fluorescein angiography (FA) has served as the gold standard for diagnosis of PSR for over 40 years. FA allows observation of areas of dye pooling, leakage, and staining, as well as dynamic visualization of blood flow. 43 This allows for documentation of sickle cell retinopathy changes in the periphery and appropriate grading on the Goldberg scale. Standard FA cameras permit images ranging from 30° to 60° in a single exposure. A 75° field of view could be achieved by using 7-standard fields. More recently, UWFA can capture up to 200° of retina in a single exposure and capture more than twice as much retina compared to conventional FA.44,45 This has allowed for more efficient imaging and reduced the need for excessive patient cooperation and technical expertise. 45 UWFA has a significant utilization in PSR screening because the disease primarily affects the peripheral vasculature. In a retrospective study done on 12 eyes of 6 SCD patients, UWFA was able to detect peripheral vascular changes missed on the classical 7-standard field photograph in all but one eye. In addition, peripheral vascular changes missed on clinical examination by experienced ophthalmologists were detected by UWFA in 25% of the eyes. 40 Despite the limited data available on comparisons of standard FA and UWFA, UWFA is likely more sensitive in detecting peripheral proliferative retinopathy.
Spectral-domain OCT and OCT angiography
SD-OCT is a non-invasive imaging modality that utilizes reflected light to produce near-histological cross-sectional and en-face images of the retina. It can highlight areas of photoreceptor loss, nerve fiber layer change, and thinning or thickening of the retina in response to ischemic insults and neovascularization. OCT-A is a more recent modality using the principle of diffractive particle movement of moving RBCs to determine vessel location through various segments of the eye without the need of any intravascular dyes. This allows for the ability to image the flow in both retinal and choroidal vasculature, which can be displayed in en-face and depth encoded slabs. This provides detailed imaging of the superficial and deep retinal vascular plexus, and choriocapillaris. Both SD-OCT and OCT-A can be essential in detecting early signs of PSR.
SD-OCT has been used recently to assess subclinical macular thinning due to macular ischemia (Figure 4). Temporal and central macular thinning, also referred to as macular splaying, has been reported in some patients with SCD.37,41,46,47,48 These discrete areas of macular thinning have been significantly linked to PSR in a 2015 retrospective study. 46 Previous case series and histopathological studies have reported inner retinal layer thinning in SCD patients with either symptomatic or asymptomatic retinopathy.49–52 The predictive ability of temporal macular thinning (atrophy) for neovascularization in PSR patients was tested in a case–control study on 38 patients. Temporal macular atrophy was found to have a positive predictive value of 83% and a negative predictive value of 13% for identifying neovascularization, demonstrating that the presence of temporal macular atrophy suggests the concurrent presence of neovascularization and PSR. 53 In addition, a recent retrospective case series correlated the degree of peripheral ischemia on UWFA to areas of macular thinning on SD-OCT. 54 Therefore, the use of SD-OCT in early diagnosis and screening of retinopathy could be useful because of the association between macular thinning and PSR.

Optical coherence tomography angiography demonstrating decreased vascular density (asterisk) in the superficial capillary plexus (a) and deep capillary plexus (b). Areas of vascular loss correspond to temporal macular thinning (white arrow) shown on spectral-domain optical coherence tomography (c).
Although the etiology behind the macular thinning in PSR patients is not entirely understood, studies have suggested it might be due to ischemia of the deep capillary plexus.55,56 Using microperimetry, a prospective study on 19 SCD patients demonstrated a significant decrease in retinal sensitivity in areas of macular thinning on SD-OCT compared to areas without thinning or controls, suggesting functional consequences of this macular thinning. 57 In addition, SCD with focal macular thinning has associated thinning in the peripapillary retinal nerve fiber layer compared to those without focal macular thinning. 39 OCT-A is likely more sensitive in detecting early areas of macular ischemia when compared to traditional FA. 58 This difference was noted in a study that reported that OCT-A was capable of demonstrating microvascular abnormalities in the macula in 18 eyes of 9 patients, while FA appeared normal in 9 out of 18 eyes. 59 In another study, 16 adolescent patients (32 eyes) with SCD were examined using biomicroscopy, UWFA, SD-OCT, and OCT-A to determine the frequency of retinopathy with the newer, more sensitive modalities. 60 Biomicroscopy demonstrated that 69% of eyes had evidence of retinopathy, in the form of salmon-patch hemorrhages, vessel tortuosity, and sunburst lesions. About 20% of eyes showed temporal macular thinning on SD-OCT and corresponding flow voids on OCT-A. In addition, UWFA detected peripheral arteriolar occlusion in 17% of eyes (Goldberg stage I) and peripheral arteriovenous anastomosis in 83% of eyes.
The above results suggest that these modalities may prove useful in the early detection of retinopathy. Preliminary data indicate that children with SCD commonly display retinal vascular abnormalities by these sensitive imaging techniques, including Goldberg stage-I and -II retinopathy identified on UWFA, temporal macular thinning on SD-OCT, and vessel dropout on OCT-A. Clinical implications of these findings are still unclear and will demand more longitudinal testing to assess what risk these patients have for serious vision threatening pathology and what future intervention may be required for future vision preservation.
Treatment
Due to the low likelihood of vision loss in SCD, sickle cell retinopathy does not usually require treatment. The peripheral lesions seen in NPSR do not require any treatment. Considering the rates of autoinfarction, small sea fans may be observed closely. However, in cases of larger or multiple areas of neovascularization or vitreous hemorrhage, scatter retinal laser photocoagulation or panretinal photocoagulation (PRP) is required to prevent the development of retinal detachment. 61 One study of 21 eyes with PSR found complete regression in 24 out of 28 sea-fan lesions treated with scatter laser photocoagulation. 62 Others have found complete regression in 30.2% of treated eyes compared to 22.4% of untreated control eyes. 61
The involution of neovascularization after the off-label intravitreal injection of anti-VEGF agents as bevacizumab has been reported. However, further studies are recommended to determine their definite role in management of PSR.63–65
As a surgical approach, vitrectomy should be considered in cases of non-clearing vitreous hemorrhage and retinal detachment. It may be combined with intraoperative scatter laser photocoagulation, as well as with intravitreal injection of anti-VEGF agent. Extra care should be taken while peeling the fibrovascular membranes from the friable, ischemic retinal tissue, which may be prone to iatrogenic tears and breaks.66,67 However, the pediatric population does not typically require treatment as the advanced stages of PSR do not generally occur until later in life.
An association between elevated HbF and lower prevalence of retinopathy has been demonstrated on a retrospective review of 123 children with SCD. 68 These data suggest that induction of HbF by hydroxyurea, along with other possible drug effects, may prevent pediatric retinopathy and the development of visual loss from proliferative disease. The only known cure for SCD is allogenic stem-cell therapy from human leukocyte antigen-matched donor. Therefore, determining whether sickle cell retinopathy stabilizes or improves following the transplantation should be further studied.
Prevention
Current recommendations by the American Academy of Pediatrics is for retinopathy screening of children by dilated fundoscopic examination with HbSS and HbSC beginning at age 10 years. 69 Other recommendations include biannual or annual examination at age 9 years.70,71 In addition, patients with complaints of sudden or gradual diminution of vision, flashes of light, or floaters should be referred for an ophthalmological check-up, as these symptoms may reflect the development of macular ischemia, retinal tears, retinal detachment, or vitreous hemorrhage.
The decision to perform imaging, such as fundus photography, angiography, SD-OCT, or OCT-A, is based on the presence and severity of retinopathy at time of examination.
SD-OCT will likely be used more often in the screening examination of SCD patients, given the ease of image acquisition in both adult and pediatric populations. The connection between temporal macular thinning on SD-OCT, proliferative retinopathy on UWFA, and microvascular changes on OCT-A will need to be explored in future studies. If correlations between the imaging modalities are found in the pediatric population as they have been found in adults, abnormal SD-OCT studies may prompt differing monitoring strategies for patients with SCD.