
Abstract
Introduction
Sickle cell disease is a genetic blood disorder in which red blood cells become sickle-shaped under conditions such as hypoxemia, acidosis, and dehydration, leading to occlusion within vessels throughout the body and potential ischemia. The retina is particularly susceptible to ischemia due to its high metabolic demand and microvasculature, which lacks collateral circulation. 1 Ischemia can lead to neovascularization (NV) and subsequent sickle cell retinopathy, with complications including vitreous hemorrhage (VH) and retinal detachment (RD). As a result, it is critical to monitor sickle cell retinopathy closely, often with annual eye examinations. Because they generally comprise a disadvantaged population, patients with sickle cell disease face many barriers, including the cost of frequent appointments, limited access to specialists, and inadequate pain management. 2 Compared with the general population, these barriers can compound and manifest as a high likelihood of loss to follow-up. 3 Furthermore, most medical centers that treat sickle cell disease are not adequately staffed or funded to assist patients and families with scheduling appointments and arranging frequent transportation. 4 Loss to follow-up has not been explored in ophthalmic care among pediatric patients with sickle cell disease and is of particular importance because the peripheral avascular retina can trigger active retinopathy, even in the absence of visual symptoms. Therefore, the purpose of the current study is to identify the rates of loss to follow-up to help inform strategies to improve retention among this patient population.
Methods
This retrospective cohort study included pediatric patients (≤18 years at the initial visit) with sickle cell disease who were referred from Boston Children’s Hospital hematology division and had an ophthalmic examination between January 2014 and January 2024. International Classification of Diseases (ICD) 10th edition codes for retinal diseases included H35 and H36; ICD 9th edition codes included 282.60 to 282.69, and D57.0 to D57.2 and D57.4 to D57.8 were used to identify the eligible population. The sickle cell genotype was confirmed by cross-checking with hematology notes.
Due to its retrospective nature, this study was determined to be exempt by the Boston Children’s Hospital Institutional Review Board, with the requirement for informed consent waived. The study adhered to the tenets of the Declaration of Helsinki.
In addition to demographic characteristics, data collected included best-corrected visual acuity (BCVA) at the initial visit, reported in Snellen and converted to logMAR, number of eye examinations over the study period, severity of sickle cell retinopathy as defined by the Goldberg classification, adverse systemic outcomes (eg, acute chest syndrome, vaso-occlusive crisis, hospitalization, asthma), and any intervention (eg, vitrectomy, laser photocoagulation). 5 Additionally, retinal imaging data were collected, specifically from optical coherence tomography (OCT). OCT images were obtained using the Spectralis device (Heidelberg OCT2, Heidelberg Engineering) and processed using Fiji software (National Institutes of Health). The definition of sickle cell maculopathy was based on qualitative analysis of retinal thinning on OCT scans. 6 Peripheral sickle cell retinopathy was defined according to the Goldberg classification: peripheral arteriolar blockage (stage 1), arteriovenous anastomosis at the retinal border (stage 2), peripheral NV with sea fan (stage 3), VH (stage 4), and RD (stage 5). Loss to follow-up was defined as the failure to attend an appointment within 6 months of the scheduled date after an initial visit with an ophthalmologist. Patients with at least 1 documented ophthalmic visit in the study period were included, and patients with incomplete medical records were excluded.
Covariates identified for statistical analysis included age and BCVA at testing, sex, race, and sickle cell genotype. Continuous variables were presented as medians and interquartile range (IQR). Categorical variables were reported as frequencies and percentages. After controlling for covariates, analyses of severity of sickle cell retinopathy and systemic conditions of the loss to follow-up and non–loss to follow-up groups were completed using the Mann-Whitney U test in SPSS (version 28, SPSS Inc.). P < 0.05 was considered statistically significant.
Results
The study sample included 255 patients, with a median of 3 (IQR, 1-5) ophthalmic visits per patient during the study period. The median age at presentation was 14.4 years (IQR, 10.2-18.8; range, 1 month-36 years) (Table 1). The median BCVA (logMAR) at presentation was 0 in both eyes, equating to 20/20 in Snellen. About half of the patients (53.5%) were male. Most patients were Black (74.9%), had Hgb SS disease (50.2%), and had no evidence of sickle cell retinopathy (77.6%) on initial examination. A total of 165 (65%) patients had at least 1 loss to follow-up event, with 66 (26%) having attended only 1 visit. These 66 patients did not follow-up after their initial visit despite documented plans for continued care. Of the patients (n = 99) who did follow-up eventually, the median duration of loss to follow-up was 17.2 months/523.5 days (IQR, 335.5-655).
Characteristics of the Study Sample.
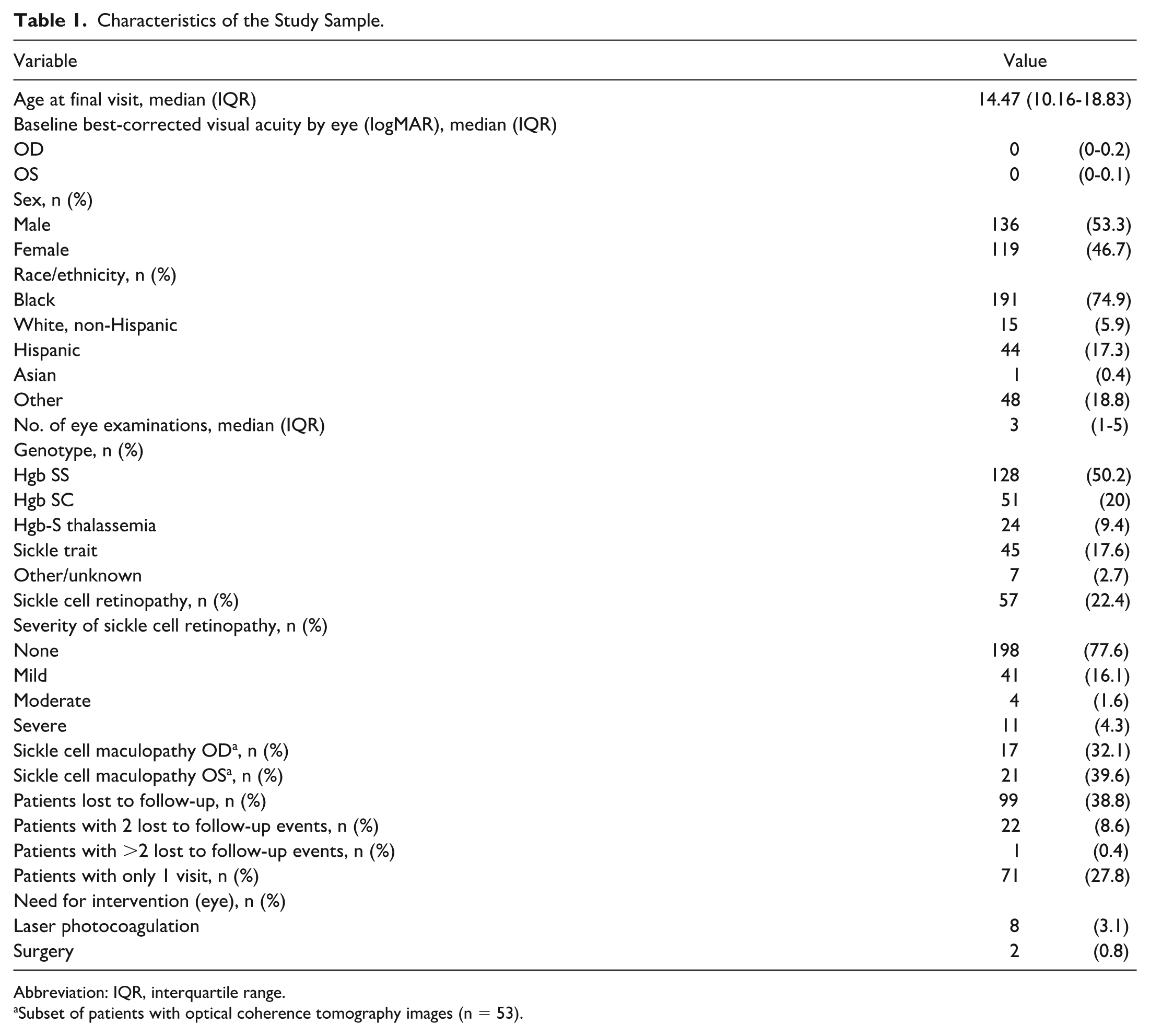
Abbreviation: IQR, interquartile range.
Subset of patients with optical coherence tomography images (n = 53).
Peripheral sickle cell retinopathy was documented in 61 eyes (24%), with assessment based on dilated fundus examination, widefield fundus photography, or adjunct imaging methods such as fluorescein angiography (FA, 28% of eyes) or OCT (21% of eyes). Sickle cell retinopathy was present in the right eye in 35 (21.9%) patients lost to follow-up and 21 (23.3%) patients in the non–loss to follow-up group (P = .410). In the left eye, sickle cell retinopathy was present in 40 (24.2%) patients lost to follow-up and 24 (26.7%) patients in the non–loss to follow-up group (P = .921).
No statistically significant differences in demographics existed between the 2 groups (Table 2). However, we did find that the loss to follow-up group attended fewer median eye examinations (3.6) compared with the non–loss to follow-up group (5.8) (P = .023). The rate of coexisting major systemic outcomes (eg, acute chest syndrome, hospitalization, vaso-occlusive crisis, stroke) was 13%. There were no statistically significant differences in the incidence of systemic outcomes other than occurrence of asthma (P < .001). Eleven patients in the loss to follow-up group (6.7%) had asthma vs 22 (24.4%) in the non–loss to follow-up group, 36 patients in the loss to follow-up group (21.8%) experienced acute chest syndrome vs 19 patients in the non–loss to follow-up group (21.1%) (P = .896), 27 patients in the loss to follow-up group (16.4%) experienced hospitalization vs 14 patients in the non–loss to follow-up group (15.6%) (P = .764), 20 patients in the loss to follow-up group (12.1%) experienced vaso-occlusive crisis vs 17 in the non–loss to follow-up group (18.9%) (P = .143), and 8 patients in the loss to follow-up group (4.8%) experienced stroke vs 6 patients in the non–loss to follow-up group (6.7%) (P = .542). In the loss to follow-up group, 4 eyes (2.4%) were treated with laser photocoagulation, and 1 eye (0.6%) required vitrectomy. In the non–loss to follow-up group, 4 eyes (4.4%) were treated with laser photocoagulation, and 1 eye (1.1%) required vitrectomy. Indications for the type of intervention were based on severity of sickle cell retinopathy and VA.
Comparison of Lost to Follow-Up and Non–Lost to Follow-Up Groups.
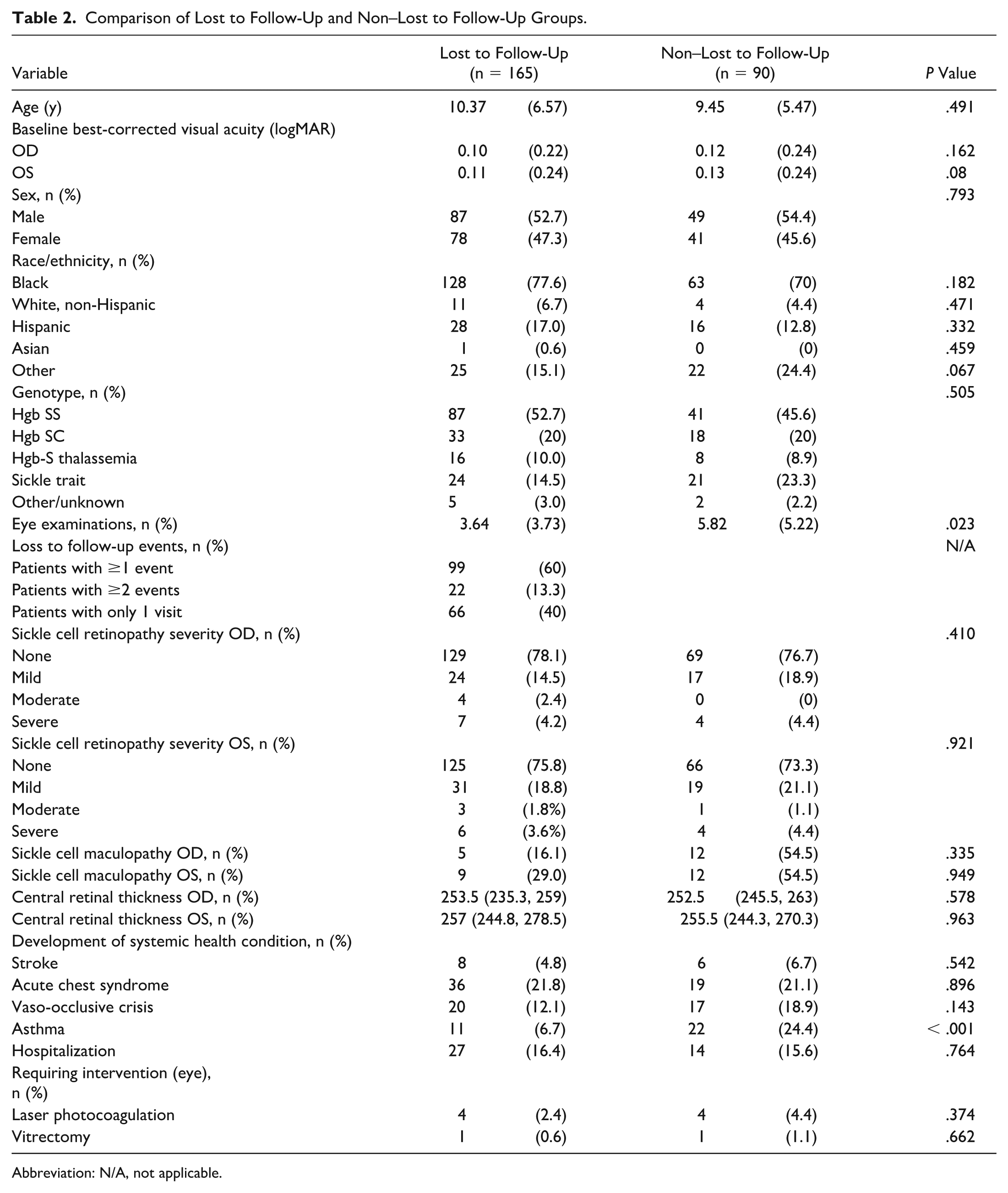
Abbreviation: N/A, not applicable.
A subset of patients (n = 31, 19% loss to follow-up; n = 22, 24% non–loss to follow-up) had OCT images. Sickle cell maculopathy was detected in the right eye in 5 patients (16.1%) in the loss to follow-up group and 12 patients (54.5%) in the non–loss to follow-up group (P = .335). Sickle cell maculopathy was found in the left eye in 9 patients (29%) in the loss to follow-up group and 12 patients (54.5%) in the non–loss to follow-up group (P = .949). Figure 1 describes a patient who presented at age 14 and was lost to follow-up for 3 years. Initial FA of the left eye in 2022 (Figure 1A) shows an avascular retina with mild late leakage. In 2025, when the patient returned, FA of the same eye demonstrated significant progression of NV temporally with traction. At this point, the decision was made to laser the avascular retina to facilitate regression of the NV, decreasing the incidence of VH. 7 The patient was then followed for 4 months after laser, with no further complications.
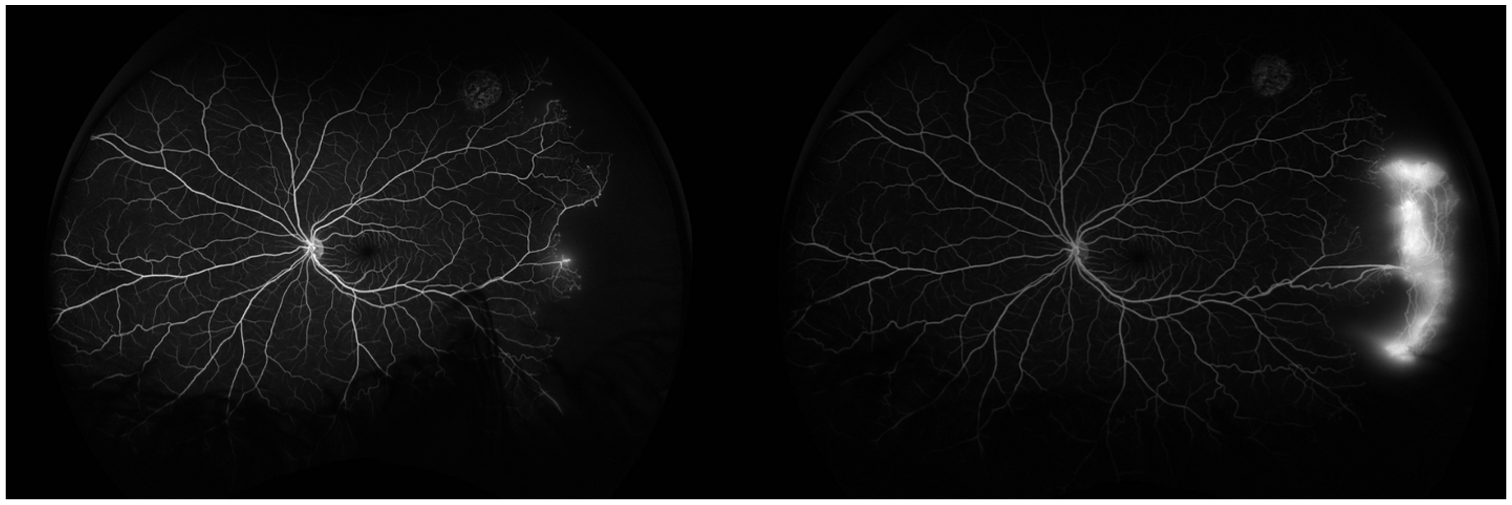
(A) Fluorescein angiography of the left eye in 2022. (B) Fluorescein angiography of the left eye in 2025, demonstrating temporal retina neovascularization.
Conclusions
In this study cohort, over half (65%) of the pediatric patients with sickle cell disease, many of whom had retinopathy or maculopathy, were lost to follow-up. Our findings emphasize that compliance is a major issue in this patient population, highlighting the need for reinforced care delivery and improved retention rates. We found a significant difference in the median eye examinations over the study period, with the loss to follow-up group attending fewer than the non–loss to follow-up group. The high rate of loss to follow-up may relate to the nature of visits for monitoring seemingly asymptomatic disease. A subset of patients had medication data (n = 217); of these, 7 were on hydroxyurea (3%). Few patients being on disease-modifying therapy may suggest that the patient population had relatively less severe sickle cell disease and felt less of a need for close follow-up.
One important consideration to keep in mind is that pediatric patients with sickle cell retinopathy, especially the proliferative form, may be completely visually asymptomatic. In some cases, peripheral NV may auto-infarct without treatment. The follow-up schedule should be as recommended, although parents/caregivers should understand that patients with proliferative sickle cell retinopathy may have no symptoms until progression from Goldberg stage III to Goldberg stage IV.
Compared with the general population, patients with sickle cell disease face many barriers that hinder medical retention. 3 It is well known that race, socioeconomic status, and health literacy can adversely affect follow-up rates in patients with sickle cell disease, often resulting in delayed care and increased complications.8–10 In a previous study, Wong et al 11 found 46% of adults with sickle cell disease were lost to follow-up at least once during the 10-year study period. The rate in our study was higher, at 65%. Wong et al also found that patients with sickle cell retinopathy who are lost to follow-up had poorer visual outcomes, and patients with nonproliferative sickle cell retinopathy more often required laser photocoagulation, with significantly worse final VA. In another study of adults with sickle cell disease, about 50% received at least 1 dilated fundoscopic examination during the study period, with 40% of patients without existing retinopathy screened. 12 In the current study, high rates of loss to follow-up may be attributed to the requirement of multiple subspecialty clinic visits. It is well known that medical appointments are not attended to the same degree as surgery-related visits, whether preoperative or postoperative. 13
Compared with similar proliferative ophthalmologic conditions, particularly diabetes, the loss to follow-up rates in our study are higher in sickle cell disease. A study by Suresh et al 14 found a loss to follow-up rate of 54.4% among patients with diabetic retinopathy (DR). Our study adopted a similar definition, with a time interval of 6 months.
Loss to follow-up in ophthalmic care for pediatric sickle cell disease has not been previously studied; however, data regarding patients with sickle cell disease and follow-up in other medical specialties do exist. At 1 academic center, more than one-third of children with sickle cell disease did not have yearly follow-ups in the first 5 years. 15 Outside of the United States, a 35% loss to follow-up rate was found in Tanzania and 28% in a tribal area of western India.16,17 The rate in our study is higher than those found previously, likely attributed to the referral aspect of appointments. 3 Patients were referred by their hematologist for evaluation of potential retinopathy; once the initial screening occurred, it was possible that patients felt that their hematologist could address minor ocular concerns, or they could be referred again later if new concerns arose. Another contributing factor is that pediatric patients are not independent and rely on caregivers, who may have a multitude of responsibilities and challenges.
Retention in the health system is particularly critical for sickle cell retinopathy due to its progression, which is often insidious. Subclinical changes may not be detected by conventional VA testing but rather by methods only available at ophthalmology appointments. Retinal manifestations in sickle cell disease sometimes precede those in other organs. 18 Vision loss from proliferative sickle cell retinopathy can be a late finding often associated with complications, including NV, VH, and tractional retinal detachment, further reinforcing the need for routine ophthalmic visits. Anecdotally, many of the patients presenting to our practice in young adulthood with tractional detachment or hemorrhage requiring vitrectomy were either lost to follow-up or did not have previous care. Throughout childhood and beyond, vision preservation is essential to ensure work productivity, quality of life, and overall health prognosis. 19 One option to consider in patients with proliferative sickle cell retinopathy is prophylactic laser photocoagulation, but further study is required. As we observed a high rate of loss to follow-up, further investigation may be required to determine whether there is a role for prophylactic laser treatment of large areas of avascular retina with high-risk features. Potential support can be made from the proliferative DR literature, in which loss to follow-up events not requiring laser have been associated with more negative outcomes. 20 Further long-term investigation is warranted to assess the number needed to treat, which may be large. However, this may be challenging due to population migration and the variable timing of complications. Improving the transition of care from pediatric to adult providers may also be important, as one study has demonstrated a link between a transition program and reduced loss to follow-up rate. 21
This study has several strengths. In a large cohort, we present findings related to both systemic and retinal outcomes over a decade, a longer time frame than previously explored. Loss to follow-up among pediatric patients with sickle cell disease has been underexplored, especially for ophthalmic care. Our definition of loss of follow-up (6 months) was established based on other studies to enable comparison of rates. Highlighting this issue may compel consideration of enhanced care and prophylaxis options for this patient population.
This study has some limitations. Only a subset of patients in this cohort had retinal imaging and data. Loss to follow-up was determined solely by ophthalmic visit notes. It is also unclear whether patients who were lost to follow-up were seen at a different practice, leading to absence of follow-up notes in their charts and overestimation of the loss to follow-up rate. Rates of surgical and laser procedures were derived from the institution’s records and may reflect interventions planned or performed either at the initial visit or during subsequent encounters. However, because some patients never returned for follow-up and others may have received care elsewhere, these comparisons should be interpreted with caution. Although patients do receive reminders about upcoming appointments, a major limitation of this study is the limited support to promote follow-up among these patients. Our study center does offer social work services, but we did not track the proportion of patients who were in touch with a social worker. Improved staffing, such as with a community health worker or integration of a hematologist in the ophthalmology clinic, to eventually form a multidisciplinary clinic, would support patients and their families more comprehensively as they face multiple barriers inside and outside of the clinic.
In conclusion, this study found a high rate (65%) of loss to follow-up among pediatric patients with sickle cell disease, including many patients with retinopathy and maculopathy. Because sickle cell disease requires a multidisciplinary team, it is essential for hematologists and pediatricians to emphasize ophthalmologic follow-up as a part of long-term care planning.
Footnotes
Acknowledgements
The authors would like to thank the patients and their families for allowing data collection.
Ethical Approval
The study was determined to be exempt by the Boston Children’s Hospital Institutional Review Board, with the requirement for informed consent waived due to the retrospective nature. This study was conducted in accordance with the Declaration of Helsinki. The collection and evaluation of patient health information was performed in a Health Insurance Portability and Accountability Act-compliant manner.
Statement of Informed Consent
Not required due to the retrospective nature of the study.
Patient consent
Not applicable for this study.
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Dr. Patel is a consultant for Alcon, Alimera, Allergan, Apellis, Atheneum, Biogen, DORC, Eye Point, Genentech, Gerson Lehrman Group, Inc, Guidepoint, Lifesciences, Regeneron, and Regenx Bio. None of the other authors declared potential conflicts of interest with respect to the research, authorship, and/or publication of the article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Dr. Patel is supported by the Retina Innovation Fund, Massachusetts Eye and Ear, the Simouran Family, and St. Vincent de Paul Foundation. The funding organizations had no role in the design or conduct of this research. Drs. Altamirano, Gonzalez, and Sorour received funding from the Children’s Hospital Ophthalmology Foundation. The remaining authors received no financial support for the research, authorship, and/or publication of this article.
Data Availability
Data will be made available upon request.