
Abstract
Background:
Ubrogepant is a novel, oral calcitonin gene–related peptide receptor antagonist for acute treatment of migraine. This study evaluated potential drug–drug interactions between ubrogepant and an oral contraceptive containing ethinyl estradiol (EE) and norgestimate (NGM).
Methods:
This open-label, single-center, two-period, fixed-sequence study enrolled healthy, postmenopausal or oophorectomized, adult women. In period 1, participants received a single oral dose of EE 0.035 mg/NGM 0.25 mg (EE-NGM) followed by a 7-day washout. In period 2, participants received oral ubrogepant 50 mg daily on days 1–14; single-dose EE-NGM was coadministered with ubrogepant on day 10. Pharmacokinetic parameters for plasma EE and norelgestromin (NGMN) were compared with and without ubrogepant.
Results:
Twenty-two participants aged 46–66 years were enrolled; 21 completed the study. Geometric mean ratios and 90% confidence intervals for the comparison of EE-NGM + ubrogepant to EE-NGM alone were contained within 0.80 and 1.25 for area under the plasma drug concentration–time curve (AUC) from time zero to infinity (AUC0–∞; 0.96 [0.91, 1.01]) and C max (0.91 [0.82, 1.004]) of NGMN and AUC0–∞ (0.97 [0.93, 1.01]) of EE, but not C max of EE (0.74 [0.69, 0.79]). Median t max of EE was delayed following EE-NGM + ubrogepant (3.0 h) versus EE-NGM alone (median of 1.5 h), whereas median t max of NGMN was unchanged (1.5 h). Geometric mean apparent terminal half-life (t ½) was similar with and without ubrogepant for EE (23 vs. 21 h) and NGMN (36 h both conditions). All ubrogepant-related adverse events were mild or moderate.
Conclusion:
Ubrogepant did not demonstrate potential for clinically meaningful drug–drug interactions with an EE-NGM oral contraceptive.
Trial registration:
Not applicable (phase 1 trial)
Keywords
Introduction
The prevalence of migraine is two to three times higher among women than men, 1 –5 with the incidence peaking in women during their reproductive years. 5,6 Migraine is also associated with greater disability among women than men, as shown by higher rates of global years lived with disability in the Global Burden of Diseases, Injuries, and Risk Factors Study 2016. 7 Currently available pharmacologic options for the acute treatment of migraine do not provide optimal treatment for many patients because of limited effectiveness, poor tolerability, or contraindications to their use. 8 –10
Inhibition of calcitonin gene–related peptide (CGRP) has emerged as a targeted approach for the treatment of migraine. 11,12 Three monoclonal antibodies to CGRP or the CGRP receptor are approved in the United States for the preventive treatment of migraine in adults, 13 –15 and a number of small-molecule CGRP receptor antagonists (i.e. “gepants”) have been explored for the acute treatment and for the prevention of migraine. 16 The CGRP receptor antagonists appear to lack vasoconstrictive effects, 17 –20 suggesting the potential for an improved safety and tolerability profile compared with the vasoactive triptans and ergotamine derivatives, which are the current standard of care for acute treatment of migraine.
Ubrogepant is a novel, small-molecule, oral CGRP receptor antagonist approved for the acute treatment of migraine with or without aura in adults. 21 The efficacy and safety of ubrogepant for the acute treatment of migraine was evaluated in one phase 2b 22 and two phase 3 clinical trials (ACHIEVE I and II). 23,24 In both phase 3 trials, ubrogepant was well tolerated and demonstrated clinically meaningful improvements in pain and most bothersome migraine-associated symptom. 23,24 Ubrogepant is rapidly absorbed after oral administration, with a median time to maximum plasma drug concentration (t max) of 1.7 h and a terminal elimination half-life (t ½) of 4.4 h for the 100-mg dose. 25 Ubrogepant has not been shown to induce or inhibit cytochrome P450 2D6 (CYP2D6), cytochrome P450 3A4 (CYP3A4), or p-glycoprotein at clinically relevant concentrations. Ubrogepant is metabolized primarily by CYP3A4, and the glucuronide conjugates (M15 and M20) of its oxidative metabolites (M9 and M8) are the primary circulating metabolites in human plasma. 26 A population pharmacokinetic study of data pooled from phase 1, 2, and 3 trials of ubrogepant determined that no dosage adjustments of ubrogepant are needed owing to differences in sex, race, weight, and age, or owing to mild or moderate hepatic or renal impairment, food consumption, or concomitant use with mild CYP3A4 inhibitors. 26
The use of oral contraceptives is common among women with migraine. A retrospective cohort study of 949 female migraine patients (aged 15–45 years) reported that 51.9% of the patients were taking an oral contraceptive. 27 Given the high prevalence of migraine among women, especially those of childbearing age, 5,6 and the high rate of oral contraceptive use in this population, 27 it is likely that ubrogepant will be used concurrently with oral contraceptives. Therefore, it is important to evaluate the potential for clinically meaningful drug–drug interactions between ubrogepant and oral contraceptives.
Combination oral contraceptives (COCs) typically include two components, an estrogen and a progestin. Ethinyl estradiol (EE), the estrogen component of most COCs, is metabolized by hydroxylation via cytochrome P450 isoenzymes CYP3A4 and CYP2C9 and by conjugation via sulfation and glucuronidation via UDP-glucuronosyltransferase 1A1. 28,29 Norgestimate (NGM), a commonly used progestin, is extensively metabolized by first-pass mechanisms in the gastrointestinal tract and/or liver. 29 Norelgestromin (NGMN), the primary active metabolite of NGM, is subsequently metabolized in the liver to norgestrel, also an active metabolite, and to various hydroxylated and conjugated metabolites. 29 Although NGMN and its metabolites have been shown to inhibit P450 enzymes in human liver microsomes, the in vivo concentrations of NGMN and its metabolites under the recommended dosing regimen are relatively low compared with its inhibitory constant (K i). 29 The COC labels also state that they may inhibit the metabolism of some medications (e.g. cyclosporine), resulting in an increase in their plasma concentrations, and that they may decrease plasma concentrations of other medications (e.g. acetaminophen, morphine, and lamotrigine). Thus, it is important to study the potential for clinically meaningful interactions between COCs and ubrogepant.
The primary objective of this study was to evaluate the effect of multiple-dose administration of ubrogepant 50 mg on the pharmacokinetics of the components of a monophasic COC (EE plus NGM). The effect of EE and NGM on the pharmacokinetics of ubrogepant and safety was also evaluated.
Methods
Study design
This was an open-label, two-period, fixed-sequence, single-site study investigating the effect of multiple doses of ubrogepant on the single-dose pharmacokinetics of Ortho-Cyclen® (Janssen Pharmaceuticals, Inc., Titusville, New Jersey, USA), a monophasic combination of EE and NGM oral contraceptive, in healthy women. In period 1, participants received a single oral tablet of EE 0.035 mg/NGM 0.25 mg (EE-NGM) on day 1. There was a 7-day washout period between the single dose of EE-NGM in period 1 and the beginning of period 2. In period 2, participants received ubrogepant 50 mg/day (five 10-mg tablets) on days 1–14, in addition to a single dose of EE-NGM coadministered with ubrogepant on day 10. Liver safety parameters (alanine aminotransferase, aspartate aminotransferase, alkaline phosphatase, and total bilirubin) were measured at follow-up visits at 28, 42, and 70 days after the first dose of ubrogepant in period 2. The study duration from screening to last follow-up was approximately 13 weeks.
This study was conducted in accordance with the ethical principles of the Declaration of Helsinki and the International Council for Harmonisation’s Guideline for Good Clinical Practice. An institutional review board, Chesapeake Research Review, Inc., Columbia, MD, approved the study protocol (Merck study MK-1602 protocol 005). All participants provided written informed consent. The trial was conducted at one center in Arizona, USA.
Participants
Eligible participants were healthy, postmenopausal or bilaterally oophorectomized, adult women (estradiol level <35 pg/mL) with normal alanine aminotransferase levels and no use of nicotine-containing products for at least 3 months. Participants were selected, recruited, and enrolled at the study center by the contract research organization (Celerion, Phoenix, Arizona, USA). This low endogenous estrogen population was selected to avoid the variable levels of sex hormone-binding proteins that can occur with cyclic endogenous estrogen levels in women of childbearing potential and alter the pharmacokinetics of hormonal contraceptives. Key exclusion criteria were an estimated creatinine clearance ≤60 mL/min and a history of stroke, chronic seizures, or major neurological disorder. Participants could not have used, within 14 days of dosing or 5 drug t ½ (whichever was longer), any compounds known to be significant inducers or inhibitors of CYP450 isozymes, P-glycoprotein, or breast cancer resistance protein; prescription or nonprescription medications, herbal remedies, or grapefruit-containing products; sex hormone-binding globulin agents (within 4 weeks) or hormone replacement therapy (within 6 months); or excessive alcohol or caffeine.
Procedures
All doses of ubrogepant and EE-NGM were administered in the clinic and taken with 240 mL of water after an overnight fast of at least 8 h. Water was restricted 1 h before and 1 h after each dose. In period 1, participants were confined to the clinic from the evening before dosing until 24 h post dose. In period 2, participants were confined to the clinic from the evening before dosing until 24 h post dose on days 1 and 10.
Blood sample collection
Blood samples for determining plasma concentrations of EE and NGMN were collected into vacutainer tubes containing potassium oxalate and sodium fluoride before dosing and at specified time points up to 96 h post dose. Blood samples for the assessment of ubrogepant pharmacokinetics were collected into vacutainer tubes containing K2 ethylenediaminetetraacetic acid before and up to 24 h post dose on days 1 and 10 of period 2. Blood samples were centrifuged to extract plasma according to standard procedures and transferred to either PPD, LP (Richmond, Virginia, USA), or WuXi AppTech (Shanghai, China) for analysis of EE/NGMN or ubrogepant, respectively.
Bioanalytical assessments
Ethinyl estradiol
Plasma samples for analysis of EE were analyzed using high-performance liquid chromatography with tandem mass spectrometry detection using 17α-ethinylestradiol-2, 4, 16, 16-d4 as an internal standard. Samples were extracted with hexane/ethyl acetate and chromatographed using an analytical column (DS-AQ, 2 × 100 mm, 3 μm) and a trapping column (Pursuit PFP, 2 × 50 mm, 5 μm). The lower limit of quantitation for EE was 2.00 pg/mL, with a linear calibration range from 2.00 pg/mL to 500 pg/mL.
Norelgestromin
Plasma samples collected for assay of NGMN were analyzed using high-performance liquid chromatography with tandem mass spectrometry detection and desacetylnorgestimate-d5 as the internal standard. Analytes were isolated by liquid–liquid extraction with ethyl acetate/hexane and separated using an analytical column (Synergi™ 4 μm, Polar-RP 80 Å, 2 × 150 mm). The lower limit of quantitation for NGMN was 0.0200 ng/mL, with a linear calibration range from 0.0200 ng/mL to 10.0 ng/mL.
Ubrogepant
Following liquid–liquid extraction of plasma samples with methyl tert-butyl ether, ubrogepant and its stable isotope-labeled internal standard, [D3]-ubrogepant, were chromatographed using reverse-phase high-performance liquid chromatography on a Waters Atlantis dc18 column. 30 Analytes were detected using tandem mass spectrometry (API 4000; Applied Biosystems, Sciex, Framingham, Massachusetts, USA) employing a turbo ionspray interface operated in the positive ionization mode. The multiple reaction monitoring ion transition of m/z 550 → 264 was used for ubrogepant and m/z 553 → 267 was used for the internal standard. The lower limit of quantitation for ubrogepant was 0.182 nM, with a linear calibration range from 0.182 nM to 181.96 nM.
Pharmacokinetic assessments
Area under the plasma drug concentration–time curve (AUC) from time zero to infinity (AUC0–∞), maximum plasma drug concentration (C max), t max, and apparent terminal t ½ of EE and NGMN were calculated after administration of EE-NGM alone and after coadministration with multiple-dose ubrogepant. The AUC from time 0 to 24 h (AUC0–24 h), C max, t max, and t ½ of ubrogepant were calculated after administration of ubrogepant alone on day 1 and after coadministration with EE-NGM on day 10 in period 2. All standard noncompartmental pharmacokinetic parameters were calculated using WinNonlin® Professional (version 5.2) (Pharsight Corporation, Mountain View, California, USA).
Safety assessments
Safety and tolerability were monitored by physical examination, vital signs (blood pressure, heart rate), routine laboratory tests (hematology, blood chemistry [fasted], urinalysis), and 12-lead electrocardiogram (ECG) throughout the study at predefined time points. Participants were monitored for adverse events throughout the study; adverse events were coded according to the Medical Dictionary of Regulatory Activities version 15.1 as modified by Merck & Co., Inc.
Statistical analyses
All statistical analyses were conducted using Statistical Analysis Software (SAS/STAT) (SAS Institute Inc., Cary, North Carolina, USA). Power calculations used the following assumptions for within-subject standard deviations obtained from previous studies of EE-NGM: for EE, 0.133 (ln-(ng·h/mL)) for ln-AUC0–∞ and 0.193 (ln-(ng/mL)) for ln-C max; for NGMN, 0.082 (ln-(ng·h/mL)) for ln-AUC0–∞ and 0.193 (ln-(ng/mL)) for ln-C max. Assuming 20 participants with available pharmacokinetic data, nonnegative correlation among the four test statistics (EE: AUC0–∞ and C max and NGMN: AUC0–∞ and C max), and true geometric mean ratios of 1.00 for all four parameters, there was at least approximately 89% probability that all four 90% CIs (confidence intervals) would fall within the 0.80–1.25 target interval simultaneously (α = 0.05).
Pharmacokinetics
Individual AUC0–∞ and C max of EE and NGMN values were natural log (ln)-transformed prior to analysis and evaluated separately using a linear mixed-effects model, with a fixed-effect term for treatment. An unstructured covariance matrix was used to allow for unequal treatment variances and to model the correlation between the two treatment measurements within each participant. The Kenward and Rogers’ method was used to calculate the denominator degrees of freedom for the fixed effects. A two sided 90% CI for the true mean geometric mean ratio (EE-NGM + ubrogepant/EE-NGM alone) was calculated for each parameter. If all four 90% CIs for the true geometric mean ratios were within 0.80 and 1.25, then the primary hypothesis, namely that the plasma AUC0–∞ and C max of EE and NGMN after coadministration of EE-NGM with multiple-dose administration of ubrogepant are not substantially altered compared with administration of a single dose of EE-NGM alone, would be supported. Other pharmacokinetic parameters were summarized using descriptive statistics.
Results
Participant disposition and demographics
From May 17, 2012 to August 1, 2012, the study enrolled 22 healthy adult postmenopausal or oophorectomized women (safety population); 21 participants completed the study (Figure 1). One participant was discontinued by the investigator on day 8 before dosing because of an adverse event of vomiting. Pharmacokinetic data collected before this participant discontinued were included in analyses. Demographics and baseline characteristics are summarized in Table 1.

Participant disposition. aOne participant discontinued on day 8 of period 2. This participant received a single oral dose of EE 0.035 mg/NGM 0.25 mg (EE-NGM) on day 1 of period 1 and oral doses of ubrogepant 50 mg once daily on days 1–7 of period 2. The participant did not receive the protocol specified oral doses of ubrogepant 50 mg once daily on days 8–14 and the single oral dose of EE-NGM on day 10 of period 2. Therefore, the plasma pharmacokinetic parameters of EE, NGMN, and ubrogepant following EE-NGM + ubrogepant on day 10 of period 2 could not be calculated for this participant. EE: ethinyl estradiol; NGM: norgestimate; NGMN: norelgestromin.
Participant demographics and baseline characteristics.
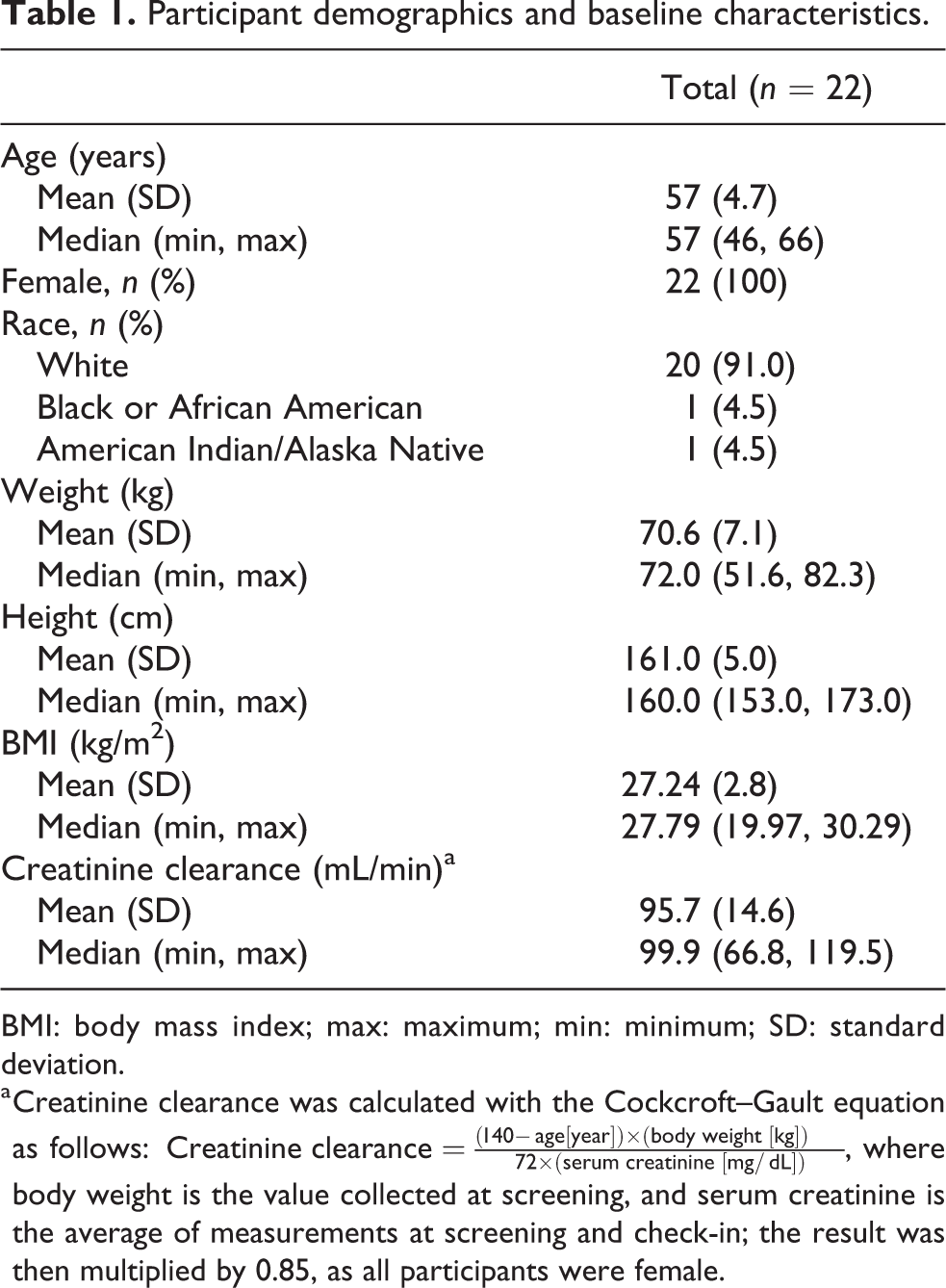
BMI: body mass index; max: maximum; min: minimum; SD: standard deviation.
a Creatinine clearance was calculated with the Cockcroft–Gault equation as follows:
Pharmacokinetics of EE and NGMN
Concentration–time profiles for plasma EE and NGMN following the administration of a single dose of EE-NGM alone or coadministered during daily dosing of ubrogepant are shown in Figure 2 (a) and (b). Pharmacokinetic parameters for EE and NGMN under both conditions are summarized in Table 2. The geometric mean ratios and 90% CIs for the comparison of EE-NGM + ubrogepant to EE-NGM alone were contained within 0.80 and 1.25 for the AUC0–∞ (0.96 [0.91, 1.01]) and C max (0.91 [0.82, 1.004]) of NGMN and AUC0–∞ (0.97 [0.93, 1.01]) of EE, but not the C max of EE (0.74 [0.69, 0.79]). The t max of EE was delayed following coadministration of EE-NGM and ubrogepant (median of 3.0 h) compared with EE-NGM alone (median of 1.5 h), whereas the t max of NGMN was unchanged (median of 1.5 h). The apparent terminal t ½ was similar between treatments for plasma EE (geometric mean of 23 h vs. 21 h for coadministration vs. alone) and plasma NGMN (geometric mean of 36 h for both coadministration and alone).
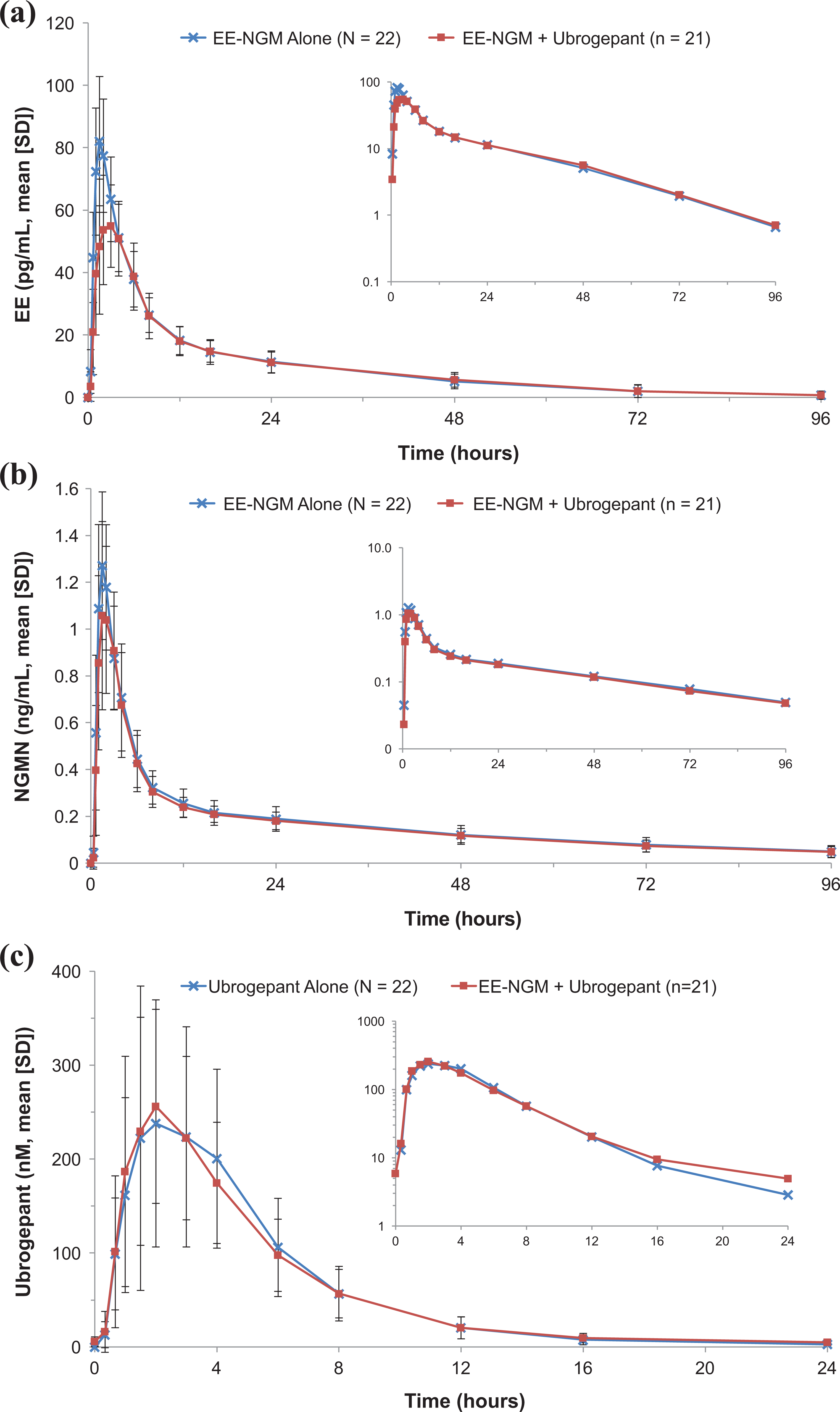
Plasma concentration versus time profiles for (a) EE and (b) NGMN following a single dose of EE 0.035 mg/NGM 0.25 mg (EE-NGM) administered alone or with multiple-dose administration of ubrogepant 50 mg and for (c) ubrogepant following a single dose of ubrogepant 50 mg administered alone and following a single dose of EE 0.035 mg/NGM 0.25 mg administered with multiple-dose ubrogepant 50 mg. The graphs inset within each panel are shown on a semi-log scale. EE: ethinyl estradiol; NGM: norgestimate; NGMN: norelgestromin; SD: standard deviation.
Plasma pharmacokinetic parameters of EE and NGMN following a single dose of EE 0.035 mg/NGM 0.25 mg (EE-NGM) alone or with multiple-dose administration of ubrogepant 50 mg.
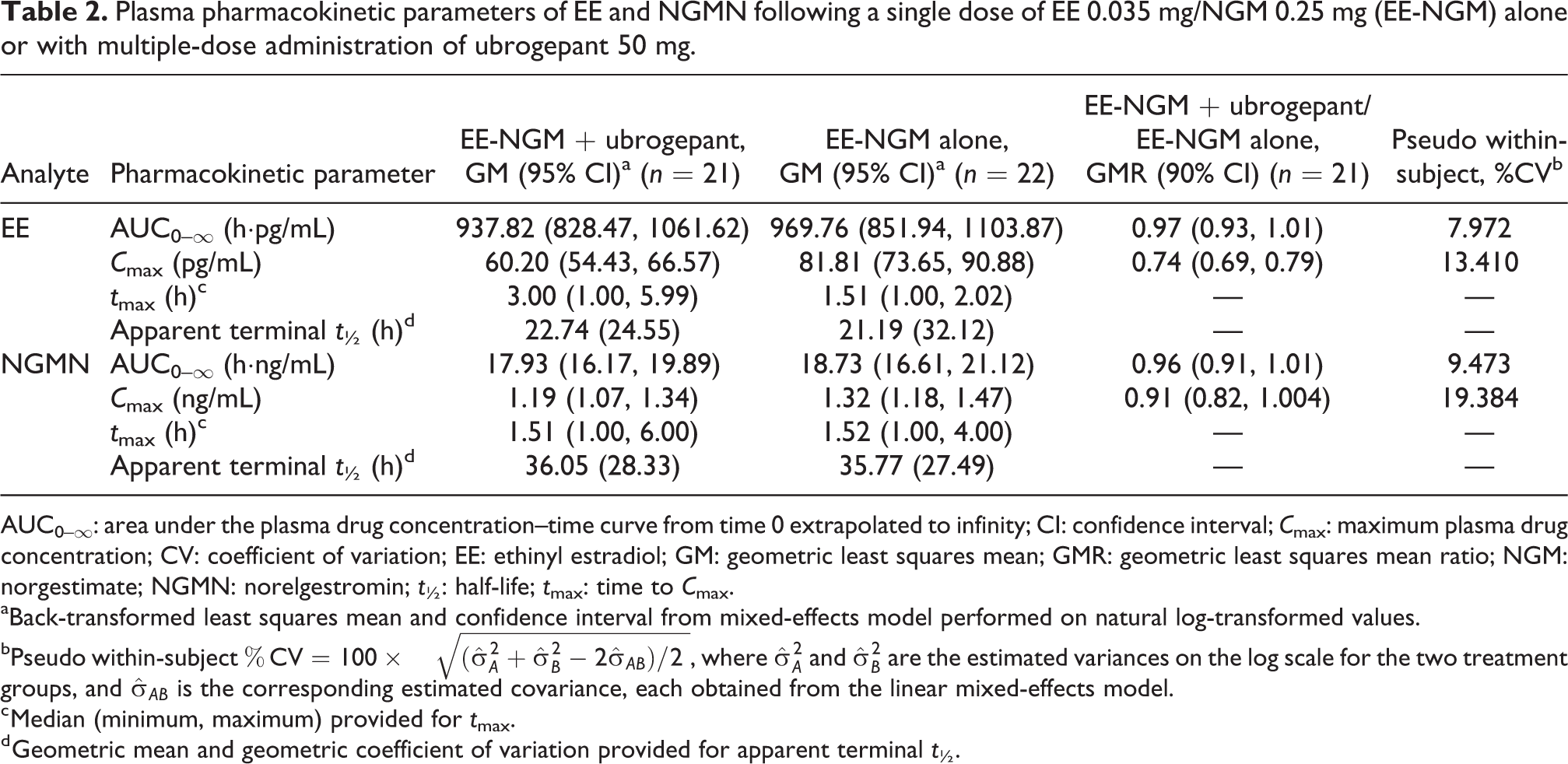
AUC0–∞: area under the plasma drug concentration–time curve from time 0 extrapolated to infinity; CI: confidence interval; C max: maximum plasma drug concentration; CV: coefficient of variation; EE: ethinyl estradiol; GM: geometric least squares mean; GMR: geometric least squares mean ratio; NGM: norgestimate; NGMN: norelgestromin; t ½: half-life; t max: time to C max.
aBack-transformed least squares mean and confidence interval from mixed-effects model performed on natural log-transformed values.
bPseudo within-subject
c Median (minimum, maximum) provided for t max.
d Geometric mean and geometric coefficient of variation provided for apparent terminal t ½.
Pharmacokinetics of ubrogepant
Concentration–time profiles for plasma ubrogepant following a single dose of ubrogepant 50 mg alone (day 1) and following multiple-dose administration of ubrogepant 50 mg with a single dose of EE-NGM (day 10) are shown in Figure 2(c). Pharmacokinetic parameters for ubrogepant are summarized in Table 3. The geometric mean ratios (EE-NGM + ubrogepant/ubrogepant alone) for ubrogepant AUC0–24 h and C max approximated to 1 (geometric mean ratios [90% CIs] of 1.02 [0.96, 1.08] and 0.97 [0.85, 1.11], respectively), indicating that the pharmacokinetic profile of ubrogepant following a single dose is similar to that observed after 10 days of daily dosing when coadministered with a single dose of EE-NGM.
Pharmacokinetic parameters for plasma concentrations of ubrogepant following a single dose of ubrogepant 50 mg alone (day 1) and following a single dose of EE 0.035 mg/NGM 0.25 mg (EE-NGM) with multiple-dose administration of ubrogepant 50 mg (day 10).

AUC0–24 h: area under the plasma drug concentration–time curve from time 0 h to 24 h; C max: maximum plasma drug concentration; EE: ethinyl estradiol; GCV: geometric coefficient of variation; GM: geometric least squares mean; NGM: norgestimate; t ½: half-life; t max: time to C max.
a Multiple oral doses of ubrogepant 50 mg once daily (days 1–14), and a single oral dose of EE-NGM with ubrogepant on day 10.
b Geometric coefficient of variation is calculated in the natural log scale with the equation:
Safety
No deaths, serious adverse events, or clinically significant laboratory findings occurred during the study. One participant was discontinued because of an adverse event of mild vomiting that occurred before dosing on day 8 and was considered related to ubrogepant alone by the investigator. This participant also reported adverse events of hypogeusia, decreased appetite, dizziness, abdominal distention, anxiety, feeling cold, feeling hot, vomiting, nausea, tremor, and headache between days 3 and 7. On the morning of day 8, the participant reported two episodes of vomiting prior to dosing. The vomiting resolved after discontinuation of ubrogepant.
Nine participants reported a total of 26 adverse events (Table 4). Twenty-one adverse events, all mild or moderate, were considered by the investigator to be related to EE-NGM and/or ubrogepant. Of these, 18 were considered to be related to ubrogepant alone; all were mild intensity. Only one adverse event that was related to ubrogepant alone occurred in more than one participant (feeling hot, n = 2). One adverse event that was considered to be related to both ubrogepant and EE-NGM occurred in more than one participant (paresthesia, n = 2). A single adverse event related to EE-NGM alone was reported (headache, n = 1). All adverse events resolved by the end of the study.
Adverse events reported by at least two participants overall.a

EE: ethinyl estradiol; NGM: norgestimate.
a Although a participant may have had two or more clinical adverse experiences, the participant is counted only once within a category. The same participant may appear in different categories.
Adverse experience terms are from the Medical Dictionary of Regulatory Activities version 15.0 as modified by Merck & Co., Inc.
b Day 1 of period 1 (reference).
c Days 1–9 of period 2 (through predose day 10).
d Day 10 concomitant dosing through end of period 2.
There were no consistent treatment-related changes in laboratory, vital sign, or ECG safety parameters. Serum transaminases and bilirubin were below twice the upper limit of normal in all participants throughout the study.
Discussion
This study did not demonstrate any clinically significant drug–drug interactions between ubrogepant and a commonly used monophasic oral contraceptive (EE 0.035 mg/NGM 0.25 mg) in healthy women. Exposure to NGM and EE (i.e. plasma AUC0–∞ and C max) following single-dose EE-NGM was similar when administered alone and with ubrogepant after 10 days of daily ubrogepant dosing. Although the C max of EE was approximately 26% lower following ubrogepant coadministration than EE-NGM alone, a decrease of this magnitude in the absence of an effect on AUC0–∞ is not considered clinically meaningful. 28
The primary mechanism of COCs, such as EE-NGM, is to lower the risk of becoming pregnant through suppression of ovulation. 31 The progesterone component (NGM in this case) is generally considered to be primarily responsible for contraceptive efficacy. 32 In addition, the estrogen component (EE in this instance) is believed to prevent breakthrough bleeding and other side effects, and doses of EE have been reduced because of the availability of the high antigonadotrophic activity of progestins. 32 Therefore, the small effect of ubrogepant on peak concentrations of EE is unlikely to impact the efficacy of EE, which is more likely to be related to overall exposure to the drug (i.e. AUC) rather than C max.
Although a number of drug classes have been shown to reduce exposure to EE and progestogen (AUC), 28 the likelihood of meaningful drug–drug interactions between ubrogepant and oral contraceptives was considered to be low because of how the compounds are metabolized and excreted. Both EE and NGM are metabolized in the liver by the CYP3A4 and CYP2C subfamilies. 28,33,34 Ubrogepant has not been shown to induce or inhibit CYP2D6, CYP3A4, or P-glycoprotein at clinically relevant concentrations (data on file, Allergan plc). The mechanism underlying the effects of ubrogepant on the C max and t max of EE is unclear. Although speculative, the effect may be mediated through substrate binding competition for CYP3A4, as both ubrogepant and EE are substrates of CYP3A4 and ubrogepant is present at much higher concentrations than EE.
The pharmacokinetic profile of ubrogepant was not affected by coadministration with a single dose of EE-NGM. The AUC0–24 h and C max of ubrogepant 50 mg were similar following a single dose alone and after multiple-dose coadministration with EE-NGM. In a series of randomized, double-blind, placebo-controlled studies in healthy volunteers, oral ubrogepant had a t max of approximately 1.5 h (range, 1–3 h) and a t ½ of approximately 5 h. No accumulation was noted after repeated once-daily dosing, and steady state was achieved within 2 days of dosing. 30 Given the short t ½ 22 and minimal accumulation of ubrogepant, exposure after the first dose is similar to that after steady state, making this a valid comparison for evaluating the effects of EE-NGM on ubrogepant pharmacokinetics.
Multiple-dose ubrogepant coadministered with a single dose of EE-NGM was safe and generally well tolerated. No serious clinical or serious laboratory adverse events, events of clinical interest, or deaths occurred, and the adverse event profile was generally consistent with that reported in larger trials. 22 –24
Sex differences in the CGRP system have been reported. One study showed that mean plasma concentrations of CGRP were significantly higher in women not taking oral contraceptives (n = 13; 50.9 pmol/L) than in men (n = 13; 25.7 pmol/L; p < 0.005). 35 Circulating levels of CGRP were even higher among women taking an oral contraceptive (n = 11; 76.5 pmol/L; p < 0.01 vs. women not taking an oral contraceptive). 35 Studies in animals and in humans have shown that fluctuations in estrogen levels modulate CGRP in the trigeminovascular system. 36 These increased levels of CGRP may contribute to the higher incidence of migraine in young women than men, but it does not appear to translate to sex differences in the efficacy of gepants. A pooled analysis of data from two phase 3 trials (ACHIEVE I and II) showed no differences between women and men in the efficacy of ubrogepant for reaching pain freedom and the absence of the most bothersome migraine-associated symptom at 2 h post dose (data on file, Allergan plc).
A potential limitation inherent in the design of this study is the possibility that some participants may not have taken all doses of ubrogepant, given that not all pill intakes were observed. Such potential deviations could, in theory, impact the observed EE/NGMN pharmacokinetics. Because ubrogepant has not been shown in vitro to be a time-dependent inhibitor or inducer of metabolic enzymes, and given its short t ½, 22 the impact of a missed dose of ubrogepant on the assessment of either interaction with EE-NGM or the pharmacokinetics of ubrogepant would be minimal.
While nearly all COCs contain EE, the progestin component varies widely. 37 The applicability of these study results extends to different dose levels of EE and NGM, which is one of the more commonly used progestin components in the United States. 37 However, the results may not be applicable to other progestins with potentially different biotransformation pathways.
Conclusions
Ubrogepant, an oral CGRP receptor antagonist for acute treatment of migraine, demonstrated no potential for clinically significant drug–drug interactions with components of a commonly used monophasic oral contraceptive. Coadministration of multiple 50 mg doses of ubrogepant with a single dose of EE 0.035 mg/NGM 0.25 mg was safe and generally well tolerated in healthy women.
Clinical implications
Ubrogepant is a small-molecule CGRP receptor antagonist for the acute treatment of migraine that is likely to be used concurrently with oral contraceptives in women. Ubrogepant demonstrated no potential for clinically significant drug–drug interactions with a commonly used oral contraceptive containing EE 0.035 mg and NGM 0.25 mg, suggesting that it may be used in combination with oral contraceptives.
Footnotes
Acknowledgements
Special thanks to Mireille Gerrits for the suggestion of population for the study. Medical writing and editorial support was provided by Peloton Advantage, LLC, an OPEN Health company, Parsippany, NJ, USA, and funded by Allergan plc, Dublin, Ireland. The results of this study were presented, in part, at the 71st Annual Meeting of the American Academy of Neurology, May 4–10, 2019, Philadelphia, PA, and at the 61st Annual Scientific Meeting of the American Headache Society, July 11–14, 2019, Philadelphia, PA.
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Chi-Chung Li, PhD, John Palcza, MS, Jialin Xu, PhD, Bob Thornton, MPH, Wendy Ankrom, PhD, and Eugene E Marcantonio, MD, PhD, were an employees of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA, and owned stock in Merck & Co., Inc., Kenilworth, NJ, USA at the time of this study. Abhijeet Jakate, PhD, is an employee and shareholder of Allergan plc, Madison, NJ, USA.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by Merck & Co., Inc., Kenilworth, NJ, USA (Merck Sharp & Dohme outside the United States and Canada).
Statement regarding accessibility of data
Data reported in this manuscript are available within the article and its supplementary materials.
Allergan may share de-identified patient-level data and/or study-level data, including protocols and clinical study reports, for phase 1 trials completed after 2008 that are registered on ClinicalTrials.gov or EudraCT. The indication studied in the trial must have regulatory approval in the United States and/or the European Union and the primary manuscript from the trial must be published prior to data sharing. To request access to the data, the researcher must sign a data use agreement. All shared data are to be used for noncommercial purposes only. More information can be found on ![]() .
.