
Abstract
Background
Rimegepant is an oral, small molecule calcitonin gene-related peptide receptor antagonist for acute treatment of migraine and migraine prevention.
Methods
This was a single-site, placebo-controlled, sequential, single and multiple ascending dose study in healthy males and females, aged 18–55 years, with no clinically significant medical history. The objectives were to assess the safety, tolerability, and pharmacokinetics of the oral capsule free-base formulation. Single oral doses of rimegepant from 25–1500 mg were evaluated in the single ascending dose phase, and 75–600 mg/day doses administered for 14 days were evaluated in the multiple ascending dose phase.
Results
No dose-related trends were observed in orthostatic systolic and diastolic blood pressure or heart rate after rimegepant administration. Rimegepant was rapidly absorbed with the median time of maximum observed plasma concentration from 1–3.5 hours. Rimegepant showed a more than dose-proportional increase in exposure from 25–1500 mg following a single dose and from 75–600 mg/day following multiple doses.
Conclusions
Rimegepant was safe and generally well tolerated at single oral doses up to 1500 mg and multiple doses up to 600 mg/day for 14 days in healthy participants in this study. Median terminal half-life ranged from 8–12 hours across the wide range of single doses studied.
Introduction
Migraine is a common, chronic, primary headache disorder associated with photophobia, phonophobia, nausea (1), and comorbidities (2,3). Migraine affects 1 in 6 persons in the United States (US) (2), 9% of the adult population in China (3), and in Europe, migraine prevalence is 11% (4). Patients with chronic migraine have increased healthcare resource utilization, including increased visits to general practitioners, psychiatrists, and neurologists (4).
Calcitonin gene-related peptide (CGRP) is an endogenous 37 amino acid peptide contained within pain signaling nociceptive afferents. Numerous studies have demonstrated that CGRP plays a key role in the pathophysiology of migraine (5–7). Anti-CGRP medications have demonstrated efficacy in the acute and preventive treatment of migraine (8).
Rimegepant (Nurtec ODT [orally disintegrating tablet], Biohaven Pharmaceuticals, New Haven, CT, USA) is an orally administered small molecule CGRP receptor antagonist approved by the US Food and Drug Administration (FDA) in 2020 for the acute treatment of migraine with or without aura in adults, and in 2021 for the preventive treatment of episodic migraine in adults. Marketing authorization for rimegepant (Vydura, Biohaven Pharmaceutical Ireland DAC, Dublin, Ireland) was granted by the European Commission in April 2022 for the acute treatment of migraine with or without aura in adults and the preventive treatment of episodic migraine in adults who have at least four migraine attacks per month. Rimegepant has demonstrated efficacy and safety in multiple randomized, placebo-controlled clinical trials (9–16).
Here, we report the results of the Phase 1 study of the safety, tolerability, and pharmacokinetics (PK) of rimegepant in healthy individuals.
Methods
Trial design and participants
This was a single-site, double-blind, placebo-controlled, sequential, single ascending dose (SAD), and multiple ascending dose (MAD) study of rimegepant in healthy participants in the US. Eligible participants were healthy males and females of non-childbearing potential, aged 18–55 years, with a body mass index of 18.0–32.0 kg/m2, and no clinically significant deviations from normal in medical history, physical examination, electrocardiogram (ECG), and clinical laboratory values. This study was conducted in accordance with Good Clinical Practice. The study protocol was approved by the Independent Investigational Review Board, Inc. (Plantation, FL) prior to initiation. Written informed consent was obtained from each participant prior to study start.
Treatment and procedures
The primary objective of the study was to assess the safety and tolerability of single and multiple ascending doses of the oral capsule free-base formulation of rimegepant. Secondary objectives were assessment of orthostatic systolic blood pressure (SBP) and diastolic blood pressure (DBP); ECG parameters including heart rate (HR) and PR, QRS, QT intervals, and QT corrected for HR (QTc) interval; maximum tolerated dose (MTD), and rimegepant PK. The effects of food, pH, and gender on rimegepant PK were also evaluated; however, results of these secondary analyses are beyond the scope of this manuscript.
The SAD phase evaluated seven sequential rimegepant dose panels of 25, 75, 150, 300, 600, 900, and 1500 mg administered after fasting for ≥10 hours. Study duration was ∼42 days/dose. Participants were randomized according to a computer-generated scheme to a single dose of rimegepant or placebo in a 3:1 ratio, stratified by gender.
Dosing in the MAD phase started after safety, tolerability, and PK data for all participants dosed through the 150 mg dose panel (SAD) were assessed and deemed safe by the Site Principal Investigator in consultation with the Sponsor’s Medical Monitor. Dose escalation did not occur until safety, tolerability, and laboratory data through Day 14 from the current dose level and Day 25 from all previous dose levels were reviewed and deemed safe.
Within each SAD and MAD panel, one female and one male were dosed (≥1 hour apart) with rimegepant or placebo. The remaining participants were dosed ≥30 minutes apart at ≥1 day (SAD) and ≥2 days (MAD) from the time the first two participants were dosed.
The MAD phase evaluated rimegepant doses of 75, 150, 300, 450, and 600 mg after fasting for ≥10 hours. Study duration was ∼62 days between dose escalations. Participants were randomized in the same manner as the SAD phase. Rimegepant or placebo was administered once daily (QD) for 14 days. In addition, 300 mg rimegepant was administered twice daily (BID), two hours apart for 14 days based on the tolerability of the 600 mg dose in the SAD phase.
Safety assessments
Safety assessments were based on reported adverse events (AE), clinical laboratory tests, ECGs, vital sign measurements, and physical examination. Participants were not discharged until the Investigator determined that any AE(s) or serious AE(s) (SAE[s]) had completely resolved or was not of clinical significance.
Clinical laboratory testing included hematology, serum chemistry, and urinalysis performed at a central/local laboratory. Blood and urine sampling was done at screening and on Days −2, 2, 5, and 12 of the SAD phase and at screening and on Days −2, 2, 7, 14, 18, 25, and 32 of the MAD phase. Participants fasted for ≥10 hours (except screening) prior to sample collection.
A single 12-lead ECG was recorded at screening and on Day −2 of the SAD and MAD phases, on Days 5 and 12 of the SAD phase, and on Days 8 and 25 of the MAD phase. Time-matched serial ECGs were obtained on Days −1 and 1 of the SAD phase and on Days −1, 1, 7, and 14 of the MAD phase.
Vital signs were recorded at screening and on Days −2, 2, 5, and 12 of the SAD phase and at screening and on Days −2, 2, 18, and 25 of the MAD phase. Vital signs included body temperature, respiratory rate, BP, and HR. BP and HR were measured after the participant had been seated quietly for five minutes. Orthostatic BP was measured on Days −1 and 1 of the SAD phase and on Days −1, 1, 7, and 14 of the MAD phase after the participant had been in a supine position for at least five minutes and then standing for at least two minutes.
Pharmacokinetic assessments
In the SAD phase, peripheral blood samples were collected pre-dose and at 0.25, 0.5, 0.75, 1, 1.5, 2, 3, 4, 6, 8, 12, 16, 24, 48, 72, and 96 hours post-dose on Day 1. In the MAD phase, blood samples were collected pre-dose; at 0.25, 0.5, 0.75, 1, 1.5, 2, 3, 4, 6, 8, 12, 16, and 24 hours post-dose on Days 1 and 14; and at 48, 72, and 96 hours post-dose on Day 14. Pre-dose samples were collected on Days 3, 5, 7, and 12. In the BID MAD dosing panel, three additional samples were drawn at 2.25, 2.75, and 3.5 hours post-first dose on Days 1 and 14.
During the SAD phase, single dose PK parameters derived from plasma concentration versus time included maximum observed plasma concentration (Cmax), time of Cmax (Tmax), area under the plasma concentration-time curve (AUC) from time zero to the last measurable plasma concentration (AUC0-T), AUC from time zero to infinity (AUCINF), plasma half-life (T-HALF), and apparent total body clearance (CLT/F).
During the MAD phase, multiple dose PK parameters derived from plasma concentration versus time included Cmax, Tmax, trough observed plasma concentration (Cmin), AUC in 1 dosing interval (time zero to 24 hours post-dose) for the QD dosing panels (AUCTAU), AUC from time zero over the entire dosing interval up to 24 hours post-first dose for the BID dosing panel, T-HALF, CLT/F, and accumulation index (AI) measured as the ratio of AUCTAU at steady state to AUCTAU after the first dose (also calculated for Cmax and AUC0–24).
Bioanalytical assay methods
All reported results with rimegepant in human potassium ethylenediaminetetraacetic acid plasma were generated in analytical runs using a chromatographic method consisting of an Acquity ultra-performance liquid chromatography BEH C18, 1.7 µm, 2.1 × 50 mm column and a gradient mobile phase and a flow rate of 600 µL/min. [13C2D4]-rimegepant was used as an internal standard. Rimegepant was monitored at m/z 535 → 256, and the internal standard was monitored at m/z 541 → 256 using a Triple Quad 5500 mass spectrometer (AB Sciex, Framingham, MA). The assay range was 0.50 to 500 ng/mL with a between-run % coefficient of variation (CV) ≤5.3 and a within-run %CV ≤4.5 (Online Supplemental Table S1).
Statistical analyses
The number of participants was not based on statistical power considerations; however, dosing of six participants with rimegepant would provide an 80% probability of observing ≥1 occurrence of any AE that would occur with 24% incidence in the sample population. All participants who received rimegepant or placebo were included in the safety data set. Participants who received placebo in the SAD and MAD phases were pooled into two separate placebo groups. All available concentration-time data from participants who received any rimegepant were reported. All available derived PK parameter values were included in the PK data set and reported, but only participants with adequate PK profiles were included in the summary statistics and statistical analysis. PK parameter values were derived by noncompartmental methods using a validated PK analysis program (KineticaTM 5.0 within eToolbox, version 2.7).
Summary statistics were tabulated for the PK parameters by dose in the SAD phase and by dose and study day in the MAD phase. The power model (PK Parameter = α*Doseβ, where α is the expected value of PK parameter at a reference dose, and β is the exponent used for assessing the proportionality) was used to assess the dose-systemic exposure relationship over the single dose range of 25–1500 mg (17,18). The effects of rimegepant on ECG parameters, HR, and BP were explored graphically and by summary statistics. All statistical analyses were carried out using SAS/STAT® Version 9.1.
Results
Participant characteristics
A total of 188 participants were enrolled with 104 entering the treatment period. The study was initiated on 3 December 2010, and completed on 5 September 2011. Of the 84 participants who did not enter the treatment period, 32 (17.0%) withdrew consent, 49 (26.1%) no longer met study criteria, and three (1.6%) did not enter the treatment period for another reason. Baseline characteristics were similar between the placebo and rimegepant groups and are described in Table 1. In the SAD phase, 56 randomized participants received either rimegepant (n = 42) or placebo (n = 14), and all 56 completed the study as planned. In the MAD phase, 48 randomized participants received either rimegepant (n = 36) or placebo (n = 12). Of these, 45 (93.8%) completed the study as planned and three (6.3%) discontinued due to AEs.
SAD and MAD population demographics.

BMI, body mass index; kg, kilogram; m, meter; MAD, multiple ascending dose; mg, milligram; SAD, single ascending dose; SD, standard deviation.
Safety of rimegepant
Adverse events
In the SAD phase, single oral rimegepant doses (25–1500 mg) were safe and well tolerated. In the MAD phase, single oral daily doses of rimegepant (75–600 mg) for 14 days were safe and well tolerated. An MTD was not reached in the SAD or MAD phase of the study.
In the SAD phase, there were no deaths, SAEs, or discontinuations due to AEs. There were no ECG-related AEs. There was no apparent dose-related pattern observed for the mean change from baseline ECG-derived HR or intervals over time. Overall, 23 (41.1%) participants had an AE in the SAD phase. There were more AEs in the rimegepant dose groups (n = 21, 50.0%) compared with placebo (n = 2, 14.3%). In the case of nausea, dizziness, and vomiting, the incidence increased with the higher 1500 mg dose. The most frequent AEs (>5%) in the rimegepant groups were nausea (n = 7, 16.7%), dizziness (n = 5, 11.9%), vomiting (n = 4, 9.5%), application site irritation due to the ECG electrodes (n = 3, 7.1%), elevated creatine kinase (CK) (n = 3, 7.1%), and headache (n = 3, 7.1%) (Table 2). Eleven (19.6%) participants had AEs that were considered related to the study drug by the Site Principle Investigator.
Adverse events occurring in ≥2 participants in the SAD or MAD populations.

CK, creatine kinase; ECG, electrocardiogram; MAD, multiple ascending dose; SAD, single ascending dose.
Four SAD participants had clinical laboratory AEs, none of which resulted in study discontinuation. There were 26 clinical laboratory abnormalities (3 hematology, 9 serum chemistry, and 14 urinalysis) with no apparent dose-related trends in the occurrence of any clinical laboratory results. Clinical laboratory abnormalities that occurred in ≥2 participants in the SAD phase included red blood cells in urine (n = 7, 26.9%), white blood cells in urine (n = 6, 23.1%), elevated CK (n = 3, 5.4%), decreased leukocytes (n = 2, 3.6%), and elevated alanine aminotransferase (ALT) (n = 2, 3.6%). One participant had an elevated ALT of 82 U/L (upper limit of normal = 55 U/L) on Day 12 that was considered unrelated to study drug and was concluded to be of skeletal muscle origin related to strenuous physical activity. The second participant had an elevated ALT of 62 U/L on Day 12 that resolved without treatment by Day 19 (30 U/L) and was not considered an AE by the Investigator.
In the MAD phase, there were no deaths or SAEs reported. There was no apparent dose relationship in either the incidence or severity of the AEs reported. There were no ECG-related AEs reported. There were no apparent dose-related trends in the occurrence of any clinical laboratory findings or vital signs. Overall, 30 (62.5%) participants had any AE, and 16 (33.3%) had AEs that were considered related to study drug in the MAD phase. The percentage of participants with AEs in the rimegepant dose groups (n = 22, 61.1%) was comparable to placebo (n = 8, 66.7%).
Four MAD participants had clinical laboratory AEs unrelated to study drug, one of which (increased creatinine) resulted in discontinuation from the study. Three participants discontinued due to AEs related to study drug following multiple dose administration: one female with moderate pruritus and urticaria in the 450 mg dose group; one female with mild rash in the 300 mg BID dose group; and one male with mild elevated serum creatinine in the 600 mg dose group.
Electrocardiograms
On Day 1 of the SAD phase of the study, no dose-related trend in mean HR change from baseline (ΔHR), mean PR change from baseline (ΔPR), mean QRS change from baseline (ΔQRS), or mean corrected QT interval by Fridericia’s formula (QTcF) change from baseline (ΔQTcF) intervals was apparent in any of the dose groups. Four participants (one each in the 75 and 150 mg dose groups and two in the placebo group) had a PR interval >200 msec. No participants had a QRS measurement of >120 msec in any rimegepant treatment group. No participants in any treatment group had a QTcF interval >450 msec. One participant each for 75 mg rimegepant and placebo had a ΔQTcF >30 but ≤60 msec. No participant in any treatment group had a ΔQTcF >60 msec.
On Day 14 of the MAD phase of the study, no dose-related trend for mean ΔHR, mean ΔQRS, or mean ΔQTcF was apparent in any of the dose groups. The rimegepant 600 mg dose group appeared to have a greater ΔPR through 16 hours relative to other doses or placebo. Otherwise, no dose-related trends in mean ΔPR on Day 14 were apparent. No participants had a PR interval of >200 msec or a QRS interval of >120 msec. An association was seen between ΔQTcF and rimegepant concentration on Day 14; however, the magnitude of this slope was small (1.148) and not statistically different than 1 (95% confidence interval [CI] [0.308, 1.988]). No participants in any treatment group had a QTcF interval >450 msec. Six participants had maximum ΔQTcF >30 but ≤60 msec (one each in the 300 and 450 mg dose groups, two in the 600 mg dose group, and two in the placebo group). One participant in the 600 mg dose group had a maximum ΔQTcF >60 msec. No other participant on any other treatment had a ΔQTcF >30 msec.
No ECG abnormalities in the SAD or MAD phase were considered to be AEs.
Vital signs and physical findings
No clear dose-related trend was apparent for mean supine, standing, or orthostatic change in SBP and DBP on Day 1 (SAD) or Day 14 (MAD). In the SAD phase, the same pattern in change from baseline in ECG-derived HR was observed in the ΔHR obtained from the vital signs taken in the supine position and was not evident in the standing position. Otherwise, no clear dose-related trend was apparent for ΔHR on Day 1 in the supine, standing, or orthostatic position.
Pharmacokinetics of rimegepant
In the SAD phase of the study, the mean rimegepant plasma concentration-time profiles are shown in Figure 1. Rimegepant exposures, measured by Cmax, AUC0-T, and AUCINF, increased with increase in dose over the single dose range of 25–900 mg under fasting conditions (Table 3). The 1500 mg dose exposure values were less than exposures following the 600 and 900 mg doses and may be related to dissolution-limited absorption due to the large number of capsules (10) dosed. The dose proportionality assessed based on the power model showed that the AUC following a single rimegepant dose was more than dose proportional for doses from 25–1500 mg (Table 4; Online Supplemental Figure 1). Median Tmax varied among doses from 1–3.5 hours, and the median terminal T-HALF ranged from 8–12 hours. Doses from 25–300 mg and 900 mg had moderate inter-subject variability (22%–68%) and 150 and 1500 mg had higher inter-subject variability (65%–85%).
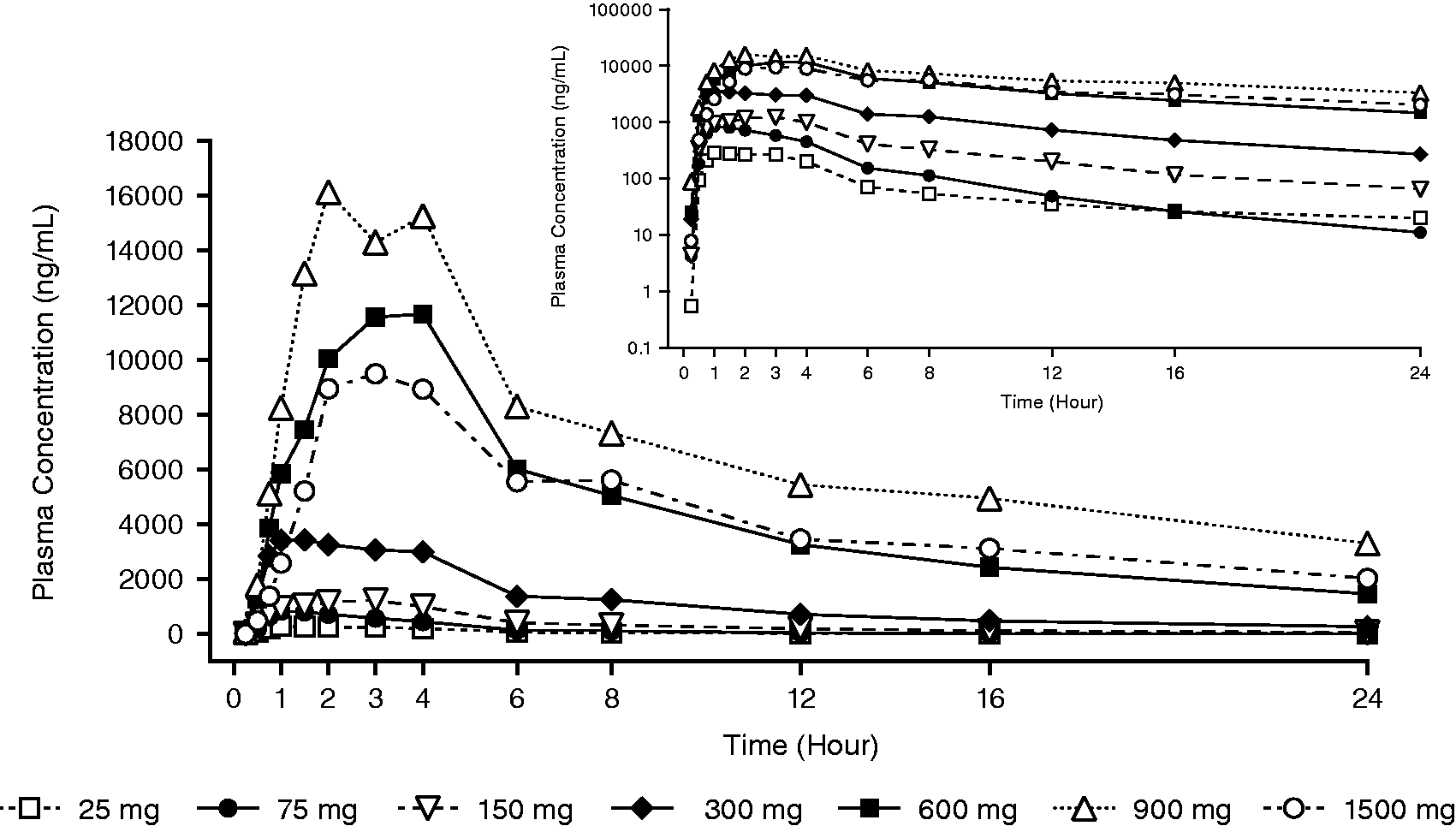
Mean plasma concentration-time profiles for rimegepant following single oral doses to healthy participants (linear scale, up to 24 hours; inset: semi-log scale). LLOQ = 0.5 ng/mL; N = 6 for all treatments.
Summary statistics for rimegepant PK parameters after single oral doses (SAD).
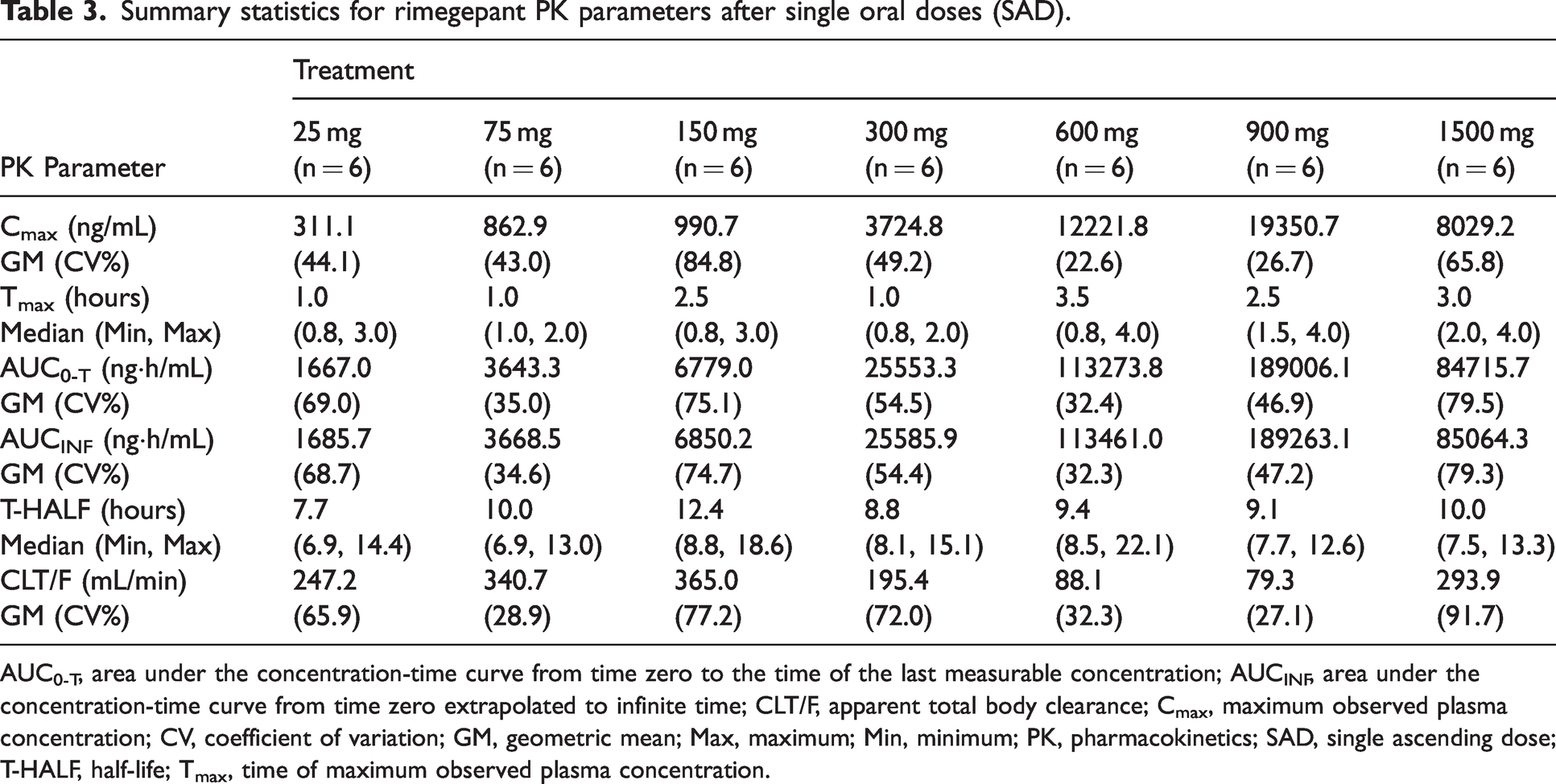
AUC0-T, area under the concentration-time curve from time zero to the time of the last measurable concentration; AUCINF, area under the concentration-time curve from time zero extrapolated to infinite time; CLT/F, apparent total body clearance; Cmax, maximum observed plasma concentration; CV, coefficient of variation; GM, geometric mean; Max, maximum; Min, minimum; PK, pharmacokinetics; SAD, single ascending dose; T-HALF, half-life; Tmax, time of maximum observed plasma concentration.
Summary of dose proportionality assessment (SAD) based on doses from 25–1500 mg.

AUC0-T, area under the concentration-time curve from time zero to the time of the last measurable concentration; AUCINF, area under the concentration-time curve from time zero extrapolated to infinite time; CI, confidence interval; Cmax, maximum observed plasma concentration; PK, pharmacokinetics; SAD, single ascending dose.
In the MAD phase of the study, the mean rimegepant plasma concentration-time profiles on Days 1 and 14 are shown in Figure 2. Selected PK parameters for rimegepant are summarized, by treatment and study day, in Table 5. Following multiple doses, rimegepant showed a more than dose-proportional increase in exposure for rimegepant doses from 75–600 mg. The AI was determined to be less than 1.5-fold across 75–300 mg doses, suggesting minimal accumulation after multiple daily dosing of rimegepant.

Mean plasma concentration-time profiles for rimegepant on days 1 (a) and 14 (b) following an oral dose to healthy participants (linear scale, up to 24 hours; inset: semi log scale). (a) LLOQ = 0.5 ng/mL; N = 6 for all treatment groups. (b) LLOQ = 0.5 ng/mL; N = 6 for 75–450 mg treatment groups, N = 5 for 300 mg BID and 600 mg QD treatment groups. BID, twice daily; LLOQ, lower limit of quantitation; QD, once daily.
Summary statistics for rimegepant PK parameters after multiple dose administration (MAD).

AI, accumulation index; AUCTAU, area under the concentration-time curve in 1 dosing interval; CLT/F, apparent total body clearance; Cmax, maximum observed plasma concentration; Cmin, trough observed plasma concentration; CV, coefficient of variation; GM, geometric mean; MAD, multiple ascending dose; Max, maximum; Min, minimum; T-HALF, plasma half-life; Tmax, time of maximum observed plasma concentration.
The increase in exposure with dose in the MAD phase was greater than dose proportional, with slope point estimates of 1.9 and 1.6 and the lower limit of the 90% CI above 1.6 and 1.3 for AUCTAU and Cmax, respectively (Table 6). No evidence of lack of fit for the power model was observed on the Day 14 Cmax or AUCTAU data based on PK exposure versus dose plots (Online Supplemental Figure 2).
Summary of dose proportionality assessment on day 14 (MAD).

AUCTAU, area under the concentration-time curve in 1 dosing interval; CI, confidence interval; Cmax, maximum observed plasma concentration; MAD, multiple ascending dose.
Steady state was reached by Day 3 for 75–450 mg rimegepant and by Day 5 for the 600 mg QD dose (Figure 3). The increase in exposure on Day 7 was within the variability of the drug exposure. The increase in exposure on Days 12–15 (600 mg) was unexplained; however, based on the T-HALF of the drug, it should be at steady state.

Geometric mean of rimegepant Cmin (ng/mL) versus study day (MAD). BID, twice daily; Cmin, trough observed plasma concentration; MAD, multiple ascending dose; QD, once daily.
Discussion
Rimegepant, presented in the oral capsule free-base formulation, was safe and generally well tolerated at single doses up to 1500 mg and multiple doses up to 600 mg/day for 14 days in healthy participants in this Phase 1, double-blind, placebo-controlled study. There were no deaths or SAEs or liver toxicity during the SAD or MAD phase. An MTD was not achieved in healthy participants in the SAD (up to 1500 mg) or MAD (up to 600 mg daily for 14 days) phases. The capsule formulation supported the characterization of rimegepant in eight early clinical studies. Subsequently, two other oral rimegepant formulations were used during the course of the development program: the two bioequivalent formulations included an ODT formulation using Zydis® technology (hemisulfate sesquihydrate salt form) and a conventional immediate release tablet formulation (hemisulfate sesquihydrate salt form). The bioequivalent tablet and ODT formulations were developed to support pivotal Phase 2 and 3 clinical studies, as well as the clinical pharmacology program that characterized rimegepant intrinsic and extrinsic characteristics after an adult clinical dose of 75 mg.
After single and multiple dosing, the increase in rimegepant exposure with dose was more than dose proportional, suggesting rimegepant demonstrates nonlinear kinetics with respect to dose level. Rimegepant is a substrate of the P-glycoprotein (P-gp) and breast cancer resistant protein transporters in vitro (19,20); therefore, a possible reason for nonlinearity in rimegepant PK is the saturation of intestinal P-gp leading to a dose-dependent increase in bioavailability with increasing dose.
A limitation of this study is that it was conducted in the US population only. It is also limited by the evaluation of rimegepant in healthy participants and not in participants experiencing a migraine attack. The small sample size and limited duration of dosing are also limitations to the assessment of clinical safety in this study.
Rimegepant has demonstrated efficacy and safety in multiple randomized, placebo-controlled clinical trials. A single dose, double-blind, randomized study tested six rimegepant doses (NCT01430442) and found that significantly more patients in the rimegepant 75 (31.4%, p = 0.0018), 150 (32.9%, p = 0.005), and 300 mg (29.7%, p = 0.0024) groups had pain freedom at two hours post-dose versus placebo (15.3%) with no incremental increase in efficacy over 75 mg. No deaths or treatment-related SAEs were reported, and no patients discontinued because of AEs in the study (12). In two double-blind, randomized, placebo-controlled Phase 3 trials, treatment of a migraine attack with a 75 mg rimegepant dose resulted in a higher percentage of patients who were free from pain and their most bothersome symptom when compared with placebo (NCT03237845 and NCT03461757). The most common AEs were nausea and urinary tract infection (9,10). In a multicenter, Phase 2/3, randomized, double-blind, placebo-controlled trial, 75 mg rimegepant, taken every other day, was effective for preventive treatment of migraine (NCT03732638). Tolerability was similar to that of placebo, and no unexpected or serious safety issues were noted (11). Rimegepant is currently approved by the US FDA and the European Commission for the acute treatment of migraine with or without aura in adults at a dose of 75 mg and for the preventive treatment of episodic migraine in adults.
Conclusions
Rimegepant was safe and generally well tolerated at single oral doses up to 1500 mg and multiple doses up to 600 mg/day for 14 days in healthy participants in this study. Rimegepant concentrations increased with dose in a more than dose-proportional manner. T-HALF ranged from 8–12 hours across the wide range of single doses studied.
Clinical implications
Rimegepant was safe and generally well tolerated at single oral doses up to 1500 mg and multiple doses up to 600 mg/day for 14 days in healthy participants. Rimegepant showed a more than dose-proportional increase in exposure from 25–1500 mg following a single dose and from 75–600 mg following multiple doses.
Supplemental Material
sj-pdf-1-cep-10.1177_03331024231179131 - Supplemental material for A placebo-controlled, randomized, single and multiple dose study to evaluate the safety, tolerability, and pharmacokinetics of rimegepant in healthy participants
Supplemental material, sj-pdf-1-cep-10.1177_03331024231179131 for A placebo-controlled, randomized, single and multiple dose study to evaluate the safety, tolerability, and pharmacokinetics of rimegepant in healthy participants by Richard Bertz, Rajinder Bhardwaj, Beth A. Morris, Eric Ashbrenner, Vladimir Coric and Robert Croop in Cephalalgia
Footnotes
Acknowledgments
Medical writing support was provided by Amy C. Porter, ISMPP CMPPTM of Certara Synchrogenix, funded by Biohaven Pharmaceuticals, Inc. which was acquired by Pfizer in October 2022.
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: RB, BAM, and VC are employees of Biohaven Pharmaceuticals, Inc. and own Biohaven Ltd. stock and/or stock options.
RC was an employee of Biohaven Pharmaceuticals, Inc. during preparation of this manuscript and owns Biohaven Ltd. stock.
RB is an employee of Certara Strategic Consulting and serves in a consultant/advisory role for Biohaven Pharmaceuticals, Inc.
EA was an employee of Navitas Data Sciences and served in a consultant/advisory role for Biohaven Pharmaceuticals, Inc. at the time of this work. He is currently an employee of Biohaven Pharmaceuticals, Inc. and owns stock options.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was funded by Bristol-Myers Squibb and Biohaven Pharmaceuticals, Inc. which was acquired by Pfizer in October 2022.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.