
Abstract
In 2011, Goldring and colleagues published a review article describing the potential safety issues of novel stem cell-derived treatments. Immunogenicity and immunotoxicity of the administered cell product were considered risks in the light of clinical experience of transplantation. The relative immunogenicity of mesenchymal stem cells, embryonic stem cells (ESCs), and induced pluripotent stem cells (iPSCs) was being addressed through in vitro and in vivo models. But the question arose as to whether the implanted cells needed to be identical to the recipient in every respect, including epigenetically, to evade immune recognition? If so, this set a high bar which may preclude use of many cells derived from iPSCs which have vestiges of a fetal phenotype and epigenetic memory of their cell of origin. However, for autologous iPSCs, the immunogenicity reduces once the surface antigen expression profile becomes close to that of the parent somatic cells. Therefore, a cell product containing incompletely differentiated cells could be more immunogenic. The properties of the administered cells, the immune privilege of the administration site, and the host immune status influence graft success or failure. In addition, the various approaches available to characterize potential immunogenicity of a cell therapy will be discussed.
Introduction
In 2011, Goldring and colleagues published a review article describing the potential safety issues of novel stem cell-derived treatments. 1 -4 Immunogenicity and immunotoxicity of the administered cell product were considered risks in the light of clinical experience of transplantation, but real-world data with cell-based therapies were limited due to the small number of trials at that time. There were 2 distinct classes of immune-related risk identified: a response by the patient against the cell therapy with the potential for reduced efficacy and the induction of an immune reaction by the product on the patient resulting in toxicity. The relative immunogenicity of mesenchymal stem cells (MSC), embryonic stem cells (ESCs), and induced pluripotent stem cells (iPSCs) was being addressed through extensive research in in vitro and in vivo models. However, assessing the risk of immunogenicity and cell therapy-induced immune-mediated toxicity can be challenging nonclinically. Extensive knowledge on the likely mode of action, product characteristics, production methods, and nonclinical assessment limitations is needed to provide the context for study findings; specifically, whether the risks will manifest clinically or indeed there is an underestimation of risk due to model limitations.
The importance of major histocompatibility complex (MHC) antigens has long been recognized in transplantation where it has been shown that the immune system is exquisitely tuned to identify cells as foreign. 5 Human leukocyte antigens (HLA), which are the human version of the MHC, are highly polymorphic genes allowing extensive allelic diversity within the population. However, this diversity of HLA class I and II loci presents a challenge when trying to prevent immune rejection by a recipient of transplanted allogeneic cells or tissues by HLA matching. In addition, there may also be an ideal level of MHC class I expression on donor cells which is low enough to reduce T-cell recognition but sufficient to prevent killing by natural killer (NK) cells. 6 The stimulus to match donor to recipient MHC has led some to develop cell banks of donor PSCs; however, there is evidence that differences in expression of minor histocompatibility antigens may still trigger an immune response. 7 Recent approaches have included engineering of iPSCs to reduce MHC I and II expression and produce hypoimmunogenic cells. 8 Similarly, others have engineered human embryonic stem cell (hESC)–derived cells to constitutively express immunosuppressive molecules cytotoxic T lymphocyte antigen 4 (CTLA4)-immunoglobulin (Ig) and programmed death ligand-1 (PD-L1), which has enabled engraftment in mice reconstituted with a functional human immune system. This is in contrast to the vigorous allogeneic immune rejection of parental hESCs in these mice. 9 Expression of both CTLA4-Ig, which disrupts T-cell co-stimulatory pathways, and PD-L1, which activates T-cell inhibitory pathways, is required to confer immune protection as neither is sufficient on their own.
Do the implanted cells need to be absolutely identical to the recipient in every respect, including epigenetically, to evade immune recognition? 2 If yes, this sets a high immunological threshold which may preclude use of many cells derived from iPSCs which have vestiges of a fetal phenotype and epigenetic memory of their cell of origin. However, for autologous iPSCs, the immunogenicity has been observed to reduce once the surface antigen expression profile becomes close to that of the parent somatic cells. 3,4 This raises the prospect that a cell product containing incompletely differentiated cells could be more immunogenic.
This publication will review the progress made since the Goldring’s paper and will identify which of the potential risks have manifested in the clinic or remain a concern in the absence of immunosuppressive therapy. The properties of the administered cells, the relative immune privilege of the site of administration, and the immune status of the host will be considered as contributors to an outcome of graft success or failure. In addition, the various approaches available to characterize potential immunogenicity of a cell therapy will be discussed.
Properties of the Administered Cells
Mesenchymal Cells
The most widely studied sources of MSCs include bone marrow, adipose, muscle, peripheral blood, umbilical cord, placenta, fetal tissue, and amniotic fluid. 10 Mesenchymal stem cells generally adhere to plastic, express cluster of differentiation (CD) markers, such as CD73, CD90, and CD105 markers, and can differentiate into adipogenic, chondrogenic, and osteogenic lineages in vitro. However, isolated MSCs have been reported to vary in their potency and self-renewal potential, and consequently, the MSCs used for clinical applications often lead to variable or even conflicting results. 10 Despite these limitations, their immunosuppressive and anti-inflammatory properties have led to MSC being used clinically to treat a broad range of disease indications including acute and chronic inflammatory disorders, autoimmune diseases, and transplant rejection. 11 The clinical use of MSCs has been supported by an extensive body of published preclinical studies showing their ability to modulate multiple immune cells. 12,13 Both autologous and allogeneic MSC therapies are in development. While allogeneic MSC were initially believed to be immune-privileged, substantial preclinical evidence now exists to prove otherwise with multiple studies documenting specific cellular and humoral immune responses against donor antigens. Although no acute clinical adverse events linked to the allogenicity of MSC have been reported, very few clinical studies have reported on evidence of sustained sensitization and development of antibodies specific to the donor HLA. 11
To address this controversy regarding immune tolerance to transplantation of allogeneic MSCs, the survival of luciferase-labeled MSCs (Luc + MSCs) was evaluated preclinically by imaging in allogeneic murine recipients. 14 This analysis showed that although MSCs exhibited longer survival compared to fibroblasts, their survival was significantly shorter compared to that exhibited in syngeneic or in immune-deficient Balb Nude or non-obese diabetic severe combined immunodeficiency (NOD-SCID) recipients. Graft rejection in re-challenge experiments infusing Luc+ fibroblasts into mice, which had previously rejected Luc + MSCs, indicated potential induction of immune memory by the MSCs. The authors concluded that MSCs are not intrinsically immune privileged and that when transplanted into an allogeneic recipient can induce rejection and result in immune memory. The formation of memory immune cells in recipients of MHC-mismatched MSCs is important since immunologic memory can lead to accelerated rejection of allogeneic cells upon reinjection. A consequence of this would be limited long-term survival of allogeneic MSCs in clinical applications.
Alofisel (TiGenix NV/Takeda) was developed to treat complex perianal fistulas in adult patients with nonactive/mildly active luminal Crohn's disease, with inadequate response to conventional or biologic therapy. This product is one of only a few allogeneic products to be approved (Table 1) and consists of donor-derived expanded adipose-derived stem cells (eASCs), which are a form of MSC. The intralesional injection of eASCs reduces proliferation of activated lymphocytes/release of pro-inflammatory cytokines at the inflammation sites. In vitro interaction studies have established that the cell viability/immunomodulatory function of Alofisel is not affected by the presence of conventional therapies for Crohn’s disease (infliximab, methotrexate, and azathioprine). Immunogenicity was evaluated in the Adipose Derived Mesenchymal Stem Cells for Induction of Remission in Perianal Fistulizing Crohn's Disease (ADMIRE-CD) clinical study, where 36% of the eASC-treated patients showed anti-donor antibody production at week 12. 15 Of patients with donor-specific antibodies (DSA) at week 12, 30% had cleared DSA by week 52. Lack of de novo DSA generation was observed between week 12 and week 52. Although DSA may have cleared by week 52, it is unclear if a second inoculum would elicit a memory B cell response. Limited data exist but there does not appear to be a detrimental effect of DSA on efficacy and safety in patients. Concomitant use of stable doses of immunosuppressants (18% of patients) or anti-TNFs (33%) or both (28%) was allowed during the study.
Status of Cell Therapy Products Approved by the European Medicines Agency.
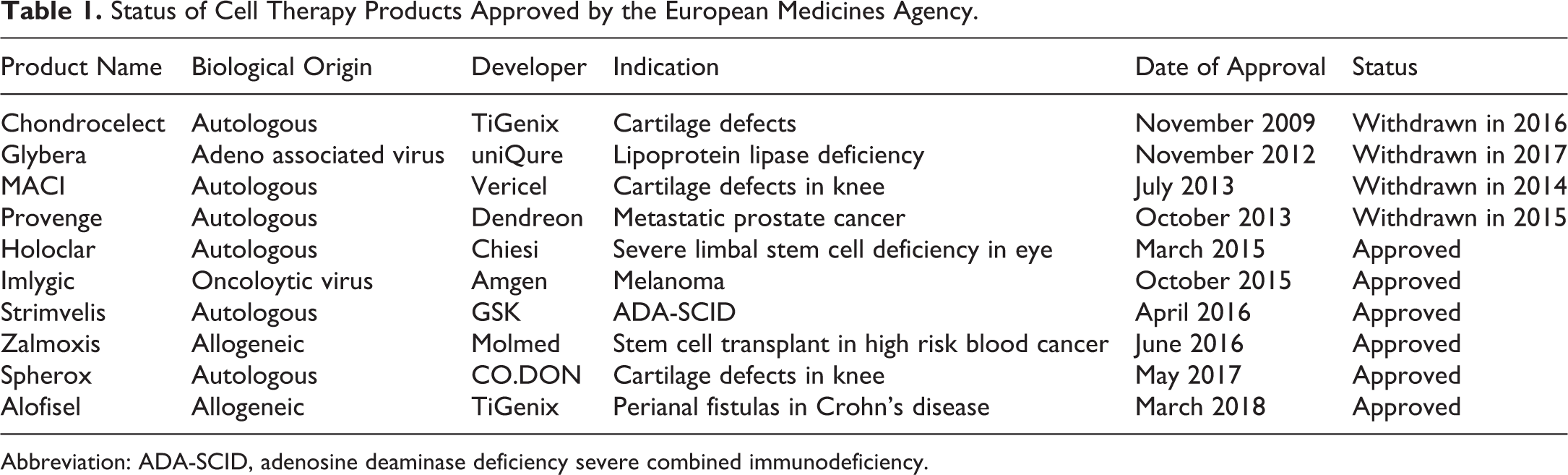
Abbreviation: ADA-SCID, adenosine deaminase deficiency severe combined immunodeficiency.
Nonclinical studies which supported development of Alofisel included perianal and intrarectal injection of human eASC in athymic rats, which demonstrated that cells were present in the rectum and jejunum and at the site of injection for at least 14 days in this immune-deficient model and were undetectable after 3 months. Expanded adipose-derived stem cells were not present in any of the tissues analyzed after 3 months or 6 months.
Human Embryonic Stem Cell–Derived Cells
Pluripotent cell-derived therapies were originally considered to be immune privileged; however, it is now accepted that the majority of the early reported data examined undifferentiated and early differentiated products. 16 Such products were likely immunologically immature, and the study findings were not truly reflecting the rate of rejection a fully differentiated and immunologically mature product could encounter in the clinical situation. Expression of immunological markers such as MHC class I increase on product maturation, and such products would be subject to an allogeneic immune response. 16
Many of the early published nonclinical in vivo studies focused on undifferentiated mouse pluripotent cells transplanted into a murine host in an allogeneic situation. Whether the findings can be extrapolated to the clinical situation with a differentiated product seems doubtful. Indeed when the impact of MHC class I upregulation upon cell maturation was investigated by comparing the survival of undifferentiated and differentiated mouse ESCs transplanted into an allogeneic host it was shown that the differentiated cells were immunologically rejected within 14 days, compared to the 28-day immunological rejection of the undifferentiated product. 17
Immunogenicity of allogeneic pluripotent cells has also been studied in a model of myocardial infarction. Administration of mouse ESCs into the myocardium of allogeneic immunocompetent mice resulted in robust progressive inflammatory responses. Cellular infiltration by both innate and adaptive components of the immune system was observed and this correlated with increased expression of MHC class I antigens on the transplanted cells. 18 If a cell therapy is antigenic, a memory immune response will be generated that could lead to a rapid and robust response on subsequent exposure, limiting the opportunity for repeat dosing.
Physical encapsulation of hESCs is one approach which has been taken to separate the grafted cells from the recipient immune system. Potential treatment of type 1 diabetes, where insulin producing hESC-derived cells are delivered, presents the most likely application of an implantable device. In most regenerative medicine indications, however, engrafted cells will need to integrate functionally into the tissue rather than merely secrete a soluble product like insulin and therefore require intimate contact with host cells.
Viacyte is in clinical development with an allogeneic ES cell-derived pancreatic progenitor cells (PEC-01) encapsulated in the Encaptra cell delivery system (PEC-Encap). In a 2-year clinical study, Safety, Tolerability, and Efficacy of VC-01™ (PEC-Encap™) Product Candidate in Type One Diabetes (STEP-ONE), the Encaptra Cell Delivery System appeared to protect against allo- and auto-immune rejection and sensitization. 19 Immune function tests showed no DSA, no increase in auto-antibody titers, no tolerance or sensitization to donor cells, and no increase in lymphocyte stimulation or antigen-specific CD8+ T cell frequency post-implantation. However, there were low levels of engraftment due to a foreign body giant cell response to the Encaptra device. Differentiation into endocrine islet cells was observed in both 12-week and 2-year explants in regions where there was good host tissue integration and vascularization. Several 2-year explants had regions containing insulin-producing β cells and glucagon-producing α cells, indicating that when engraftment occurs, cells can persist for long durations without the need for immunosuppression. Other companies, such as Semma Therapeutics (Cambridge, MA), are also evaluating alternative approaches for delivering a cure for type 1 diabetics and addressing the challenge of long cell survival required for effective therapy. A full list of clinical trials of human pluripotent stem cells, which includes hESC, is provided by Guhr et al. 20
If there is MHC mismatch, unencapsulated donor stem cells could present directly to patient’s T cells if they also expressed co-stimulatory molecules (eg, CD86, CD80, CD11c). Expression levels of co-stimulatory factors are low on human ESC and iPSC source cells; therefore, direct allogenic stimulation is probably of limited significance. However, for activation of memory T cells, lower levels of co-stimulation are required. This is a risk if stem cells express molecules recognized by host memory cells and if these cells are readministered to a patient. Preexisting antibodies cross-reactive for donor MHC molecules or alloantibodies produced following activation of B cells by cognate alloantigens can also contribute to rejection of allogeneic cells.
One approach to address the allogenic rejection bottleneck limiting wider application of hESC-based cell replacement therapies has been to genetically knock into the hESC CTLA4 and PD-L1, which are critical immune inhibitory molecules. Using humanized mice reconstituted with a functional immune system, Rong and colleagues demonstrated that both CTLA4-Ig and PD-L1 were required to protect hESC-derived teratomas, fibroblasts, and cardiomyocytes from immune rejection by the host. 9
Induced Pluripotent Stem Cell–Derived Cells
Autologous iPSC lines are genetically identical to the donor and therefore immunologically matched. As such cellular derivatives are not expected to be immunologically rejected (Table 2). However, studies by Zhao et al raised the possibility that iPSC-derived cells may have the potential to induce an autologous immune response, 21 and these initial findings were supported by further studies. 3,22 Conversely, other studies in mice have shown negligible inherent immunogenicity. 3,23,24 Additionally, in nonhuman primates, transplanted autologous rhesus monkey iPSC-derived neural progenitors have been shown to survive for up to 6 months and differentiate into neurons, astrocytes, and myelinating oligodendrocytes in the brains of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine–induced hemi-parkinsonian rhesus monkeys with a minimal presence of inflammatory cells and reactive glia. 25 Similarly, autologous transplantation of iPSC-derived midbrain dopamine neurons into a cynomolgus monkey model of Parkinson disease provided a gradual onset of functional motor improvement contralateral to the side of dopamine neuron transplantation and increased motor activity, without a need for immunosuppression for up to 2 years. 26 This is in comparison with a study of transplantation in nonhuman primates where it was found that while autologous transplantation of iPSC-derived neurons elicited only a minimal immune response on the brain, allografts caused an acquired immune response with the activation of microglia and infiltration of leukocytes. 27 Despite conflicting reports, the majority of studies reported to date show minimal or no immunogenicity of autologous iPSC-derived cellular derivatives in transplanted animals. However, there may exist differences in the immunogenicity profiles of different derived somatic cells highlighting the need to develop efficient models to systematically address this issue. 28
The Relative Risk of Rejection of Different Type of Cells or Tissues Used in Regenerative Medicine.

As with other cellular products, allogeneic iPSC-derived therapies are likely to induce immunological recognition, and immune suppression is likely required to facilitate clinical use. Shiba et al 29 described an allogeneic transplantation model using the cynomolgus monkey (Macaca fascicularis) which has a comparable MHC structure to that of humans. Monkeys were subjected to myocardial infarction followed by direct intramyocardial injection of iPSC-CMs. The grafted allogeneic cardiomyocytes survived for 12 weeks with no evidence of acute immune rejection in monkeys when treated with clinically relevant doses of methylprednisolone and tacrolimus. 29
Preclinical studies in primates have also explored the utility of matching the MHC antigens between the donor and the recipient. 30 In this study, green fluorescent protein (GFP)–labeled iPSC-CMs donated from a macaque with homozygous MHC haplotypes were transplanted into the subcutaneous tissue and hearts of macaques which had heterozygous MHC haplotypes (MHC-matched) or without identical MHC alleles (nonmatched) in conjunction with immune suppression. Major histocompatibility complex–matched animals displayed a higher GFP intensity and less immune-cell infiltration in the graft than nonmatched animals. However, MHC-matched transplantation with single or no immune-suppressive drugs still induced a substantial host immune response to the graft. 30 In the absence of immunosuppression, regardless of MHC status, there was severe rejection. Therefore, although the immunogenicity of allogeneic iPSC-CMs could be reduced by MHC-matched transplantation, there remained a need for appropriate immune suppression for successful engraftment.
Several groups are exploring options to reduce the risk of immune recognition. It has been shown that disruption of the Beta-2 Microglobulin (β2M) gene will eliminate surface expression of all class I molecules; however, this will leave the cell products vulnerable to lysis by NK cells. 31,32 One option being explored is the use of forced expression of minimally polymorphic HLA-E molecules. 33 Adeno-associated virus (AAV)-mediated gene editing is employed to knock in HLA-E genes at the β2M locus. The HLA-engineered PSCs and their differentiated derivatives are not recognized as allogeneic by CD8+ T cells, do not bind anti-HLA antibodies, and are resistant to NK-mediated lysis. 33
Use of Hypoimmunogenic Derivatives of Induced Pluripotent Stem Cells
Autologous iPSCs provide an unlimited cell source for patient-specific cell-based tissue repair. However, the production of differentiated specific cells or tissues of sufficient quality is a time-consuming process which is not suitable for those medical conditions requiring acute treatment. The recipient immune response against allogeneic histoincompatible cells has, in the absence of strong immunosuppression, prevented use of prefabricated, “off the shelf,” nonautologous cell products or tissues. However, recent work has shown that mouse and human iPSCs lose their immunogenicity when MHC class I and II genes are inactivated and CD47 is overexpressed. 8 These hypoimmunogenic iPSCs retain their pluripotent stem cell potential and differentiation capacity. Endothelial cells, smooth muscle cells, and cardiomyocytes derived from hypoimmunogenic mouse or human iPSCs reliably evade immune rejection in fully MHC-mismatched allogeneic recipients and survive long-term without the use of immunosuppression. 8 This work has demonstrated the importance of CD47 in inhibiting the innate immune response which was responsible, in previous studies, for reducing the life span of MHC class 1 knockdown cells through NK cell killing. This approach suggests that cardiac tissue, produced from hypoimmunogenic iPSCs, may act as a universal transplant for heart failure patients without the requirement for immunosuppression.
One of the safety concerns associated with transplanting hypoimmunogenic iPSCs products is the associated evasion of the host immune response and the potential for either uncontrolled malignant transformation or an impaired ability to remove differentiated cells if they become viral-infected. 8 The likelihood that cells lacking MHC expression could serve as a safe haven for viral pathogens that cannot be detected or targeted by the immune system is potentially a significant risk of this approach.
The Immunogenicity of CRISPR-Cas9 Genome–Edited iPSCs
CRISPR-Cas9 is a popular genome editing technology with wide medical application, including editing iPSCs prior to cell therapy, for example, to repair mutated alleles causing monogenic disorders, such as muscular dystrophy or sickle cell anemia. However, the 2 most commonly used sources of Cas9, an RNA-guided nuclease, are Staphylococcus aureus (SaCas9) and Streptococcus pyogenes (SpCas9), which colonize humans and can cause disease. More than 80% of healthy individuals have evidence of preexisting neutralizing antibodies or cellular immunity against these bacteria.
While the preexistence of an adaptive immune response to Cas9 may not prove to be a major barrier for the implementation of therapies wherein cells are treated ex vivo and implanted after the complete degradation of the Cas9 protein, it could pose a significant threat to the development of in vivo therapies. 34 For example, Wang et al, used an adenovirus vector carrying a Cas9 system to disrupt phosphatase and tensin homolog (PTEN) to produce a model of nonalcoholic steatohepatitis in mice, consistent with the phenotype following Cre-lox P-induced PTEN deficiency in mouse liver. 35 In addition to the desired hepatic changes, evidence of both humoral and cellular immunity against SpCas9 following CRISPR/Cas9 delivery was observed. It was hoped that the delivery of Cas9 genes using viral vectors, as opposed to intravenous dosing of Cas9-guided RNA complexes, might help reduce the impact of humoral immunity; however, expression of Cas9 epitopes in engineered target cells left the cells vulnerable to cytotoxic T cell killing.
There are a number of known strategies which could be used to minimize the chance of an adverse immune response after CRISPR-Cas9 gene therapy: structural modification of Cas9 proteins to mask immunogenic epitopes, 36 using Cas9 orthologs from nonpathogenic bacteria, inducing immune tolerance/immune suppression or targeting immune privileged organs, such as the eye. 37
The Relative Immune Privilege of the Site of Administration
Immune privilege although widely recognized is often misunderstood. One common misconception is that immune privilege is a universal exemption from immune recognition and immunological attack. Instead immune privilege is defined as the reduced incidence and extent of immune rejection. For example, skin allografts transplanted across various MHC or minor histocompatibility barriers will undergo rejection in approximately 100% of the hosts. By contrast, orthotopic corneal allografts experience long-term survival in 50% to >90% of the hosts, depending on the histocompatibility barriers that confront the host. 38 The capacity to evade immune rejection is attributable to multiple anatomical, physiological, and immunoregulatory conditions that in concert prevent the induction and expression of alloimmunity. 38,28 Immune privileged sites, including those which have been used as administration sites for cell therapies, are illustrated in Figure 1. Immune privilege will cease in virtually any condition in which inflammation, neovascularization, or trauma is present and this provides a challenge for many cellular therapies, since although they may be used in tissues which can exhibit immune privilege, many are treating highly inflammatory conditions and thus immune privilege will likely cease.
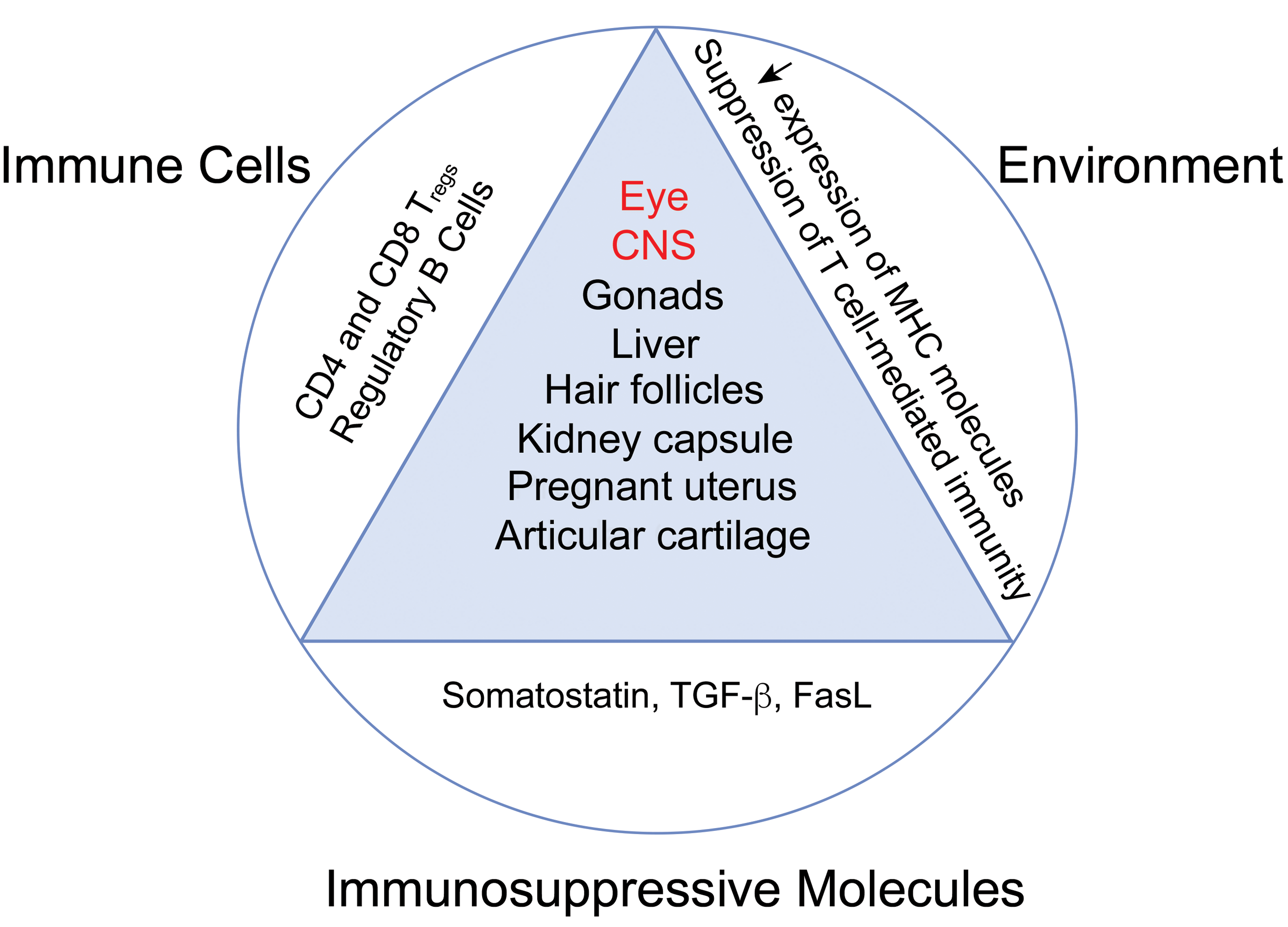
Immune privileged sites. Immune privilege occurs within several tissues. In healthy individuals, these tissues are recognized as less subject to immune responses than other tissues in the body. Immune privilege has been considered a protective response to prevent damage and preserve tissue integrity and maintain homeostasis to the extent possible, without compromising host defence. Immune privilege is controlled by multiple factors including an immunosuppressive environment which downregulates expression of MHC molecules and suppresses T cell-mediated immunity. This is facilitated by the presence of immunosuppressive cell populations such as regulatory T and B cells, as well as the expression of immunosuppressive molecules. Immune privileged sites are attractive targets for cellular therapy treatments although immune privilege is often lost in the diseased state. Both the eye and the CNS (labeled in red) have been anatomic sites of delivery of pluripotent stem cell therapies in clinical trials. CNS indicates central nervous system; MHC, major histocompatibility complex.
It is instructive to consider why the eye, for example, has been a leading organ for stem cell–derived therapy. The eye is highly accessible for surgery and there typically exists an untreated control. Its accessibility also facilitates monitoring of the clinical effects of the therapy and indeed the ability remains to remove an eye in the case of a severe adverse event. Typically, only small numbers of cells are needed simplifying scale up manufacture and robust protocols have been generated to differentiate pluripotent cells for use in the eye. While the site may be immune-privileged (see below for mechanisms), this may be impacted by disease state, and therefore localized, time-limited immune suppression may be required during disease treatment.
The subretinal space (SRS) is maintained as an immune privileged site by the retinal pigment epithelium (RPE) layer. 39 Immunosuppressive drugs are not necessary if the blood–retinal barrier is not breached during surgery. However, in a recent Pfizer trial for their ES cell–derived therapy for macular degeneration, slow release steroids were surgically implanted in the eye. 40 Despite low rates of graft rejection in animal models, the long-term clinical survival rates for ESCs, MSCs, and iPSCs in the retina remain to be determined as long-term follow-up progresses, but the possibility remains that they may be lost over time due to resident microglia activated by cell transplantation. Further, the subretinal microenvironment in adult mammals appears to possess suppressive or inhibitory cues that prevent neural lineage cell proliferation. For example, rat embryonic day-17-derived photoreceptor progenitor cells can quickly switch off mitogenic signals, committing to postmitotic fate within a week upon transplantation in the SRS in retinal degenerative rat model. 41 Graft survival is a prerequisite for RPE functionality in the host SRS and represents the most formidable technical challenge.
The mechanisms conferring immune privilege in the eye are many. These include the suppression of T-cell activation by release of cytokines from the RPE, for example, TGF-β and the production of other immunosuppressive factors by RPE cells that suppress innate immune activity, including pigment epithelium–derived factor and somatostatin. In addition, there is the surface expression of programmed cell death ligand-1 (PD-L1) and Fas ligand by RPE cells and the conversion of CD8+ and CD4+ T cells into regulatory T cells. Finally, the intact physical barrier of the RPE layer is critical to ocular immune privilege.
Many factors (active and passive) contribute to the maintenance of immune privilege. Sites of immune privilege may have selective entry of immune cells due to the presence of physical barriers and it is often these physical barriers that are compromised in the diseased state. One cell type that is actively recruited is T regulatory cells which form the basis of “acquired” rather than innate immune privilege. Cells within the immune privileged site often have specific characteristics: low expression of classical MHC class I molecules, increased expression of surface molecules that inhibit complement activation and the constitutive expression of Fas ligand that controls the entry of Fas-expressing lymphoid cells. Immune-privileged sites often show reduced lymphoid drainage and with this the concomitant reduction in immune surveillance. There is also a localized production of immunosuppressive cytokines such as TGF-β and presence of neuropeptides and in addition a local depletion of essential amino acids (tryptophan and arginine). This local environment modulation results in the suppression of pro-inflammatory and immune stimulatory responses to a variety of tissue insults.
However, surgical implantation of cells can breach the blood: organ barrier and induce inflammation which can increase risk of immune rejection. For example, the injection of MHC-mismatched MSCs into a relatively immune-privileged area like the central nervous system did not prevent immune responses against the cells. 42
Another interesting aspect is that the epigenetic signature of iPSC derived from cells originating from sites of immune privilege, for example Sertoli cells from the testis, was found to be significantly less immunogenic when transplanted to syngeneic murine recipients than conventional iPSC derived from dermal fibroblasts. 43
The Immune Status of the Host
Regenerative therapies that use allogeneic cells will need to overcome immunological barriers similar to those that occur with transplantation of solid organs and allogeneic hematopoietic stem cells (HSCs). In addition, when allogeneic immune cells, for example, T cells, are transplanted into an immunocompromised recipient, the donor cells can react against the recipient and this is termed graft-versus-host disease (GvHD). Immunodeficient mice (SCID, NOD/SCID, and NOG-SCID) transplanted with human peripheral blood mononuclear cells show many features of GvHD and enable high engraftment of the bone marrow and peripheral blood. 44
In modern transplant medicine, efforts to match recipient and donor in terms of immunological compatibility are still generally accompanied by the need to induce tolerance to “non-self” by suppressing the host immune system using a pharmacological and/or immunotherapy approach. 45 While transplantation of solid organs requires life-long immunosuppression due to the host immune system remaining intolerant to the allogeneic tissue, in allogeneic HSC transplantation, the host immune system is eventually replaced by the donor immune system. Local immunosuppression or immunotherapy which may reduce the need for systemic immunosuppression is being evaluated. 46,47 It seems clear that the clinical need and the type of immune suppression required will require careful consideration and will be product- and disease-specific. 45
Assays to Assess Immunogenicity of Administered Cells
The MHC haplotype of donors, recipients, stimulators, and responders should be determined to understand whether donor or stimulator MSCs are full or partial mismatches to recipients or responder cells. One of the well-established methods to evaluate potential immunogenicity is to use enzyme-linked immune absorbent spot (ELISPOT) to assess MHC-mismatched MSCs, by measuring IFN-γ and IL-4 in vitro, in order to assess mixed lymphocyte responses (MLRs), cytotoxicity, and indirect allorecognition. 42 Responder splenocytes or peripheral blood lymphocytes (PBLs) are cocultured with stimulator allogeneic MSCs in 1-way in-vitro MLRs to measure the immunogenicity of MSCs. However, several studies have demonstrated that in-vitro MLR assays are poor predictors of in-vivo immunogenicity. 42,48 In contrast, preformed T-cell responses measured using ELISPOTs accurately predict graft rejection in organ transplantation cases and may be able to predict the in vivo cell-mediated immunogenicity of donor MSCs. 49 Cytotoxicity assays can be used to measure direct lysis of MSCs by MHC-specific cytotoxic T lymphocytes (CTLs). An alternative approach are microcytotoxicity assays where an eosin or fluorescent dye is used to detect antibody-mediated complement-dependent cytotoxicity following incubation of sera from animals injected with MHC-matched or MHC-mismatched MSCs with donor PBLs or MSCs and rabbit complement.
Strategies to Reduce Stem Cell Immunogenicity
Several different approaches exist which may reduce stem cell immunogenicity and enable wider use of this therapeutic approach. These are the following: The creation of human ESC cell banks with a diversity of HLA antigens to provide a match for patients Knockdown of MHC expression of stem cells; however, this may leave therapeutic cells vulnerable to NK cell killing The use of stem cells which produce immunomodulatory molecules, for example, iPSC mesoangioblasts Immunosuppression by drugs. However, long-term use increases the risk of cancer, infection, and organ toxicity (eg, kidney). Short-term use may allow host immune regulation to enable graft survival Use of nondepleting antibodies to block co-stimulatory molecules, for example, CTLA-4 or CD40L Ex vivo expansion of induced, autologous regulatory T cells to control CD8+ T cells and/or macrophage responses Use of mechanisms which induce and maintain central and peripheral tolerance to donor alloantigens, as is used in transplant medicine. This could include mixed or full chimerism (donor and recipient hematopoietic cells) in an HLA-mismatched recipient. However, this requires significant conditioning using immunosuppressive pharmaceuticals Use of same stem cells to differentiate into both source of cells to induce tolerance (eg, immature dendritic cells) and fully differentiated tissue for therapy
Conclusion
Although there have been few successful marketing authorizations for stem cell-based therapies since the Goldring’s paper, the clinical trial evidence suggests that experimental stem cell products are generally safe, but particularly for allogenic therapies, their efficacy may be short lived. One of the key reasons for the short life span of the allogeneic transplanted cells is the host immune response. Sustained therapeutic benefit cannot be achieved through repeat dosing since development of an adaptive immune response with immune memory will limit the ability to repeat dose allogeneic recipients with the same cell product.
Experimental nonclinical work has increased our understanding of the relative immunogenicity of different stem cell–derived transplants and is becoming more clinically relevant. For example, differentiation of hESC enhances their immunogenicity and hypoimmunogenic iPSCs can evade immune rejection in allogeneic recipients. Currently, although genetic modification of allogeneic iPSCs can reduce their immunogenicity, there is still generally a need for local or systemic immune suppression of the recipient for successful engraftment. Some investigators have tried to address this issue through introduction of a physical barrier, as opposed to a pharmacological intervention, between administered allogeneic cells and the recipient cellular immune response. However, the clinical experience has shown that the capsules are often the target of the innate immune system which manifests as a foreign body giant cell response.
We anticipate that increased understanding and modification of immunogenic epitopes of administered allogeneic cells, alongside improved delivery techniques which do not breach the immune privilege of administration sites in the recipient will extend clinical successes beyond short-term MSC therapy.
Footnotes
Acknowledgments
The authors thank Paul Fairchild for his useful suggestions on the scope of this manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.